
Viral and Host Small RNA Response to SARS-CoV-2 Infection

 , ,
, ,
Abstract
:1. Introduction
2. Results
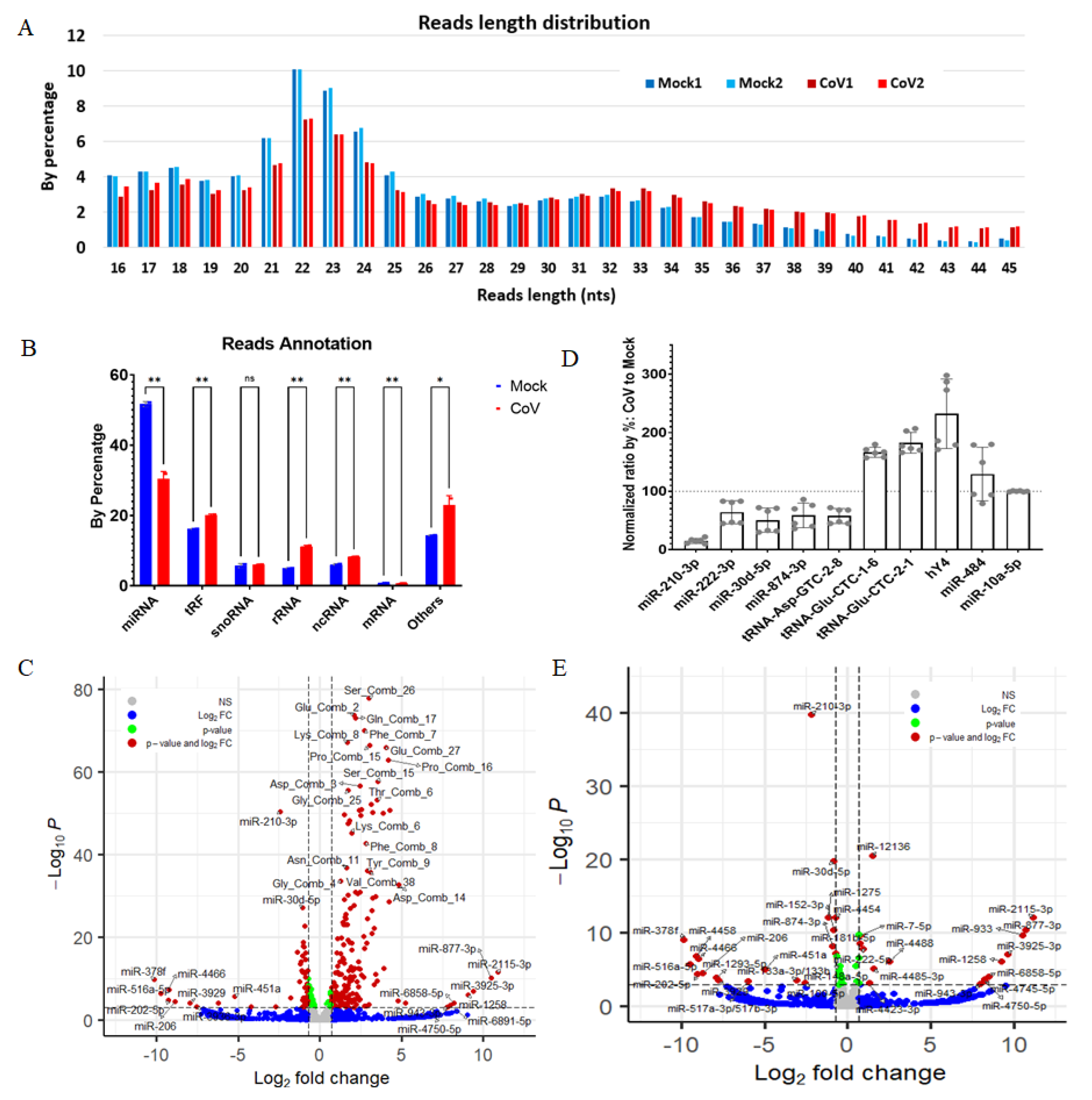
2.1. SARS-CoV-2 Infection Dysregulates Host Small RNAs in HEK293T-hACE2 Cells
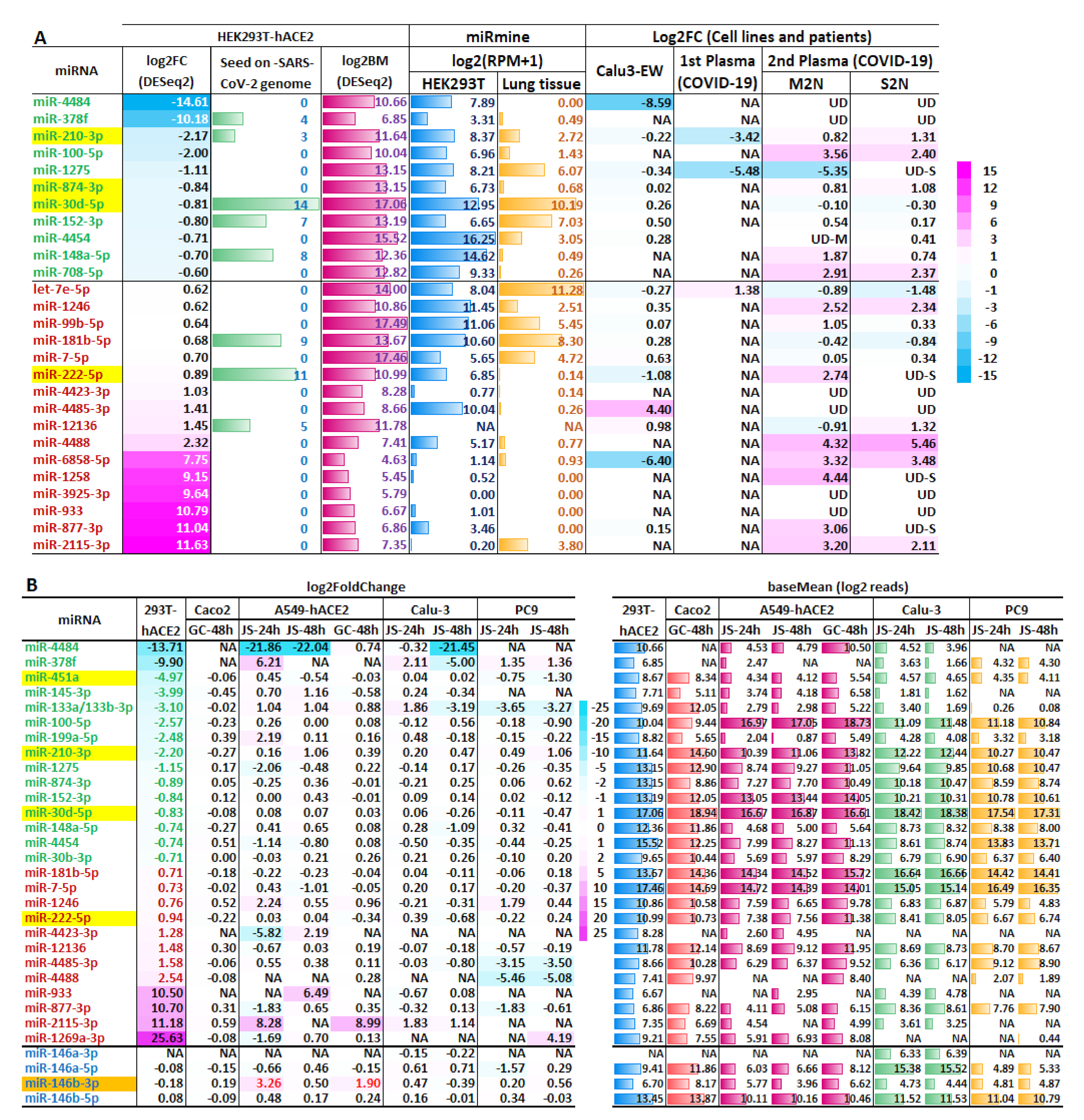
2.2. Host miRNAs Respond to SARS-CoV-2 Infection in HEK293T-hACE2 Cells
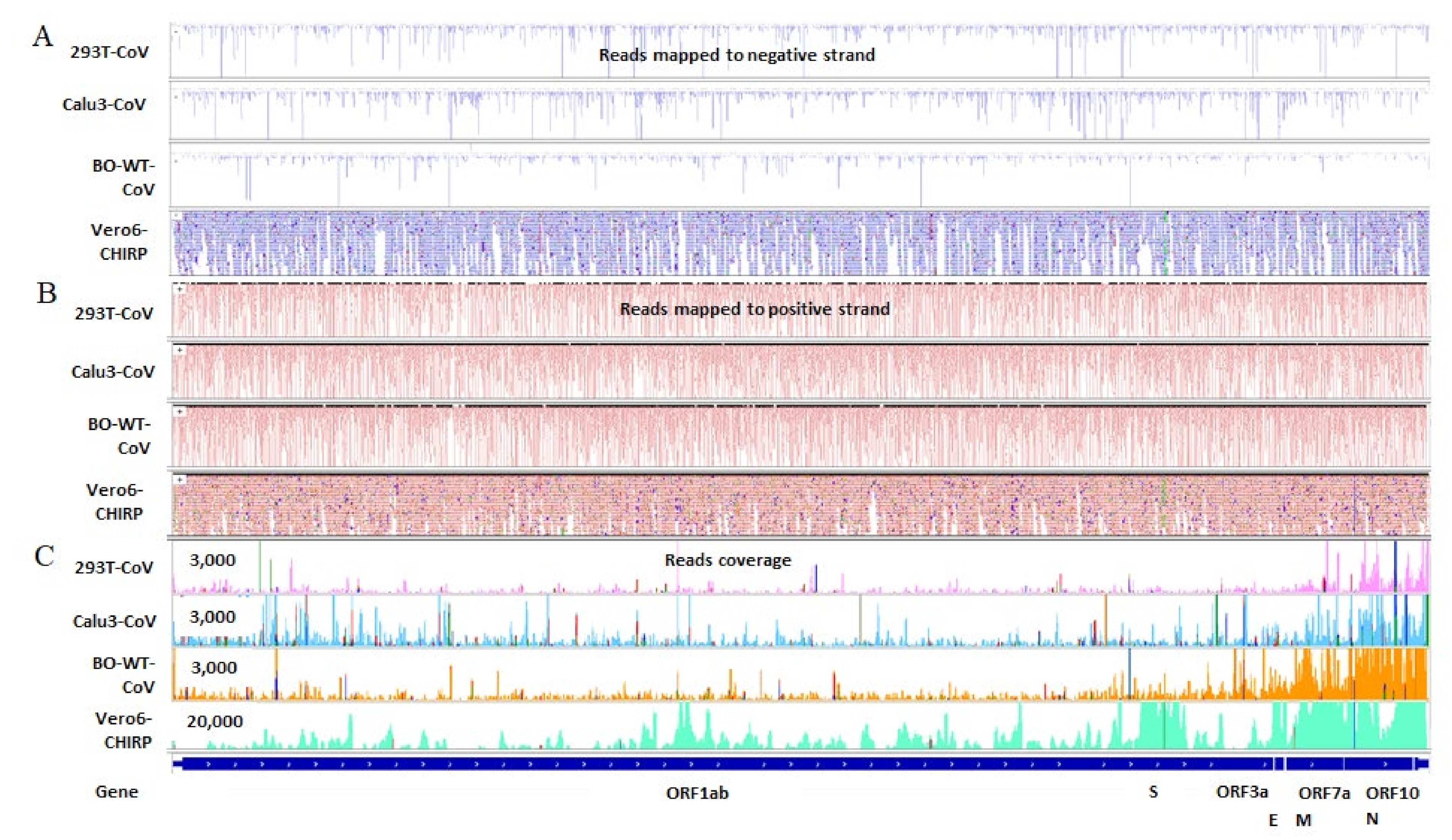
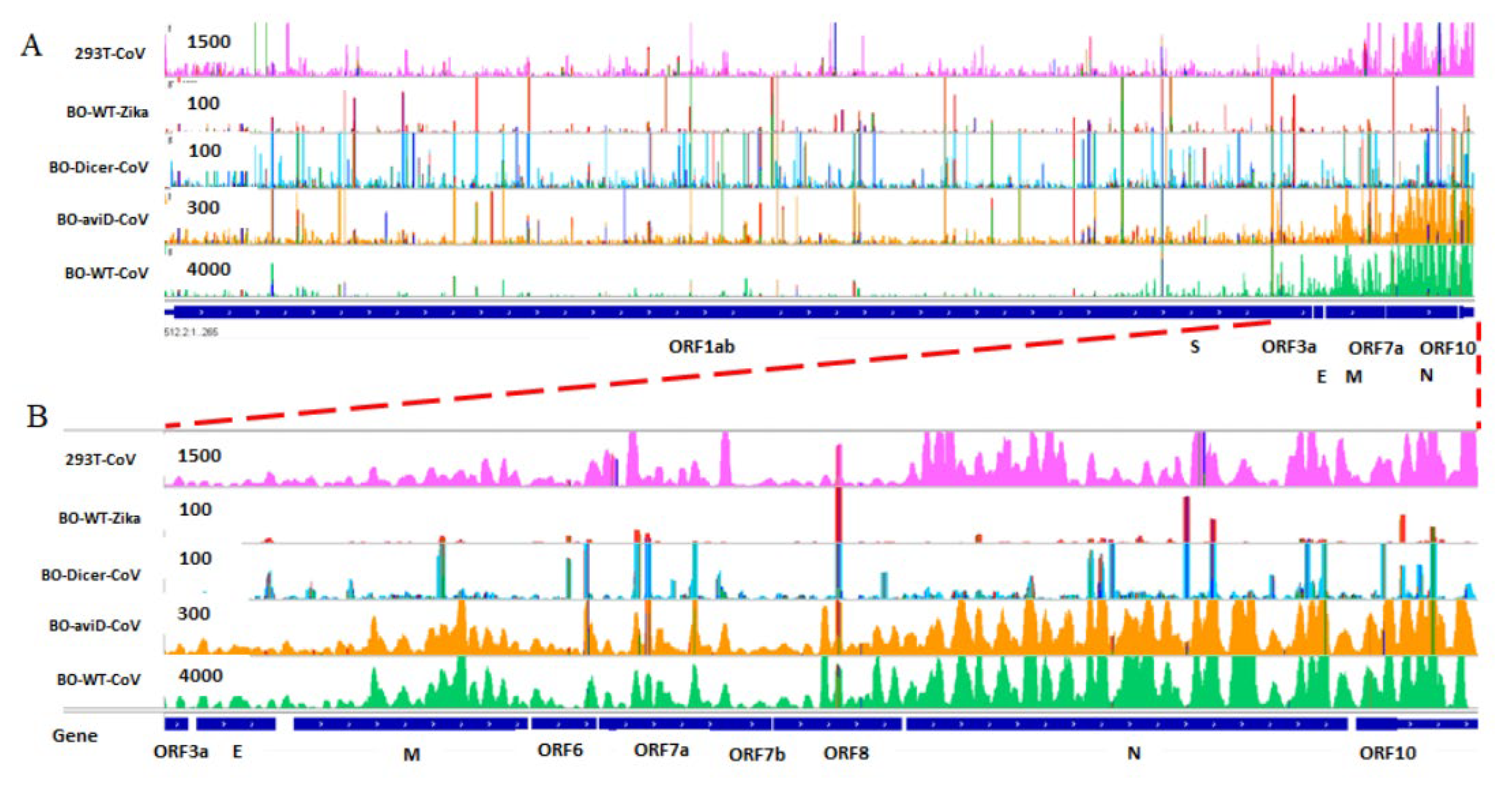
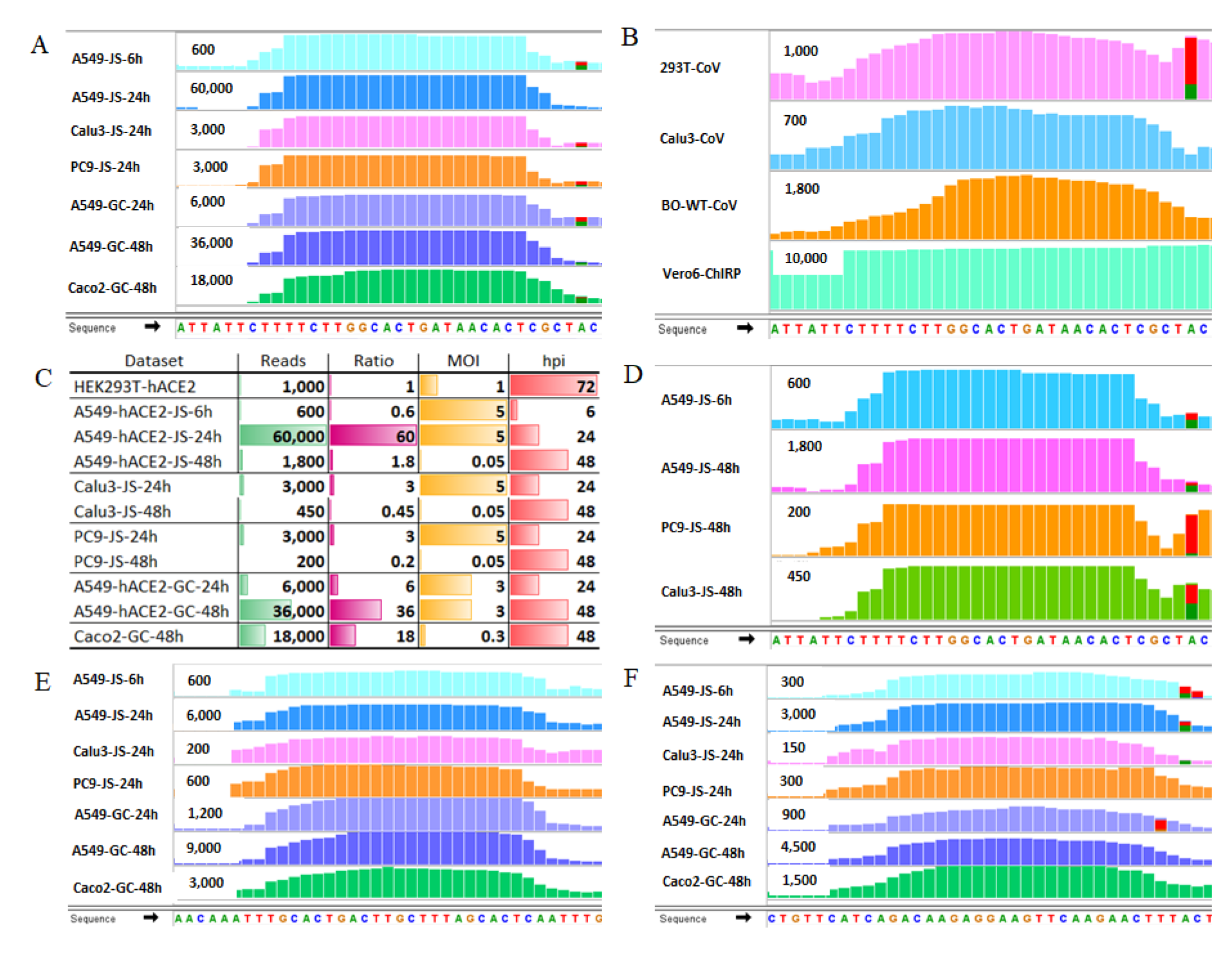
2.3. SARS-CoV-2 Infection Produces vsmRNAs in HEK293T Cells
2.4. The Biogenesis of vsmRNAs and vmiR-ORF7a
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. SARS-CoV-2 Infection, Expansion, and Quantification of Viral RNA
4.3. Plasmids and In Vitro Transcripts Were Used for Reporter Assay
4.4. Small RNA Deep Sequencing Dataset and Raw Data Processing
4.5. smRNAseq Data Analysis (Length Distribution, Reads Count, Annotation)
4.6. Small RNA qRT-PCR
4.7. SARS-CoV-2 ChIRP Data Analysis
4.8. DESeq2 Analysis of Differtially Expressed Small RNAs
4.9. IGV View of vsmRNAs
4.10. Published Small RNA Profile Datasets
4.11. miRDB Identification of miRNA Seed Complementary Sequences in SARA-CoV-2 Genome
4.12. Other Bioinoformatics Tools Used for Data Analysis and Plots
5. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maranini, B.; Ciancio, G.; Ferracin, M.; Cultrera, R.; Negrini, M.; Sabbioni, S.; Govoni, M. microRNAs and Inflammatory Immune Response in SARS-CoV-2 Infection: A Narrative Review. Life 2022, 12, 288. [Google Scholar] [CrossRef]
