1. Introduction
Worldwide, more than 750 million people have no access to safe drinking water [
1]. More than 250 million inhabitants are regularly exposed to water contaminated with arsenic [
2,
3], mostly in south east Asia [
4] and in Europe [
5]. In addition, a big part of world population is consuming drinking water with significant Cr(VI) concentrations [
6,
7], while on many occasions arsenic and chromate have been found to occur simultaneously in water or wastewater sources [
8].
Arsenic is a very toxic and carcinogenic element and its removal from drinking water is of crucial concern because chronic exposure of humans to arsenic contaminated drinking water can cause several forms of cancer and other diseases [
9,
10]. Arsenic is present mainly in groundwaters with its inorganic forms of arsenate, the pentavalent form or arsenic, As(V) and of arsenite, which is the trivalent form of arsenic, As(III) [
11]. In anoxic waters reducing conditions prevail and arsenic speciation is dominated by the presence of As(III). In oxic waters the oxidized form or arsenic, As(V), is mainly present [
12,
13]. The WHO, EU and US-EPA concentration limit of arsenic in drinking water is 10 μg/L.
The removal of arsenic is mainly accomplished by a pre-oxidation step to transform As(III) to As(V) and then conventional technologies are applied to remove As(V) [
14]. The main technologies applied worldwide are coagulation with Fe(III) or Al(III) salts and adsorption on iron oxide based adsorbents such as granular ferric hydroxide, bayoxide, aquAszero, zero valent iron. Aluminium based adsorbents are also applied, such as activated alumina or novel Al-based coagulants [
5,
15,
16].
Another major contamination problem is the presence of Cr(VI) in groundwaters worldwide [
17]. Among the affected countries belongs Greece [
6]. The limit for total chromium in drinking water is 100 μg/L in the USA and 50 μg/L in EU. However, in 2014, the Environmental Protection Agency of California has set a new limit, specifically for Cr(VI), at 10 μg/L, (which has been for the moment withdrawn and will be re-discussed because it was judged that the cost for implementing this limit was unpredictable and could be prohibitive) [
18]. At the same time, the European Union is planning to adopt a limit of 25 μg/L for Cr(VI) in drinking water, from 50 μg/L total chromium in drinking water, which is still now active [
19]. Besides, in Greece, there is a specific limit of 3 μg/L in surface inland waters, as it is written clearly in the national law (ΦΕΚ 1909/Β/08-12-2010, Annex I, Part B). The result will be that many waters will need to be subjected to additional treatment, in order to contain lower, than the limit, concentration of Cr(VI) in the finished water.
Chromium is present in waters in two forms. The first form is the hexavalent chromium anions (i.e. CrO
42− or Cr
2O
72−) which are extremely toxic to humans and very soluble in waters. The second species is the trivalent chromium (Cr(OH)
3) which is not toxic [
20] and has limited solubility in waters (Log Ksp of CrOH
3 = −32.2) [
21], mainly at circumneutral pH values [
6]. The main form of chromium found in groundwaters is the hexavalent species, which is mostly present in waters where oxidizing conditions prevail. Therefore, in Cr(VI) containing waters, when arsenic is also present, it is mostly found with the oxidized form of As(V) [
8].
Chemical reduction of Cr(VI) to Cr(III) followed by precipitation is the most commonly applied technology for the removal of Cr(VI) from natural waters or wastewaters [
22,
23]. In particular, when Cr(VI) reacts with Fe(II) salts, Cr(VI) is reduced to Cr(III) and Fe(II) is oxidized to Fe(III). Under oxygenated conditions and in pH values relevant to drinking water treatment (i.e., 6.5 to 8) Fe(III) forms insoluble hydroxides and Cr(III) forms Cr
(III)(OH)
3 species, which can either precipitate due to low solubility of Cr
(III)(OH)
3 or adsorb and precipitate on the insoluble iron hydroxides [
24,
25]. The following reactions describe well the removal of Cr(VI) by Fe(II) [
23].
These reactions, show that a molar stoichiometry [Fe]/[Cr] of 3 is needed in order to reduce all Cr(VI) in solution. However, because at neutral pH values and in oxygenated waters Fe(II) is oxidized quite fast by oxygen as well, usually a much higher molar ratio is required in order to outcompete the reaction of Fe(II) with oxygen and achieve very efficient removal of Cr(VI). As already well established, Fe(II) kinetics of oxidation by O
2 is very much pH dependent and increases as the pH increases [
26].
The above information however shows that different technologies are generally applied for arsenic and chromate removal, if they are not present simultaneously in waters. Ventura-Hoyle et al., [
8] showed that there are cases, where arsenic and chromate are simultaneously present in water.
Previous studies have examined the simultaneous removal of As(V) and Cr(VI) with several technologies, most of which are still in the research phase, such as layered double hydroxides intercalated with MoS
42− [
27], metal organic frameworks [
28], malachite nanoparticles [
29], or the use of electrocatalysis in acid aqueous solutions [
30]. As of now, only one study, conducted in two parts [
31,
32] has investigated the simultaneous removal of As(V) and Cr(VI) by the so called Fe(II) coagulation method. This study found that Fe(II) can well remove both of these contaminants under certain conditions. In that study authors have mainly examined the effects of solution pH, Fe(II) dosage and initial Cr(VI)/As(V) ratio in batch tests [
31] and conducted spectroscopic investigations [
32]. However, in these works the applied contaminant concentrations were much higher than those normally found in groundwaters (i.e., for arsenic the examined concentrations were in based on concentrations of about 10 to 20 μmol/L, which corresponds to about 750–1500 μg/L) and chromate at concentration of 10 μmol/L, corresponding to about 520 μg/L. Furthermore, in the studies [
31,
32] the experiments were conducted in pure water with background electrolyte and therefore, data on applicability of this technology in real water applications was missing, whereas in our work we have examined the application of this technology in real waters, corresponding to ground and surface waters.
Therefore, in our work, we bring the research forward by examining the simultaneous removal of As(V) and Cr(VI) starting from much lower concentrations, i.e., for both arsenate and chromate 50 μg/L (i.e., 0.67 and 0.96 μmol/L for As(V) and Cr(VI) respectively) and focusing on achieving the very low limits in drinking water. We tested the method in real natural waters and compared our findings with results obtained from the use of deionized water. In addition, by conducting experiments in the presence and absence of chromate and by using Fe(II) or Fe(III) salts, we shed light on the mechanism of arsenic removal by Fe(II) under various scenarios, which are realistic in drinking water treatment.
Consequently, this is the first study to investigate in detail the simultaneous As(V) and Cr(VI) removal from both real surface- and ground- waters focusing also on achieving the anticipated new limit for Cr(VI).
3. Results and Discussion
3.1. As(V) and Cr(VI) Removal from Drinking Water by Fe(II) and Comparison with the Use of Conventional Fe(III) Salts at Various Water Matrices
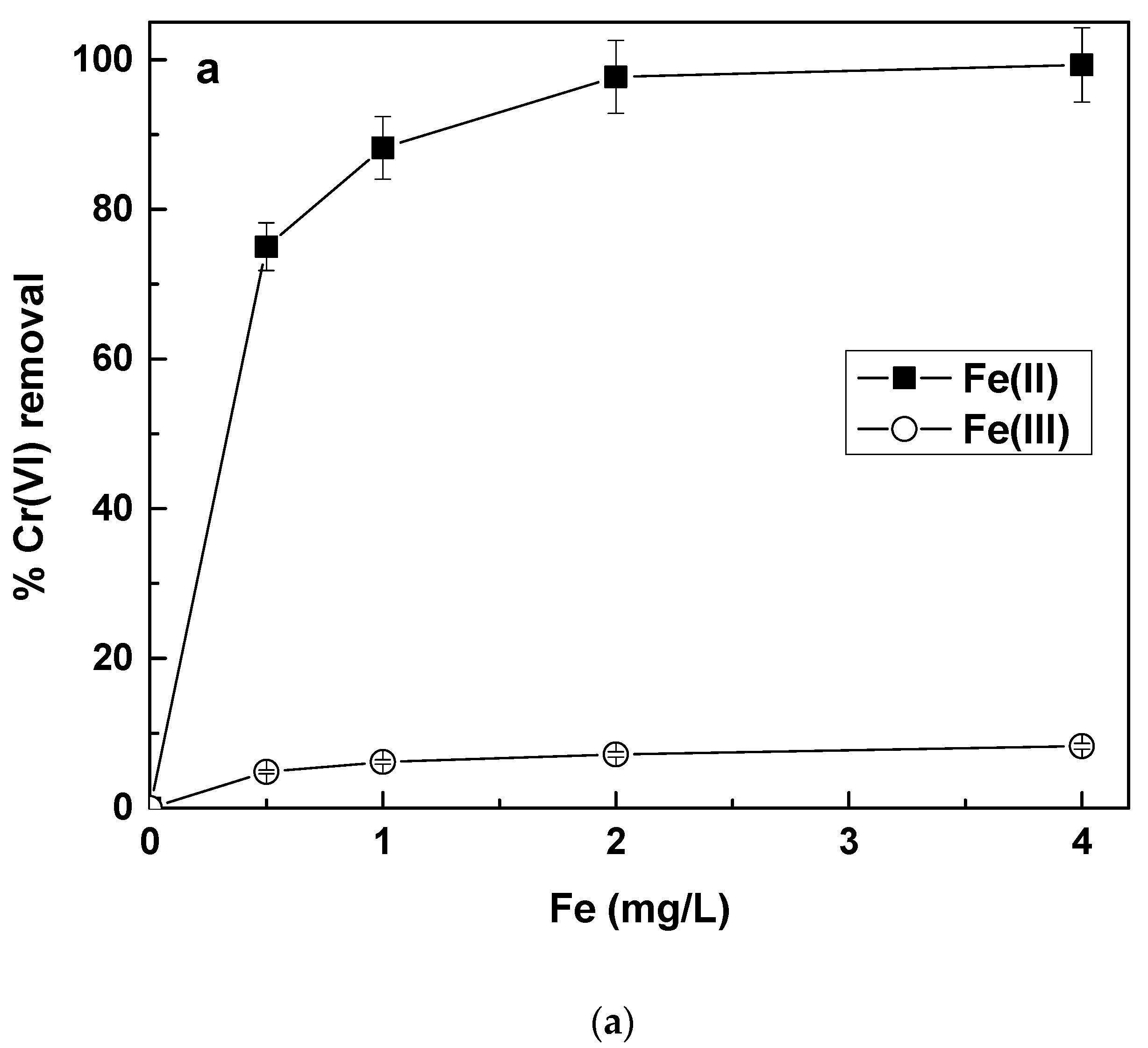
The results regarding the simultaneous removal As(V) and Cr(VI) from natural waters are shown in
Figure 1, for both As(V) and Cr(VI) removal, by application of Fe(II) or Fe(III) salts.