- Arghiani, N.; Nissan, T.; Matin, M.M. Role of microRNAs in COVID-19 with implications for therapeutics. Biomed. Pharmacother. 2021, 144, 112247. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Vazquez, L.A.B.; Reyes-Perez, P.R.; Estrada-Meza, C.; Alburquerque, R.A.A.; Pathak, S.; Banerjee, A.; Bandyopadhyay, A.; Chakraborty, S.; Srivastava, A. The role of microRNAs in solving COVID-19 puzzle from infection to therapeutics: A mini-review. Virus Res. 2022, 308, 198631. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Li, Y.; Ding, S.W. Small RNA-based antimicrobial immunity. Nat. Rev. Immunol. 2019, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Cullen, B.R. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 2009, 23, 1151–1164. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, J. The pandemic pipeline. Nat. Biotechnol. 2020, 38, 523–532. [Google Scholar] [CrossRef]
- Uludag, H.; Parent, K.; Aliabadi, H.M.; Haddadi, A. Prospects for RNAi Therapy of COVID-19. Front. Bioeng. Biotechnol. 2020, 8, 916. [Google Scholar] [CrossRef]
- Mu, J.; Xu, J.; Zhang, L.; Shu, T.; Wu, D.; Huang, M.; Ren, Y.; Li, X.; Geng, Q.; Xu, Y.; et al. SARS-CoV-2-encoded nucleocapsid protein acts as a viral suppressor of RNA interference in cells. Sci. China Life Sci. 2020, 63, 1413–1416. [Google Scholar] [CrossRef] [Green Version]
- Wyler, E.; Mosbauer, K.; Franke, V.; Diag, A.; Gottula, L.T.; Arsie, R.; Klironomos, F.; Koppstein, D.; Honzke, K.; Ayoub, S.; et al. Transcriptomic profiling of SARS-CoV-2 infected human cell lines identifies HSP90 as target for COVID-19 therapy. iScience 2021, 24, 102151. [Google Scholar] [CrossRef]
- Tang, H.; Gao, Y.; Li, Z.; Miao, Y.; Huang, Z.; Liu, X.; Xie, L.; Li, H.; Wen, W.; Zheng, Y.; et al. The noncoding and coding transcriptional landscape of the peripheral immune response in patients with COVID-19. Clin. Transl. Med. 2020, 10, e200. [Google Scholar] [CrossRef] [PubMed]
- Poirier, E.Z.; Buck, M.D.; Chakravarty, P.; Carvalho, J.; Frederico, B.; Cardoso, A.; Healy, L.; Ulferts, R.; Beale, R.; Reis, E.S.C. An isoform of Dicer protects mammalian stem cells against multiple RNA viruses. Science 2021, 373, 231–236. [Google Scholar] [CrossRef]
- Farr, R.J.; Rootes, C.L.; Rowntree, L.C.; Nguyen, T.H.O.; Hensen, L.; Kedzierski, L.; Cheng, A.C.; Kedzierska, K.; Au, G.G.; Marsh, G.A.; et al. Altered microRNA expression in COVID-19 patients enables identification of SARS-CoV-2 infection. PLoS Pathog. 2021, 17, e1009759. [Google Scholar] [CrossRef] [PubMed]
- Grehl, C.; Schultheiss, C.; Hoffmann, K.; Binder, M.; Altmann, T.; Grosse, I.; Kuhlmann, M. Detection of SARS-CoV-2 Derived Small RNAs and Changes in Circulating Small RNAs Associated with COVID-19. Viruses 2021, 13, 1593. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Siu, G.K.; Mok, B.W.; Sun, J.; Fung, K.S.C.; Lam, J.Y.; Wong, N.K.; Gedefaw, L.; Luo, S.; Lee, T.M.H.; et al. Viral MicroRNAs Encoded by Nucleocapsid Gene of SARS-CoV-2 Are Detected during Infection, and Targeting Metabolic Pathways in Host Cells. Cells 2021, 10, 1762. [Google Scholar] [CrossRef]
- Kim, D.; Kim, S.; Park, J.; Chang, H.R.; Chang, J.; Ahn, J.; Park, H.; Park, J.; Son, N.; Kang, G.; et al. A high-resolution temporal atlas of the SARS-CoV-2 translatome and transcriptome. Nat. Commun. 2021, 12, 5120. [Google Scholar] [CrossRef]
- Singh, M.; Chazal, M.; Quarato, P.; Bourdon, L.; Malabat, C.; Vallet, T.; Vignuzzi, M.; van der Werf, S.; Behillil, S.; Donati, F.; et al. A virus-derived microRNA targets immune response genes during SARS-CoV-2 infection. EMBO Rep. 2021, 23, e54341. [Google Scholar] [CrossRef] [PubMed]