The results show that Fe(II) was quite efficient in removing both As(V) an Cr(VI), but the use of Fe(III) was effective only for the removal of As(V), because Cr(VI) removal by Fe(III) was almost negligible. The reason for the poor Cr(VI) removal by Fe(III) is that Cr(VI), although present in water as an oxyanion (i.e., CrO
42− or Cr
2O
72−) [
23], is not efficiently adsorbed on the positively charged surfaces of iron oxides and it is for this reason that generally needs to be reduced to the more insoluble Cr(III) species. The contrary happens with arsenate oxyanions, which are removed very efficiently by both Fe(II) or Fe(III), as shown in
Figure 1b. In this point, it must be noted that we did not observe any significant pH changes at the end of the experiments, as compared to the initial values.
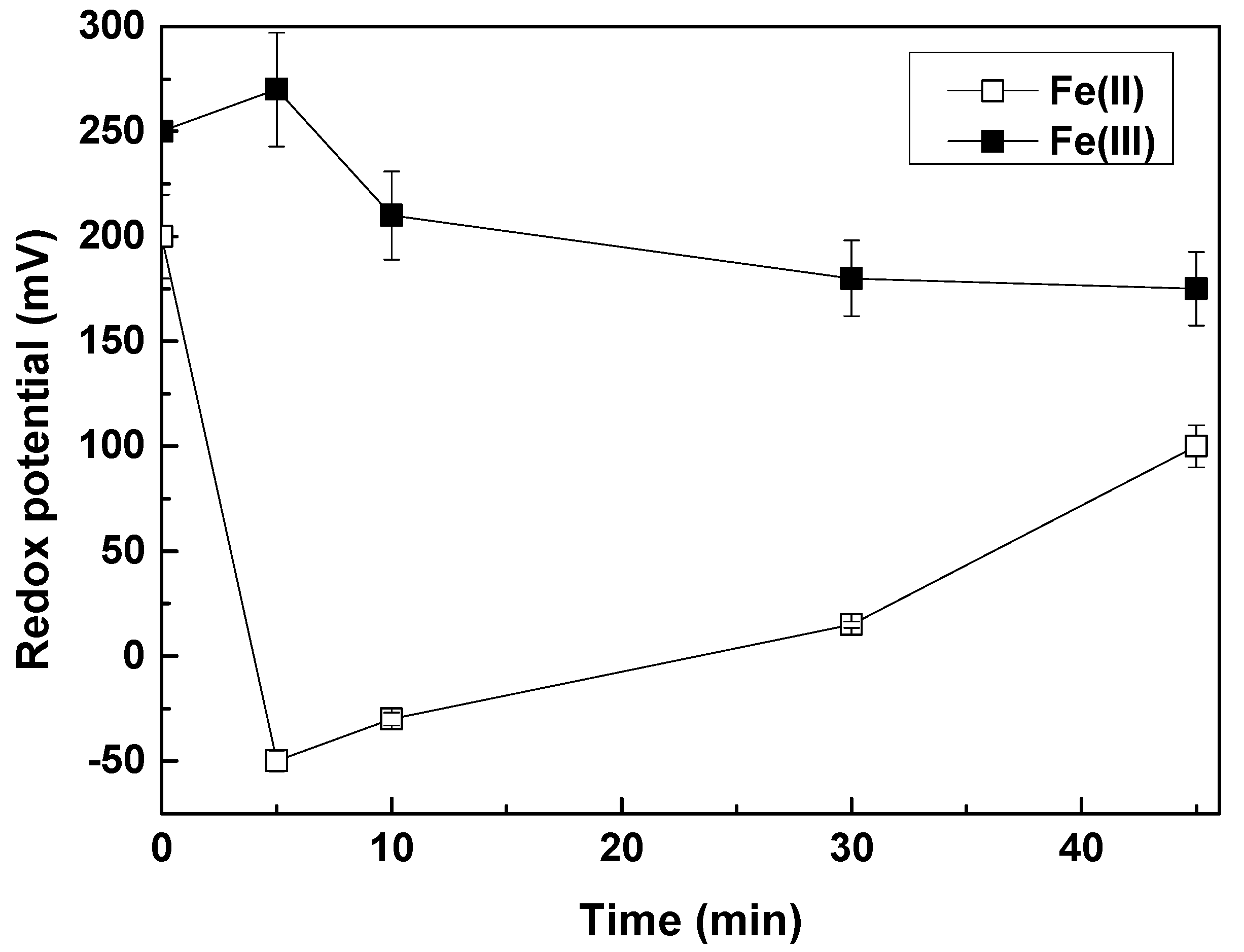
Figure 1a,b show that by applying a concentration of 2 mg/L Fe(II), 90% removal of As(V) and Cr(VI) can be achieved, from 50 μg/L. Final concentrations for both contaminants were far below 10 μg/L. The effectiveness of Fe(II) against Fe(III) for Cr(VI) can be explained by observing the redox potential variations of the water, upon adding Fe(II) or Fe(III) in the water, which are shown in
Figure 2.
The addition of Fe(II) in the water, caused an immediate sharp decrease of redox potential of the water, revealing the favorable conditions enabling the Cr(VI) reduction to Cr(III), as a result of its reaction with Fe(II). In contrast, Fe(III) cannot react with Cr(VI), is not a reductant and this is clearly seen in the redox potential variations, after Fe(III) addition in the solution. After that, and as Fe(II) starts to get oxidized to Fe(III), redox potential starts again to increase.
Furthermore, these results show that in the presence of Cr(VI), Fe(II) is slightly more efficient in removing As(V) than Fe(III), especially at the low Fe doses. This is shown in literature for first time, because researchers up to now did not investigate the application of Fe(III) as compared to the use of Fe(II) in the presence of Cr(VI) for As(V) removal. The presence of Cr(VI) accelerates Fe(II) oxidation and the iron oxides formed in situ by Fe(II) are known to have greater adsorbing capacity than the Fe(III) hydroxides, created by the hydrolysis of iron(III) salts [
35].