- Pawlica, P.; Yario, T.A.; White, S.; Wang, J.; Moss, W.N.; Hui, P.; Vinetz, J.M.; Steitz, J.A. SARS-CoV-2 expresses a microRNA-like small RNA able to selectively repress host genes. Proc. Natl. Acad. Sci. USA 2021, 118, e2116668118. [Google Scholar] [CrossRef] [PubMed]
- Farr, R.J.; Rootes, C.L.; Stenos, J.; Foo, C.H.; Cowled, C.; Stewart, C.R. Detection of SARS-CoV-2 infection by microRNA profiling of the upper respiratory tract. PLoS ONE 2022, 17, e0265670. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hu, X.; Li, L.; Li, J.H. Differential microRNA expression in the peripheral blood from human patients with COVID-19. J. Clin. Lab. Anal. 2020, 34, e23590. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Wu, X.; Wang, J.; Li, H.; Li, X.; Gao, H.; Rossi, J.; Yen, Y. A bias-reducing strategy in profiling small RNAs using Solexa. RNA 2011, 17, 2256–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, G.; Cheng, Y.W.; Lai, L.; Huang, T.C.; Wang, J.; Wu, X.; Wang, Y.; Huang, Y.; Wang, J.; Zhang, K.; et al. Signature miRNAs in colorectal cancers were revealed using a bias reduction small RNA deep sequencing protocol. Oncotarget 2016, 7, 3857–3872. [Google Scholar] [CrossRef] [Green Version]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef] [Green Version]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, G.; Cui, Q.; Garcia, G., Jr.; Wang, C.; Zhang, M.; Arumugaswami, V.; Riggs, A.D.; Shi, Y. Comparative transcriptomic analysis of SARS-CoV-2 infected cell model systems reveals differential innate immune responses. Sci. Rep. 2021, 11, 17146. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e910. [Google Scholar] [CrossRef]
- Shapiro, J.S.; Langlois, R.A.; Pham, A.M.; Tenoever, B.R. Evidence for a cytoplasmic microprocessor of pri-miRNAs. RNA 2012, 18, 1338–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouha, H.; Thurner, C.; Mandl, C.W. Functional microRNA generated from a cytoplasmic RNA virus. Nucleic Acids Res. 2010, 38, 8328–8337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, R.A.; Belk, J.A.; Qi, Y.; Yasumoto, Y.; Wei, J.; Alfajaro, M.M.; Shi, Q.; Mumbach, M.R.; Limaye, A.; DeWeirdt, P.C.; et al. Discovery and functional interrogation of SARS-CoV-2 RNA-host protein interactions. Cell 2021, 184, 2394–2411.e2316. [Google Scholar] [CrossRef]
- Panwar, B.; Omenn, G.S.; Guan, Y. miRmine: A database of human miRNA expression profiles. Bioinformatics 2017, 33, 1554–1560. [Google Scholar] [CrossRef]
- Huang, X.; Le, Q.T.; Giaccia, A.J. MiR-210--micromanager of the hypoxia pathway. Trends Mol. Med. 2010, 16, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitsure, M.; Sarangi, B.; Shankar, G.H.; Reddy, V.S.; Walimbe, A.; Sharma, V.; Prayag, S. Mechanisms of Hypoxia in COVID-19 Patients: A Pathophysiologic Reflection. Indian J. Crit. Care Med. 2020, 24, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Hennig, T.; Prusty, A.B.; Kaufer, B.B.; Whisnant, A.W.; Lodha, M.; Enders, A.; Thomas, J.; Kasimir, F.; Grothey, A.; Klein, T.; et al. Selective inhibition of miRNA processing by a herpesvirus-encoded miRNA. Nature 2022, 605, 539–544. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Castelli, G.; Labbaye, C. miR-146 and miR-155: Two Key Modulators of Immune Response and Tumor Development. Noncoding RNA 2017, 3, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parameswaran, P.; Sklan, E.; Wilkins, C.; Burgon, T.; Samuel, M.A.; Lu, R.; Ansel, K.M.; Heissmeyer, V.; Einav, S.; Jackson, W.; et al. Six RNA viruses and forty-one hosts: Viral small RNAs and modulation of small RNA repertoires in vertebrate and invertebrate systems. PLoS Pathog. 2010, 6, e1000764. [Google Scholar] [CrossRef] [Green Version]
- Ziv, O.; Price, J.; Shalamova, L.; Kamenova, T.; Goodfellow, I.; Weber, F.; Miska, E.A. The Short- and Long-Range RNA-RNA Interactome of SARS-CoV-2. Mol. Cell 2020, 80, 1067–1077.e1065. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, J.; Cheng, H.; Ke, X.; Sun, L.; Zhang, Q.C.; Wang, H.W. Cryo-EM Structure of Human Dicer and Its Complexes with a Pre-miRNA Substrate. Cell 2018, 173, 1549–1550. [Google Scholar] [CrossRef]
- Mousavi, S.R.; Sajjadi, M.S.; Khosravian, F.; Feizbakhshan, S.; Salmanizadeh, S.; Esfahani, Z.T.; Beni, F.A.; Arab, A.; Kazemi, M.; Shahzamani, K.; et al. Dysregulation of RNA interference components in COVID-19 patients. BMC Res. Notes 2021, 14, 401. [Google Scholar] [CrossRef]
- Sharma, A.; Garcia, G., Jr.; Wang, Y.; Plummer, J.T.; Morizono, K.; Arumugaswami, V.; Svendsen, C.N. Human iPSC-Derived Cardiomyocytes Are Susceptible to SARS-CoV-2 Infection. Cell Rep. Med. 2020, 1, 100052. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, M.; Garcia, G., Jr.; Tian, E.; Cui, Q.; Chen, X.; Sun, G.; Wang, J.; Arumugaswami, V.; Shi, Y. ApoE-Isoform-Dependent SARS-CoV-2 Neurotropism and Cellular Response. Cell Stem Cell 2021, 28, 331–342.e335. [Google Scholar] [CrossRef]