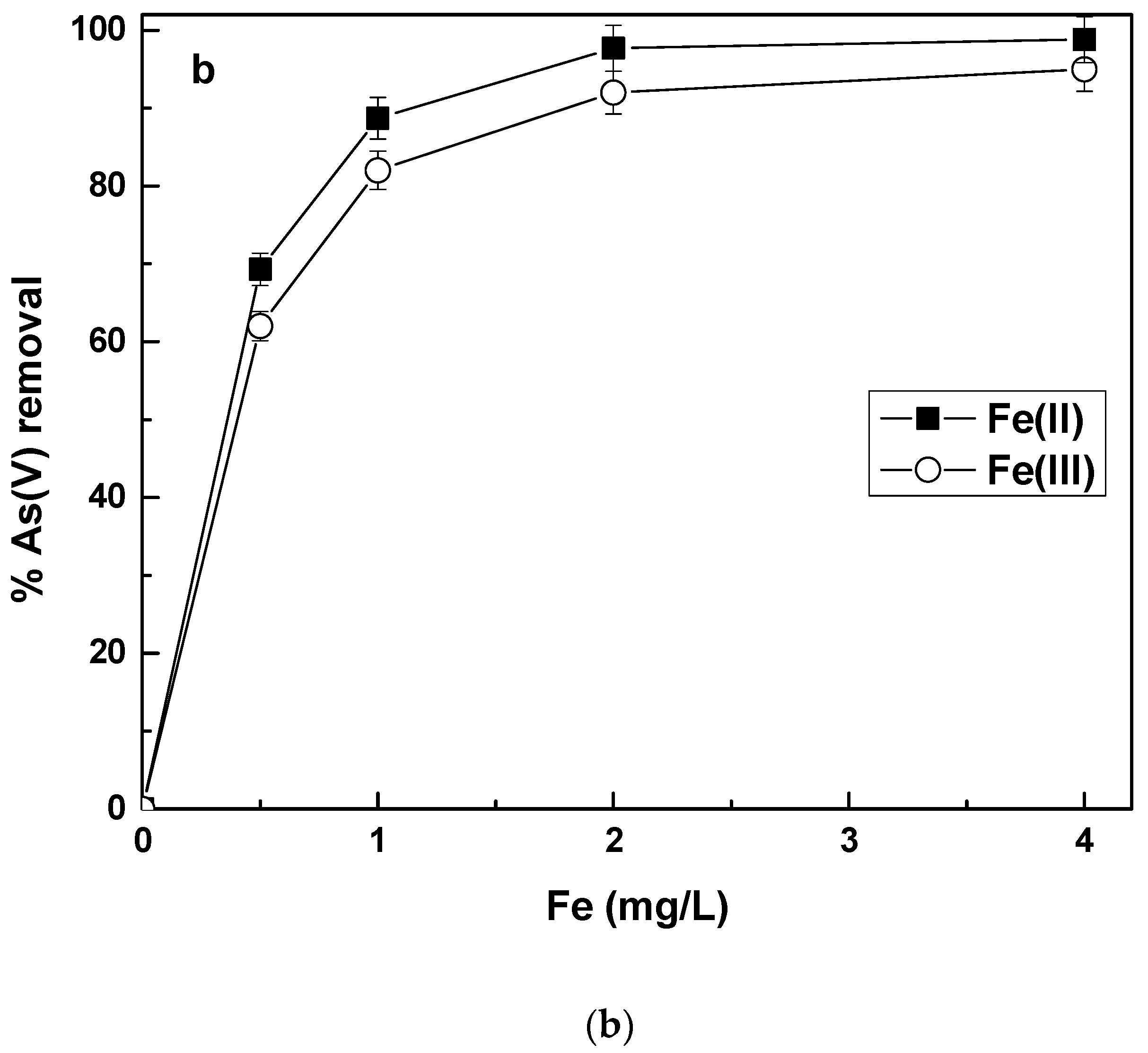
To investigate this finding in more detail, experiments were conducted to remove As(V) by Fe(II) or Fe(III) in the absence of Cr(VI) in solution. The results are shown in
Figure 3.
These results show that in the absence of Cr(VI), Fe(III) was slightly more efficient than Fe(II), especially at the low Fe-doses. (i.e., 0.5 mg/L), which is the opposite of the results obtained in the presence of Cr(VI). Again however, at higher Fe-doses, Fe(II) and Fe(III) were almost equally efficient.
As(V) removal by Fe(II) in the absence of Cr(VI) is based on Fe(II) oxidation caused by oxygen, which is forming subsequently Fe(III) hydroxides able to adsorb and remove As(V) from the water. At neutral pH values, Fe(II) oxidation kinetics by oxygen is fast enough to be completed during the time frame of the experiment and therefore, As(V) removal is almost equally efficient as in the presence of Cr(VI), at the higher Fe(II) doses, but not at the low doses.
At low doses, if some Fe(II) remains unoxidized or is oxidized very late, there are not enough adsorption sites and thus As(V) is not so efficiently absorbed. In the presence of Cr(VI), Fe(II) oxidation kinetics gets faster, because Cr(VI) causes additional Fe(II) oxidation through its reduction to Cr(III), and Fe(III) hydroxides are formed faster.
Therefore, the first conclusion, is that Fe(II) at doses below 1 mg/L, removes more efficiently As(V) than Fe(III) does, when Cr(VI) is present in the water, for contaminant concentrations relevant to drinking water treatment (i.e., both As(V) and Cr(VI) were present at initial concentrations of 50 μg/L). In the absence of Cr(VI), Fe(III) was more efficient than Fe(II) in removing As(V). This difference was eliminated with the application of higher Fe(II) dosages, i.e., at 2 mg/L, which is required for achieving less than 10 μg/L concentrations for both As(V) and Cr(VI). It has to be noted, that these results correspond to experiments conducted at ambient temperatures of 22 °C. Previous studies [
23,
36] have shown that temperature can affect slightly the efficiency of both Cr(VI) reduction and of As(V) removal, since both reactions are endothermic and therefore, increase in temperature increases the efficiency of the processes.
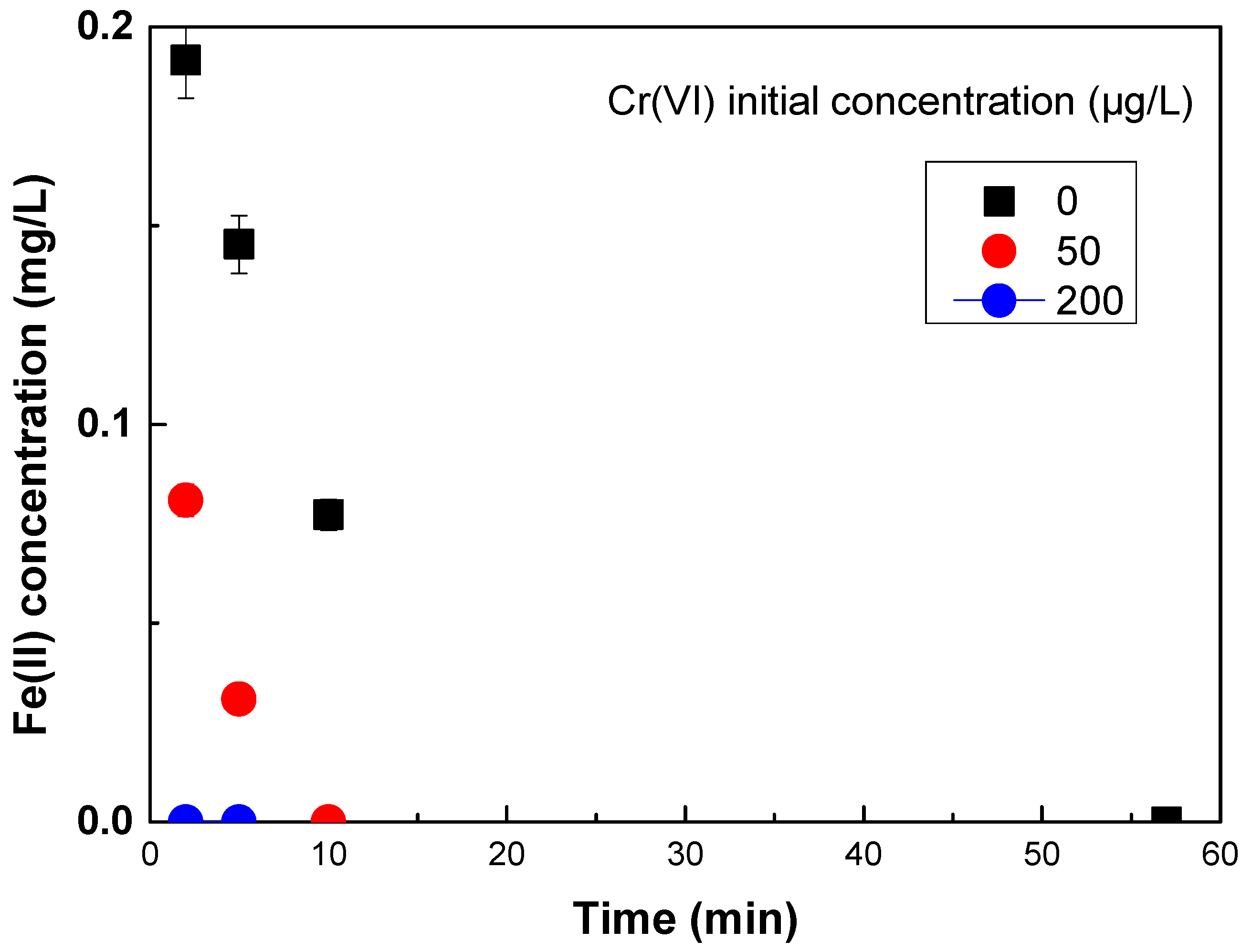
Further on, experiments were conducted in this study to investigate the kinetics of Fe(II) oxidation in the presence or absence of varying concentrations of Cr(VI). The results are shown in
Figure 3 and show that increasing concentrations of Cr(VI) result in faster Fe(II) oxidation at pH 7, which in turn can explain the improved removal of As(V) by Fe(II) in the presence of low concentrations of Cr(VI) at the low Fe(II) doses.