- Abbott, T.R.; Dhamdhere, G.; Liu, Y.; Lin, X.; Goudy, L.; Zeng, L.; Chemparathy, A.; Chmura, S.; Heaton, N.S.; Debs, R.; et al. Development of CRISPR as an Antiviral Strategy to Combat SARS-CoV-2 and Influenza. Cell 2020, 181, 865–876.e812. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Lu, Y.; Baras, A.S.; Halushka, M.K. miRge 2.0 for comprehensive analysis of microRNA sequencing data. BMC Bioinform. 2018, 19, 275. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.H.; Halushka, M.K. miRge3.0: A comprehensive microRNA and tRF sequencing analysis pipeline. NAR Genom. Bioinform. 2021, 3, lqab068. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Zhang, X.; Liu, H.; Wang, Z.; Zhong, J.; Huang, Z.; Peng, X.; Zeng, Y.; Wang, Y.; Yang, Y.; et al. A novel real-time PCR assay of microRNAs using S-Poly(T), a specific oligo(dT) reverse transcription primer with excellent sensitivity and specificity. PLoS ONE 2012, 7, e48536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Samples of Infection | MOI | hpi | GEO | Reference |
|---|---|---|---|---|---|
| HEK293T-Mock * | Mock | N/A | 72 | GSE189920 | This study |
| HEK293T-CoV * | SARS-CoV-2 | 1 | 72 | ||
| Calu3-CoV/Calu3-EW | SARS-CoV-2 | 0.33 | 12 and 24 | GSE148729 | [10] |
| Vero6-ChIRP | SARS-CoV-2 | 0.01 | 48 | GSE167341 | [29] |
| BO-WT-CoV | SARS-CoV-2 | ** | 96 | GSE173946 | [12] |
| BO-aviD-CoV | SARS-CoV-2 | ||||
| BO-Dicer-CoV | SARS-CoV-2 | ||||
| BO-WT-Zika | Zika virus | *** | 96 | ||
| A549-JS-Mock * | Mock | N/A | N/A | GSE183280 | [18] |
| A549-JS-6 h * | SARS-CoV-2 | 5 | 6 | ||
| A549-JS-24 h * | SARS-CoV-2 | 5 | 24 | ||
| A549-JS-48 h * | SARS-CoV-2 | 0.05 | 48 | ||
| Calu3-JS-Mock | Mock | N/A | N/A | ||
| Calu3-JS-24 h | SARS-CoV-2 | 5 | 24 | ||
| Calu3-JS-48 h | SARS-CoV-2 | 0.05 | 48 | ||
| PC9-JS-Mock | Mock | N/A | N/A | ||