The results shown in
Figure 4, show a quick initial oxidation phase in all cases. From initial Fe(II) concentration of about 0.5 mg/L, immediately after added into the water, even in the absence of Cr(VI), significant oxidation occurs after 1 min and then slows down. In the presence of increasing concentrations of Cr(VI), Fe(II) oxidation gets faster and in the presence of 200 μg/L of Cr(VI), Fe(II) was almost completely and very quickly oxidized to Fe(III), even before the first measurement. These results can explain that the more efficient removal of As(V) for low Fe doses, was observed when Fe(II) was used in the presence of 50 μg/L Cr(VI), attributed to the faster Fe(II) oxidation, which caused faster Fe(III)/oxides formation, with greater sorption capacities than Fe(III) oxides, thus faster and hence more efficient As(V) removal.
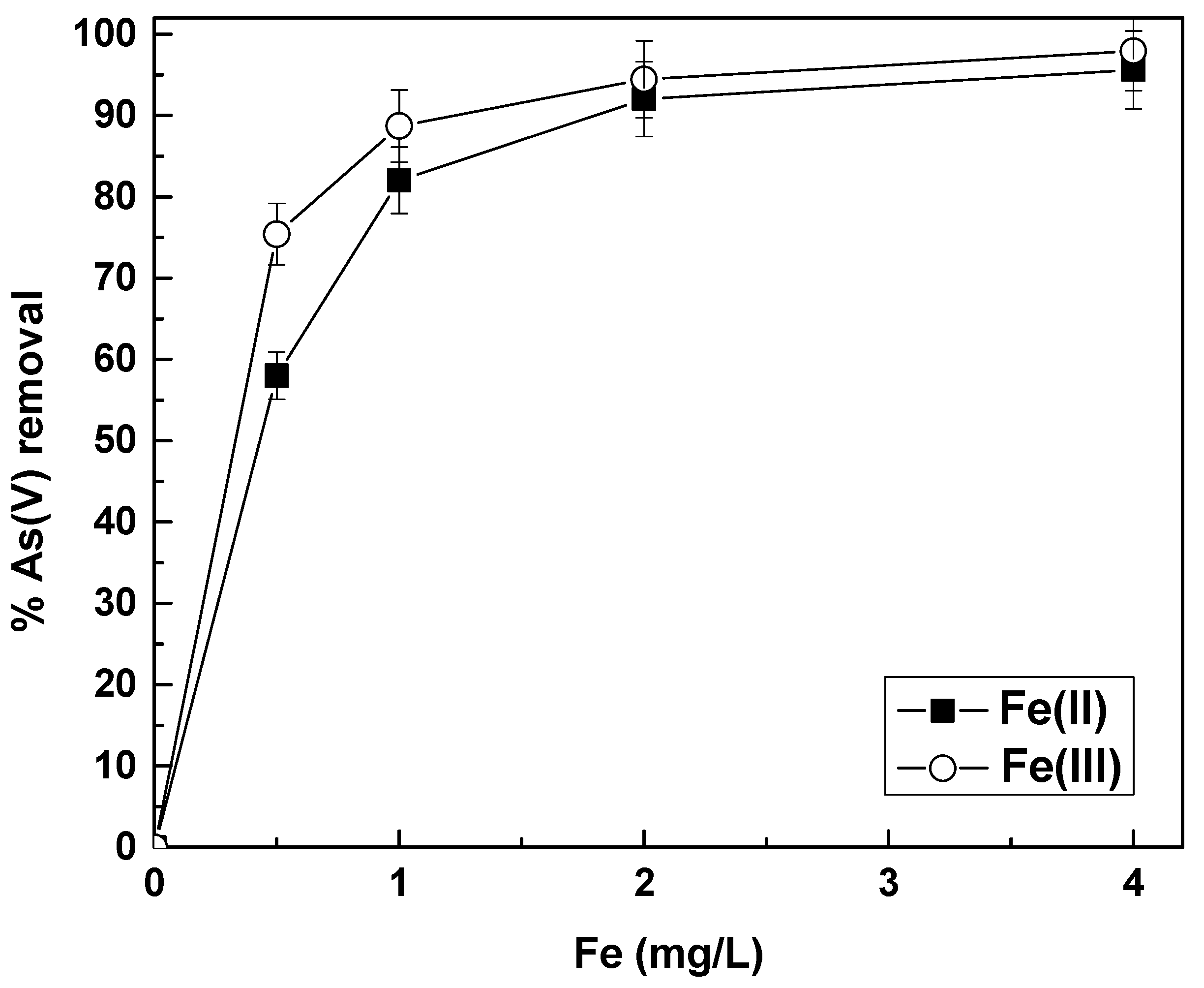
Roberts et al., [
37] have investigated the removal of As(V) by Fe(II) and Fe(III), but not in the presence of Cr(VI), and have shown that Fe(II) was slightly more efficient than Fe(III) in lowering As(V) concentration from 500 to 50 μg/L. In particular, they showed that 90% removal of As(V) was achieved by 2 mg/L of Fe(II) and about 2.5 mg/L of Fe(III). In the present study, we investigated the removal of As(V) from 50 μg/L and in order to achieve 90% removal, about 2 mg/L of iron (both Fe(II) or Fe(III)) were enough to lower arsenic below 5 μg/L, but Fe(III) was slightly more efficient than Fe(II). This can be attributed either to the fact that As(V) concentration was much lower (50 μg/L vs. 500 μg/L) or to the water composition of the examined waters. For example, the synthetic water that Roberts et al. 2004 used for their studies contained about 8 mM HCO
3−, whereas the tap water from Thessaloniki had a bicarbonate concentration of about 4 mM (240 mg/L) (
Table 1). Previous studies [
38] have shown that there is a strong dependence of Fe(II) apparent oxidation rate on bicarbonate concentration, and it gets significantly increased by doubling the bicarbonate concentration in the range from 0.01 to 0.1 M., because at pH values above 6, iron(II)-carbonate complexes prevail, which are oxidized rapidly by oxygen. Positive effect of alkalinity on As(III) removal by poly-ferric sulfate (PFS) (a mixed Fe(II)/Fe(III)) coagulant was observed in a recent study [
39]. Increasing alkalinity from 1 to 8 mM, caused an As(III) removal increase from 74 to 86% by a PFS dose of 5 mg/L.
3.2. Effect of Initial pH on As(V) and Cr(VI) Removal by Fe(II)
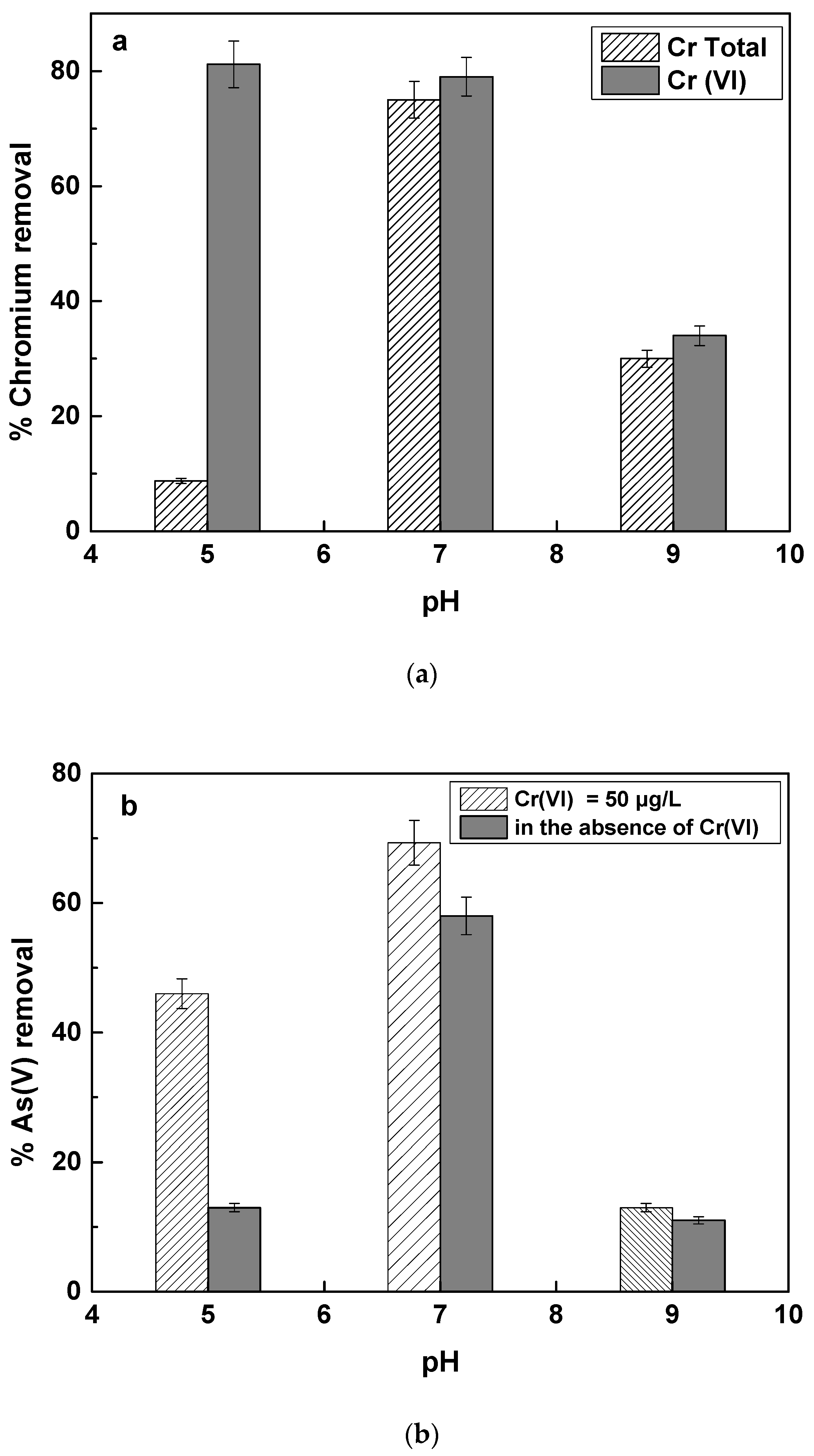
The effect of initial pH of the water on As(V) and Cr(VI) removal was examined at three different pH values, 5, 7 and 9, corresponding to the acidic, neutral and basic values. The results are shown in
Figure 5a,b corresponding to the removal of Cr(VI) and As(V) respectively.