| PC9-JS-24h | SARS-CoV-2 | 5 | 24 | ||
| PC9-JS-48h | SARS-CoV-2 | 0.05 | 48 | ||
| A549-GC-Mock * | Mock | N/A | 24 & 48 | GSE162318 | [17] |
| A549-GC-24 h * | SARS-CoV-2 | 3 | 24 | ||
| A549-GC-48 h * | SARS-CoV-2 | 3 | 48 | ||
| Caco2-GC-Mock | Mock | N/A | 24 & 48 | ||
| Caco2-GC-24 h | SARS-CoV-2 | 0.3 | 24 | ||
| Caco2-GC-48 h | SARS-CoV-2 | 0.3 | 48 | ||
| COVID-19 # | Plasma of COVID-19 | N/A | N/A | N/A | [13] |
| Non-CoV (n-CoV) ## | Plasma of non-COVID-19 | N/A | N/A | N/A, Web download | [14] |
| Mild-CoV (m-CoV) ## | Plasma of mild COVID-19 | N/A | N/A | ||
| Severe-CoV (s-CoV) ## | Plasma of severe COVID-19 | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, G.; Cui, Q.; Garcia, G., Jr.; Lizhar, E.M.; Arumugaswami, V.; Shi, Y.; Riggs, A.D. Viral and Host Small RNA Response to SARS-CoV-2 Infection. Microbiol. Res. 2022, 13, 788-808. https://doi.org/10.3390/microbiolres13040056
Sun G, Cui Q, Garcia G Jr., Lizhar EM, Arumugaswami V, Shi Y, Riggs AD. Viral and Host Small RNA Response to SARS-CoV-2 Infection. Microbiology Research. 2022; 13(4):788-808. https://doi.org/10.3390/microbiolres13040056
Chicago/Turabian StyleSun, Guihua, Qi Cui, Gustavo Garcia, Jr., Elizabeth M. Lizhar, Vaithilingaraja Arumugaswami, Yanhong Shi, and Arthur D. Riggs. 2022. "Viral and Host Small RNA Response to SARS-CoV-2 Infection" Microbiology Research 13, no. 4: 788-808. https://doi.org/10.3390/microbiolres13040056
APA StyleSun, G., Cui, Q., Garcia, G., Jr., Lizhar, E. M., Arumugaswami, V., Shi, Y., & Riggs, A. D. (2022). Viral and Host Small RNA Response to SARS-CoV-2 Infection. Microbiology Research, 13(4), 788-808. https://doi.org/10.3390/microbiolres13040056