Figure 5a shows that at pH 5, the removal of Cr(VI) was as efficient as at pH 7, but the total chromium (Cr Tot) removal was much less efficient than at pH 7. This practically means that when Fe(II) is used for the removal of chromium at acidic pH values, it is efficient in causing the reduction of Cr(VI) to Cr(III), but apparently Cr(III), which mainly forms hydroxides, remains in the solution at a big part. The higher Cr(VI) reduction by Fe(II) at acidic pH values, is due to the lower competition caused by O
2 for Fe(II) oxidation and thus more Cr(VI) reacts with Fe(II), at constant Fe(II) dosage. The formed Cr(III) however remains in big parts in the solution, firstly because the solubility is higher at lower pH values [
40] than it is at pH 7 and also that the Fe(III) formation caused by the Fe(II) oxidation is not so efficient at acidic pH values, while Fe(III) precipitation is also not so effective at acidic pH values.
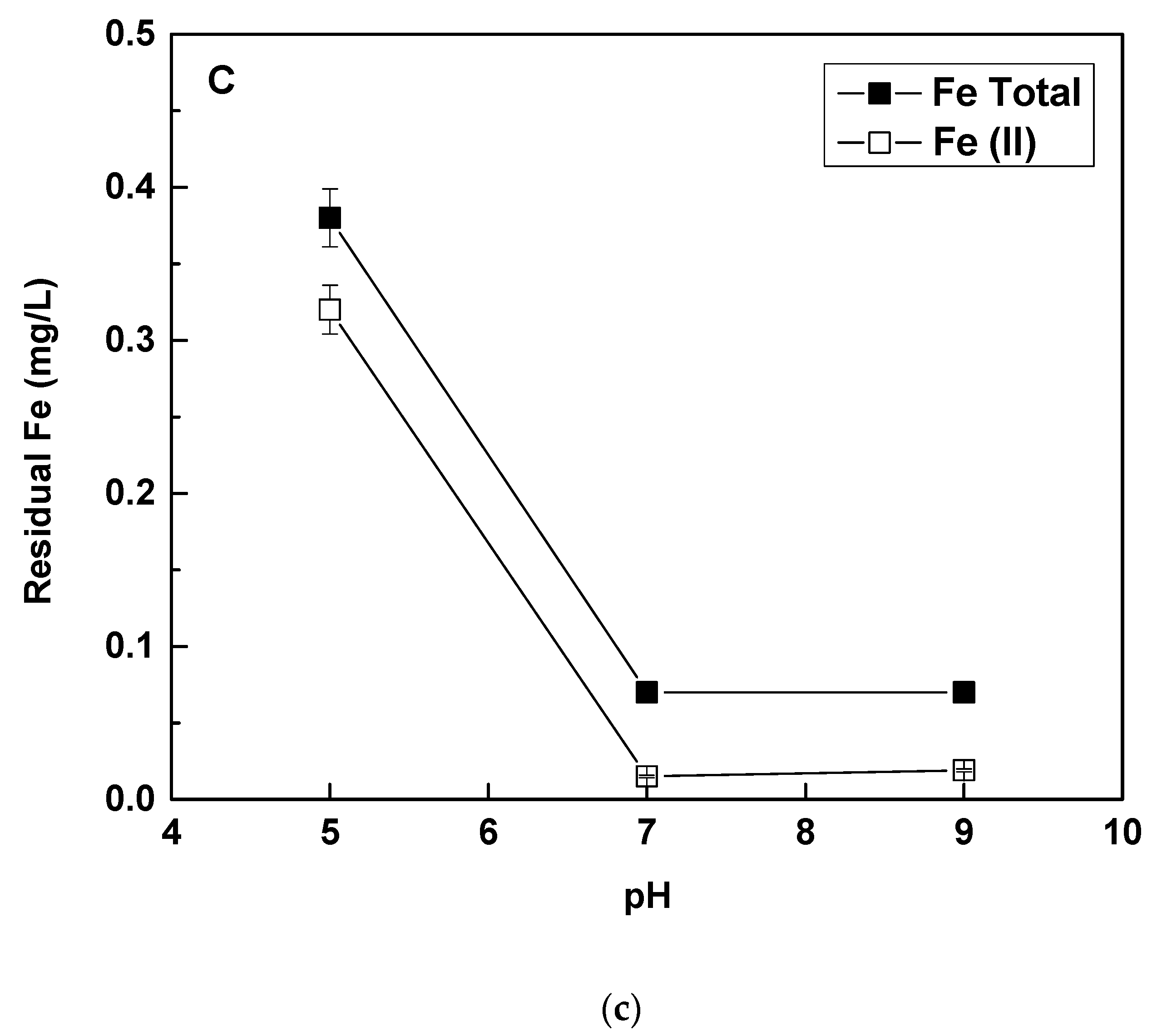
The latter is shown in
Figure 5c, in which it can be observed that at pH 5, Fe(II) is only by about 40% oxidized, and a big part of total iron remains in solution, accounting for the unoxidized Fe(II) and the Fe(III), which is not fully precipitated. At pH 5 and even more at lower pH values, Fe(II) oxidation by dissolved oxygen is very slow, as it has been reported by previous researchers [
26] and has been also measured in the present study (
Figure 6c). Thus, it can be concluded that at pH 5, Fe(II) is mainly oxidized by the presence of Cr(VI), which cannot cause complete Fe(II) oxidation, because stoichiometrically in the present experiment, the molar Fe(II) concentration was roughly 10 times higher than the molar Cr(VI) concentration. The reaction of Fe(II) with Cr(VI) requires stoichiometry of about 3, i.e., 3 moles of iron(II) react with 1 mole of Cr(VI) to achieve the required reduction of Cr(VI) to Cr(III).
These results are in agreement with previous studies, which examined the removal of chromate by Fe(II) and showed that at lower pH values, the removal of total chromium was much lower than the removal of Cr(VI) by Fe(II) [
23,
24]. Mitrakas et al., [
23] have examined this reaction at the pH range 6 to 8 in artificial groundwater. They showed that at pH 6, from 100 μg/L of Cr(VI) by using Fe(II) concentration of 0.5 mg/L, the removal of total Chromium was only 40%, while Cr(VI) reduction was about 80%. In our study, we show that this effect becomes even stronger, as the pH decreases further, and only about 8 to 9% removal of total chromium was accomplished at pH 5, while Cr(VI) removal was higher than 80%.
At alkalic pH values, both Cr(VI) and total chromium removal was very low, accounting roughly for 30%. This can be attributed to the fact that as the pH increases, so does the kinetics of Fe(II) oxidation by oxygen [
26]. Therefore, the reaction of Fe(II) with Cr(VI) is strongly competed by oxygen and thus less Cr(VI) can be reduced to Cr(III). Therefore, although all iron is oxidized and precipitates out of the solution, chromium remains in the solution. Mitrakas et al., [
23] studied this effect up tot pH 8 and also concluded that the removal of Cr(VI) and Cr total was almost similar, and accounted for about 40%.
Figure 5b shows the removal of As(V) by Fe(II) in the presence or absence of Cr(VI) at different pH values. It is shown that as the pH decreases, As(V) removal by Fe(II) decreased substantially and was even much less when Cr(VI) was not present in solution. This observation is ascribed to the fact, that Fe(II) oxidation kinetics is very slow as the pH is decreasing to pH values below 7 and as it is shown
Figure 6c. Therefore, Fe(II) remains mostly soluble and not able to adsorb As(V), thus As(V) removal was very low. In the presence of Cr(VI), some more Fe(II) is oxidized to Fe(III), due to the reaction with Cr(VI), producing Fe(III) hydroxides faster, which have strong affinity with As(V) and therefore As(V) removal by Fe(II) at acidic pH values is more efficient in the presence of Cr(VI). These results are in agreement with the study by Guan et al., who reported improved arsenate removal by Fe(II) in the presence of Cr(VI).
The overall conclusion however by the results obtained by pH variation, is that Fe(II) redox assisted coagulation is an efficient treatment technology for simultaneous As(V) and Cr(VI) removal mainly at circumneutral pH values. At acidic and alkaline pH values, arsenic and chromate removal was much less efficient. Besides, at lower pH values, significant residual iron concentrations remained in the water. This is an important finding, because it demonstrates the need for development of other types of reagents, which could be proven efficient at removing total arsenic and chromium at acidic pH values, because there are natural waters or wastewaters which have pH values far below neutral.
3.3. Effect of Initial As(V) and Cr(VI) Concentrations on Arsenic and Chromate Removal by Fe(II) Treatment
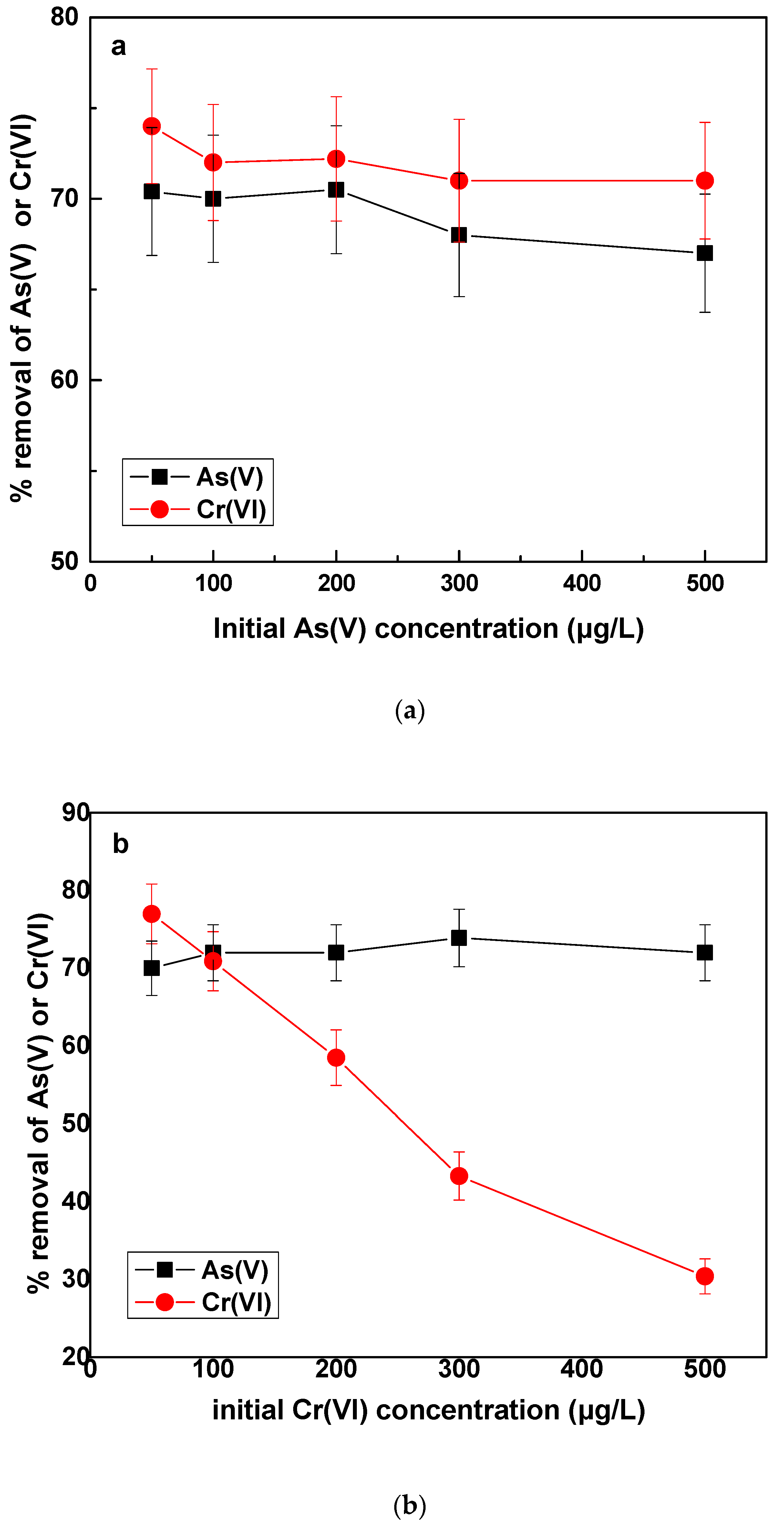
In this section, the effect of As(V) or Cr(VI) in increasing concentrations on the removal of As(V) and Cr(VI) by Fe(II) was investigated. The increase of arsenic concentration from 50 to 500 μg/L, shown in
Figure 6a, did not have any significant effect on either As(V) or Cr(VI) percentage removal. For the case of Cr(VI), since its concentration remained stable, the residual concentration was almost constant. The interesting point was however, that increasing the As(V) concentration and keeping the Fe(II) dose constant, the percentage As(V) removal remained almost stable, despite the increased residual As(V) concentrations. For example, when the initial As(V) concentration was 200, 300 and 500 μg/L, the corresponding residual concentrations were 53.5, 120 and 175 μg/L, all however corresponding to similar percentage As(V) removal of about 65—70%. This observation could be ascribed to a better coverage of adsorption sites on the created iron oxides by the increased As(V) concentration is solution, and therefore on absolute numbers, a higher quantity of arsenic could be adsorbed by the same dose of coagulant. This has been reported in the past also by other researchers [
41].
Figure 6b shows the effect on removal of As(V) and Cr(VI), by increasing the initial concentration of Cr(VI) in the water. Firstly, As(V) removal was not significantly affected. A slight improvement was noticed, which however was very little to be interpreted. In previous paragraphs, it has been shown that As(V) removal was slightly improved in the presence of Cr(VI) 50 μg/L, as compared with experiments conducted in the absence of Cr(VI) was in the water.
Figure 6b shows that additional concentration of Cr(VI) did not enhance accordingly the removal of As(V), probably because as it has been shown in
Figure 4, iron was fully oxidized within 10 min and the results shown in
Figure 6c correspond to residual arsenic concentration after about 60 min, which is adequate time for arsenic removal to occur, either with 50 μg/L Cr(VI) or more.
With regard to the removal of Cr(VI), as affected by increasing Cr(VI) initial concentration, it was noted that the percentage removal was reduced, by constant Fe(II) dose. This effect has been also observed in a previous study of our group [
42] in experiments carried out in a continuous operation pilot plant. It can be attributed to the fact that because Fe(II) is kept constant but Cr(VI) concentration increases, the molar ratio Fe(II)/Cr(VI) becomes lower, thus Fe(II) is not enough to reduce all the Cr(VI) which is present in the water. Therefore, more Fe(II) would be needed to reduce the additional Cr(VI) concentration.
The remarkable finding of this test was that even though the percentage of Cr(VI) removal decreased and the residual concentrations of Cr(VI) increased, the amount of Cr(VI) removed calculated in μg/L, per mg/L of Fe(II) added, increased. For example, when the initial concentration was 50, the amount of Cr(VI) removed was 38 μg/L, whereas for initial concentration of Cr(VI) of 500 μg/L, the amount of Cr(VI) removed was roughly 150 μg/L. Both results were obtained by the same dose of Fe(II) of 0.5 mg/L. The increased amount of Cr(VI) removed by the same Fe(II) dose, could be attributed to the fact, that in the presence of over stoichiometric Cr(VI) concentrations, the kinetics of Fe(II) oxidation by Cr(VI) can be increased and partly outcompete oxygen. Thus higher molar amount of Cr(VI) will react with the molar amount of Fe(II) added and hence more Cr(VI) will be reduced/mg of Fe(II) added. These results are in agreement with a previous study [
43] in which Cr(VI) removal by ferrous iron was investigated applying under-stoichiometric conditions, i.e., representing a deficient amount of Fe(II).
This finding might have a positive effect in achieving efficient Cr(VI) removal by lower iron doses, i.e., by dosing Fe(II) in multiple steps and kept always under-stoichiometric. Dosing of Fe(II) in multiple steps, than all in one dosing, has resulted in more efficient arsenic removal in Bangladesh groundwaters [
37]. Therefore, multiple dosing of Fe(II) might achieve efficient arsenic and chromate removal by overall less iron doses. This would result in less iron sludge production, which will reduce the overall costs for handling and disposal of toxic sludge.
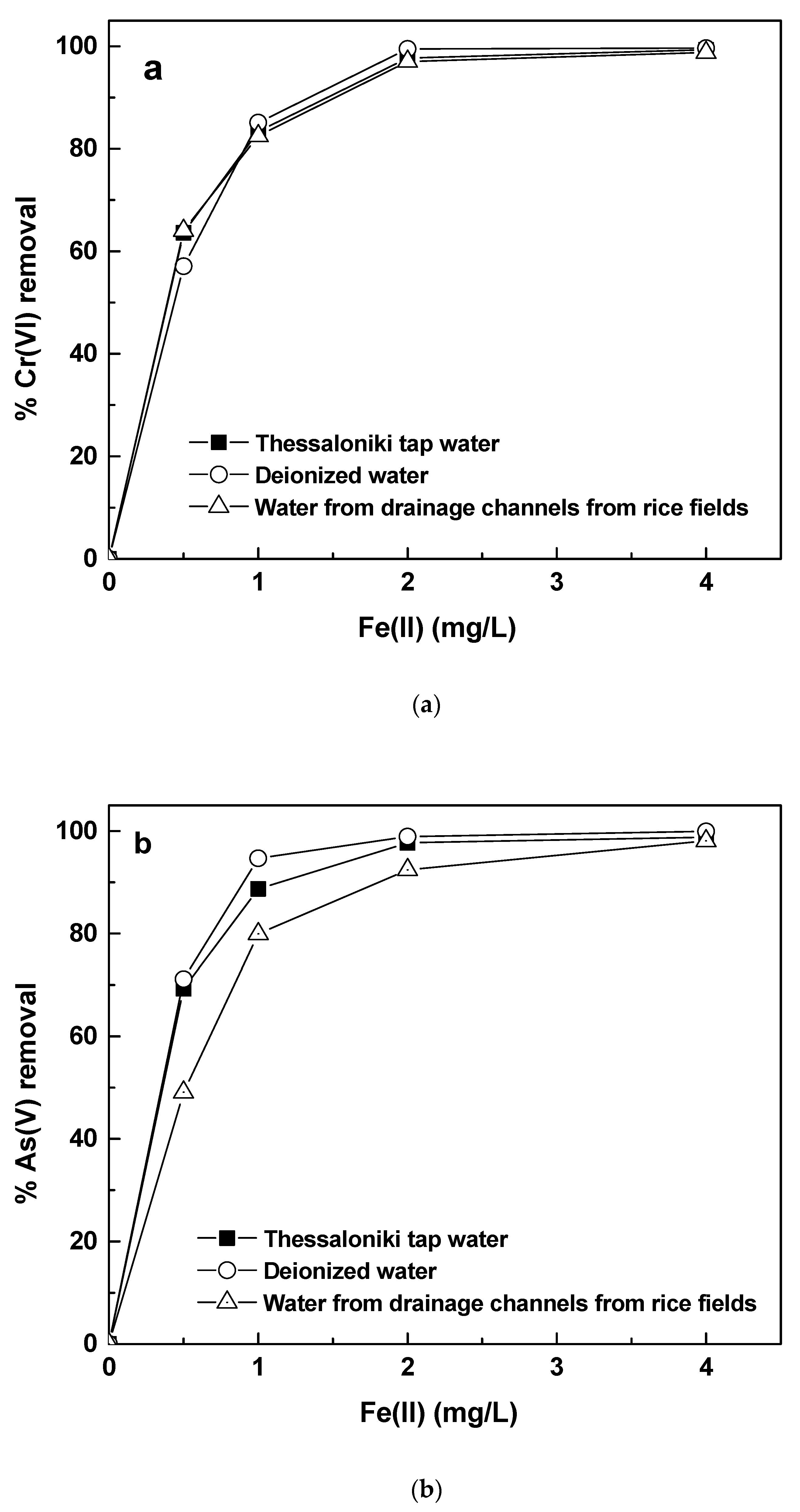
3.4. As(V) and Cr(VI) Removal by Fe(II) in Different Water Matrices
We investigated the removal of As(V) and of Cr(VI) in different water matrices, namely deionized water, Thessaloniki tap water (which originates from groundwater wells) and also surface waters from drainage channels from rice fields, collecting the irrigation waters used for rice production. The basic water composition of Thessaloniki tap water and Drainage channel surface waters is given in
Table 1.
Figure 7 shows the results of arsenic and chromate removal by Fe(II) in the three different water matrices. The main differences between these waters were the conductivity and the organic matter and phosphate concentration. In surface waters or our study, manganese was also present in trace quantities of about 50 μg/L. However, from the ICP-MS analysis, it was noticed that Mn concentration remained almost unaffected after the end of experiment. Therefore, we believe that under these conditions, Mn did not play any role in the whole experiment. The results regarding As(V) and Cr(VI) removal are shown in
Figure 7a,b.
The results demonstrated that the removal of Cr(VI) (shown in
Figure 7a) was not affected by the change in water matrix, which means that Cr(VI) removal by Fe(II) is most likely independent of the differences in the water composition. The tap water of Thessaloniki has a conductivity of about 600 μS/cm and hardness of about 300 mg CaCO
3/L. Cr(VI) removal was shown to be independent of these constituents, most likely, because its removal is based on the reduction of Cr(VI) by Fe(II), and this reaction is competed only by oxygen, i.e., oxygen is the limiting factor determining the efficiency of Cr(VI) removal by Fe(II). Even in the case that the experiments were conducted in drainage water from rice production, Cr(VI) removal was almost equally efficient. The drainage water contains some increased phosphate and nitrate concentration compared with tap water of Thessaloniki and a natural organic matter concentration (measured as TOC), much higher than the Thessaloniki tap water, as it can be seen in
Table 1. This shows that even the quite high concentrations of TOC did not affect the Cr(VI) removal, nor the total chromium removal, since these measurements correspond to total chromium concentrations.
A previous study has revealed that in artificial waters spiked with humic acids [
44], Cr(VI) removal by Fe(II) was adversely affected by the presence of 3 mg/L of humic acids. However, in the present work, such differences were not observed, which is most likely attributed to the fact that these results were obtained in waters with much more complex water composition, which might have prevented organic matter to create soluble complexes with Cr(III).
On the contrary, As(V) removal was quite influenced by the water composition, especially when the experiments were conducted in the drainage channel surface water. This can be explained by the fact that the surface water contains concentrations of competing substances, such as phosphate and organic matter. Phosphate is a well-established competitor for arsenic removal, whereas organic matter is not expected to have had a major effect on arsenic removal, as some previous studies have presented [
45,
46], which demonstrated that As(V) removal by iron oxides was affected by humic acids only when the latter were above 10 and 40 mg/L respectively. Furthermore, NOM concentration in groundwaters is usually low and therefore is usually not a limiting factor for efficient As(V) removal. Therefore, most likely the reduced As(V) removal in drainage channel surface water was due to phosphate presence. Therefore, in the following section, phosphate effect on As(V) and Cr(VI) removal by Fe(II) will be separately investigated.
The effect on As(V) removal was however visible only for the low doses up to 1 mg/L of Fe(II). At higher doses the presence of other constituents, which might affect the removal of arsenic by competing for the same adsorption sites, was obviously covered by the additional adsorption sites offered to the system by the higher Fe(II) dosage and As(V) removal was notable above 90%. This corresponded to residual As(V) concentrations far below the limit of arsenic in drinking water, which is 10 μg/L. At these doses, Cr(VI) residual concentration was also below 10 μg/L, which is the proposed limit for Cr(VI) in drinking water by the USEPA and far below the 25 μg/L, which is the limit currently under discussion by the European Commission.
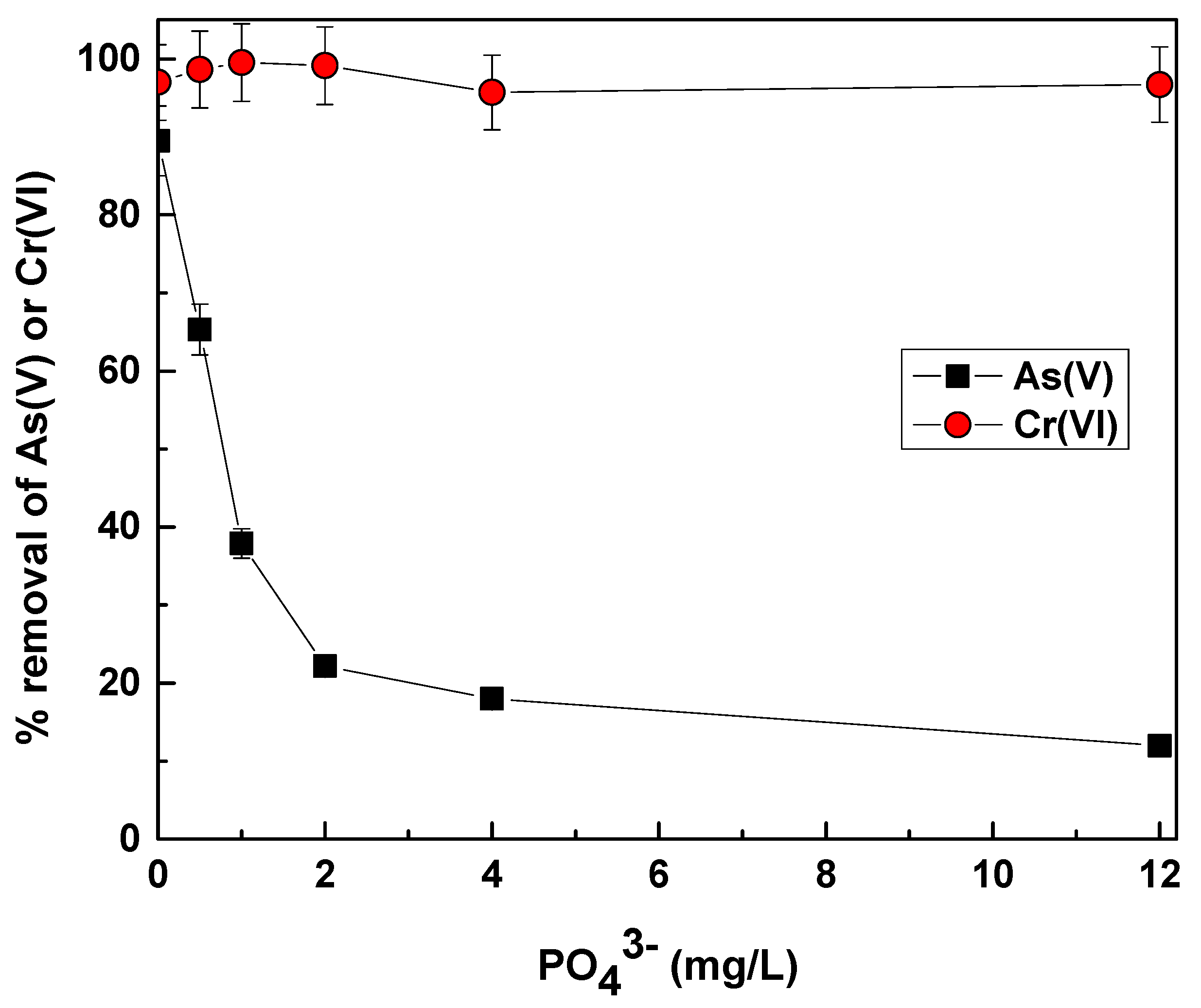
3.5. Effect of Phosphate Concentration on As(V) and Cr(V) Removal by Fe(II)
Phosphate effect on As(V) and Cr(VI) removal was investigated in the following section of this study, in order to investigate the efficiency of this technology in the presence of relevant competitors. The results are presented in
Figure 8 with respect to both As(V) and Cr(VI) removal in the presence of increasing phosphate concentrations, in a range relevant to groundwater occurrences.
Figure 8 illustrates that Cr(VI) removal was not practically affected by the increase in phosphate concentration and this agrees with results displayed in
Figure 7, which depicted that Cr(VI) removal was equally efficient in all water matrices.
On the other hand, As(V) removal was considerably affected by the presence of phosphate. As(V) removal is shown to decrease significantly as phosphate concentration increased. For example, even 0.5 mg/L phosphate can cause a reduction of As(V) removal from about 90% to nearly 70%. In the presence of 4 mg/L phosphate, As(V) removal was much less and accounted only for 20–25% and was further decreased by increasing phosphate concentration up to 12 mg/L. This competition has been reported in several studies for arsenic removal [
37] and seems to be relevant even in the case of the surface waters of the present study. As(V) removal by 1 mg/L of Fe(II) in surface waters containing about 0.5 mg/L of phosphate, was 20% lower from that observed in corresponding experiment conducted in the Thessaloniki tap water. This is most likely attributed to the presence of phosphate, but a combining synergistic effect could play a further role in the case of the results taken in the drainage channel waters. It has been shown in past studies [
47] that phosphate has a moderate effect on As(V) removal by iron hydroxides, while when in the presence of bicarbonate and silicate the adverse effect of phosphate on As(V) adsorption was magnified. Meng et al., [
46] reported that the residual As(V) concentration after iron hydroxide treatment from an initial concentration of 300 μg/L, increased from less than 13 μg/L in separate bicarbonate (2.2 mM) and phosphate (0.062 mM) solutions to 110 μg/L in the solution containing both anions.
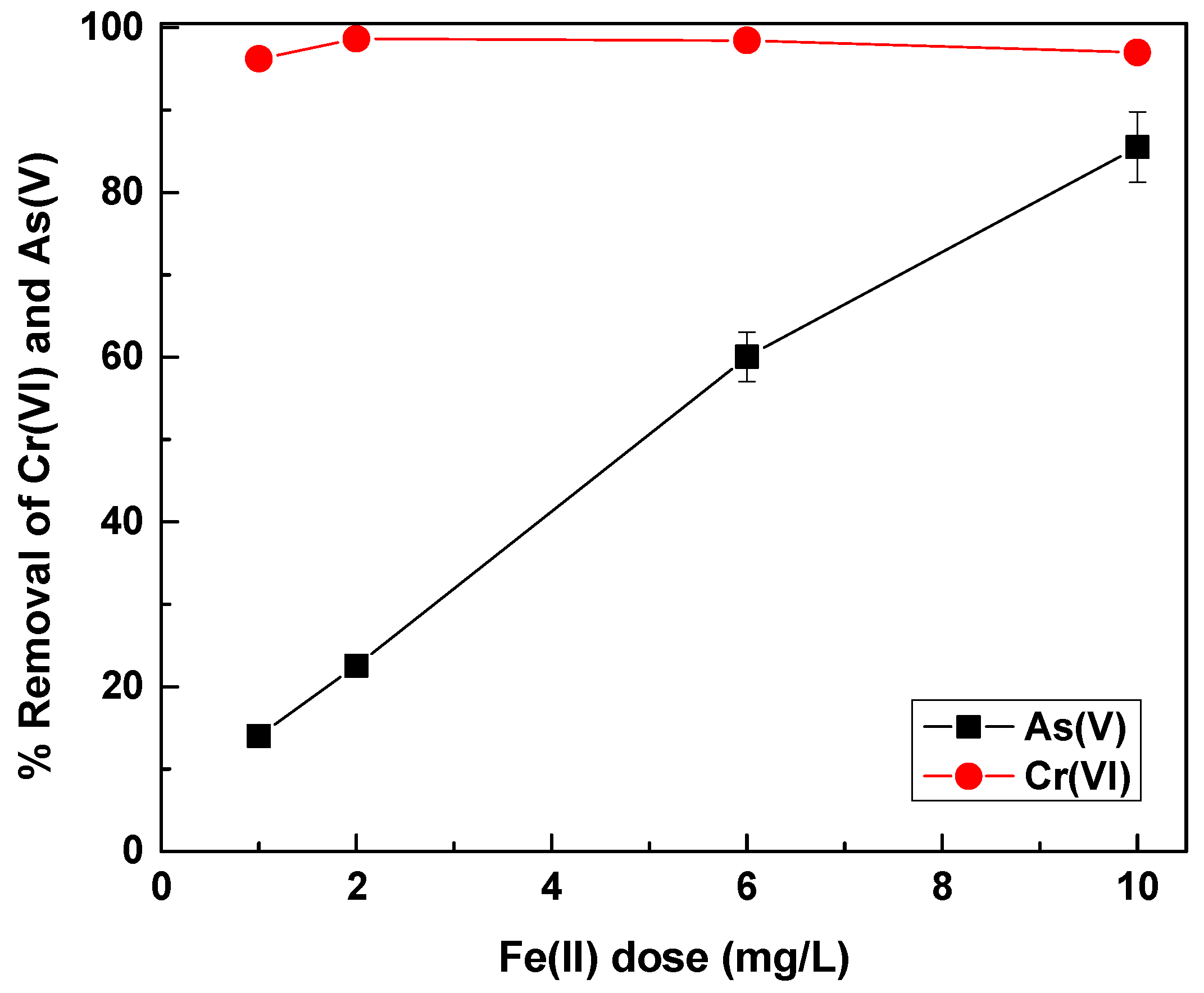
In order to eliminate the effect of phosphate, increasing concentrations of Fe(II) were applied in water containing 12 mg/L of phosphate, in order to investigate the demand for additional coagulant doses. The results are shown in
Figure 9.
Figure 9 demonstrates that in waters containing Cr(VI), As(V) and a much higher phosphate concentration (i.e., 12 mg/L), a dose of about 10 mg/L of Fe(II) was needed in order to achieve satisfying As(V) removal. This dose also achieved roughly 90% phosphate removal (data not shown), which shows also the efficiency of Fe(II) for phosphate removal. This has been demonstrated in the past. It was also shown that Fe(II) was more efficient for phosphate removal than Fe(III) [
35]. In addition, recent studies have shown that the presence of phosphate in waters treated by Fe(II), lead to the formation of amorphous Fe(II)-phosphate precipitates [
48], which leads to phosphate removal, in a mechanism similar to As(V) removal during Fe(II) oxidation [
49]. These results indicated the potential efficiency of Fe(II) in simultaneously removing arsenic, chromate and phosphate, which can represent a real case for water treatment.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}