
Food Resources Biodiversity: The Case of Local Cattle in Slovakia

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source and Cleaning Procedure
2.2. State of Intra-Population Genetic Diversity
2.3. State of Inter-Population Genetic Diversity
3. Results
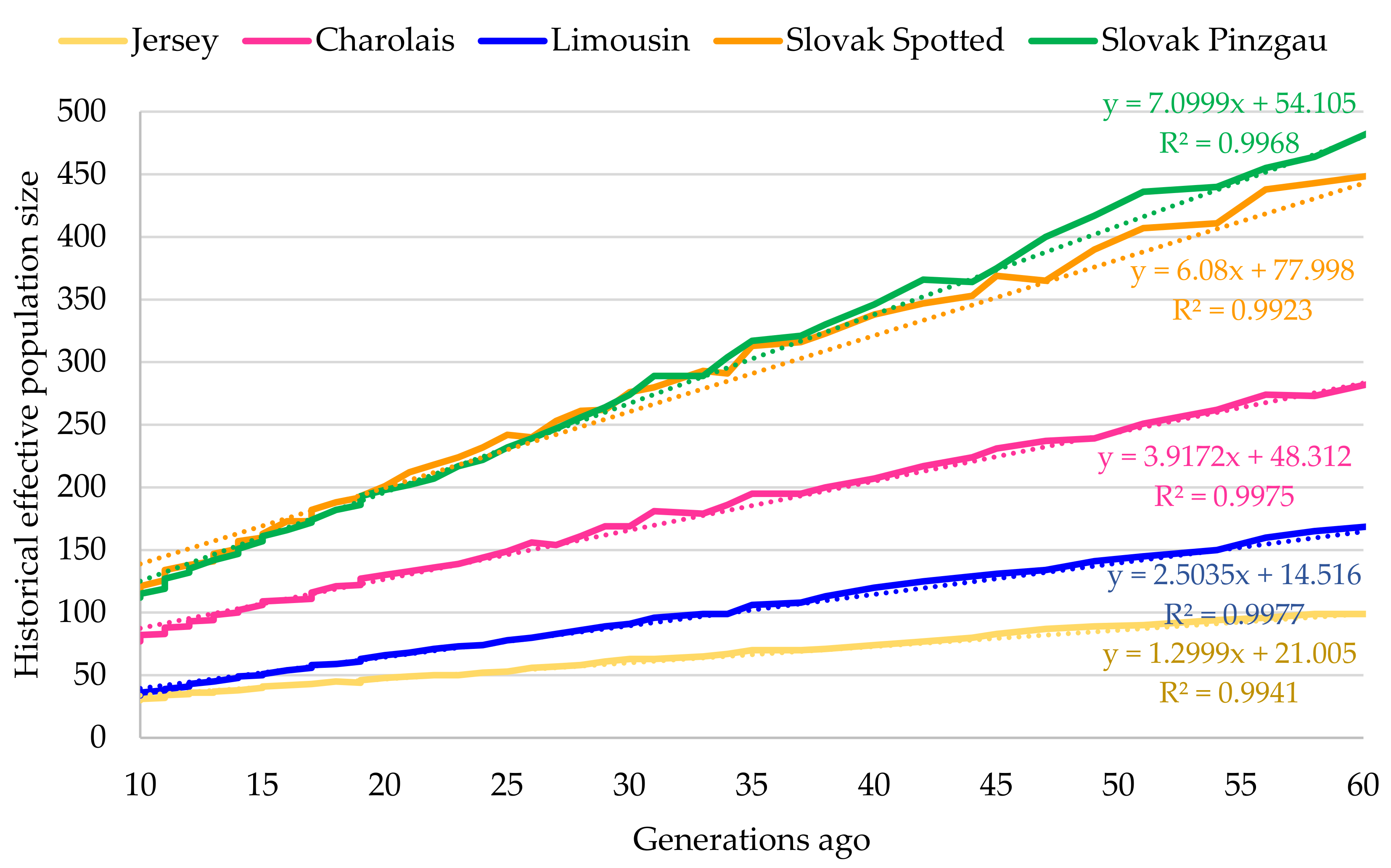
3.1. State of Intra-Population Genetic Diversity
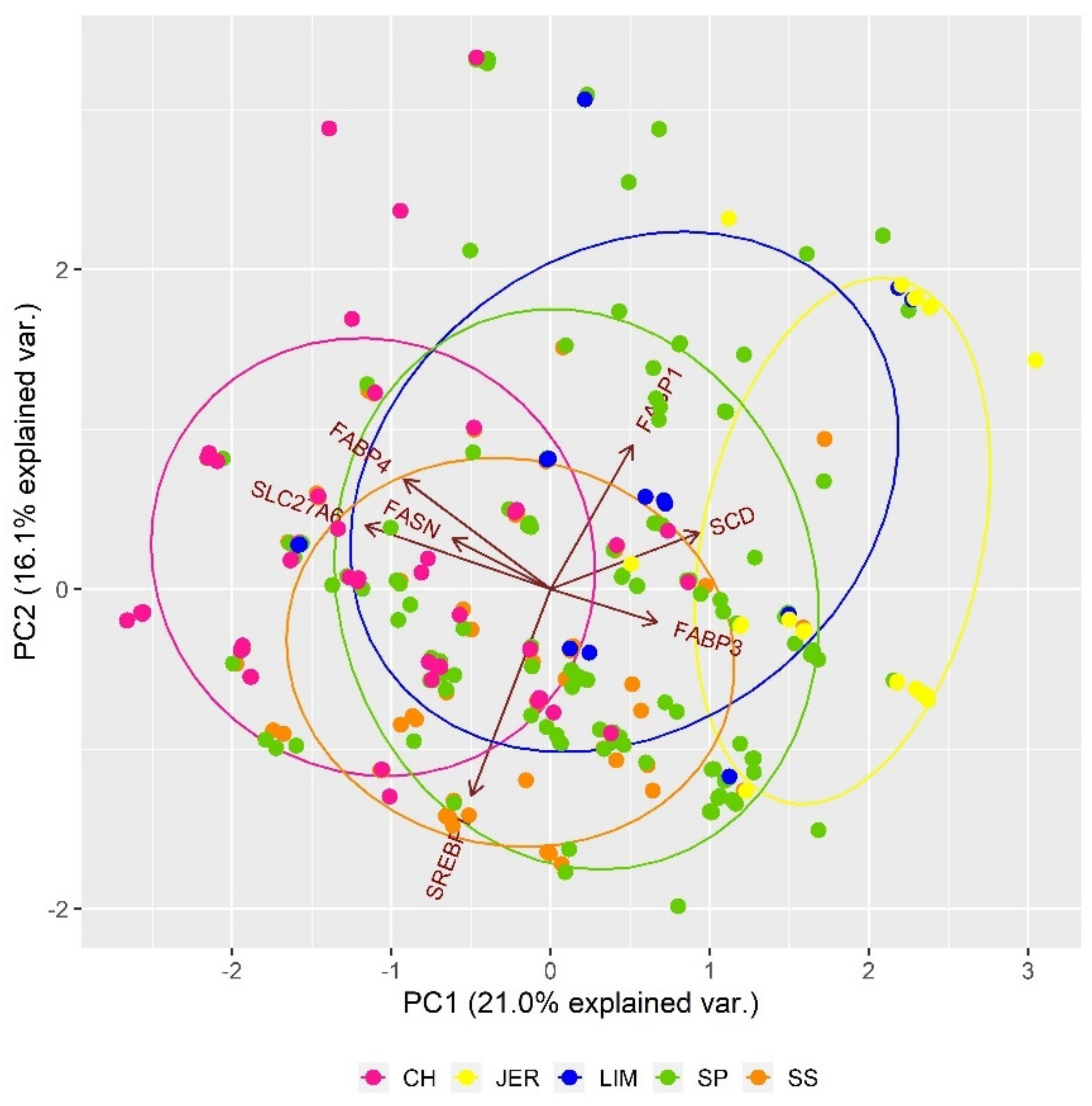
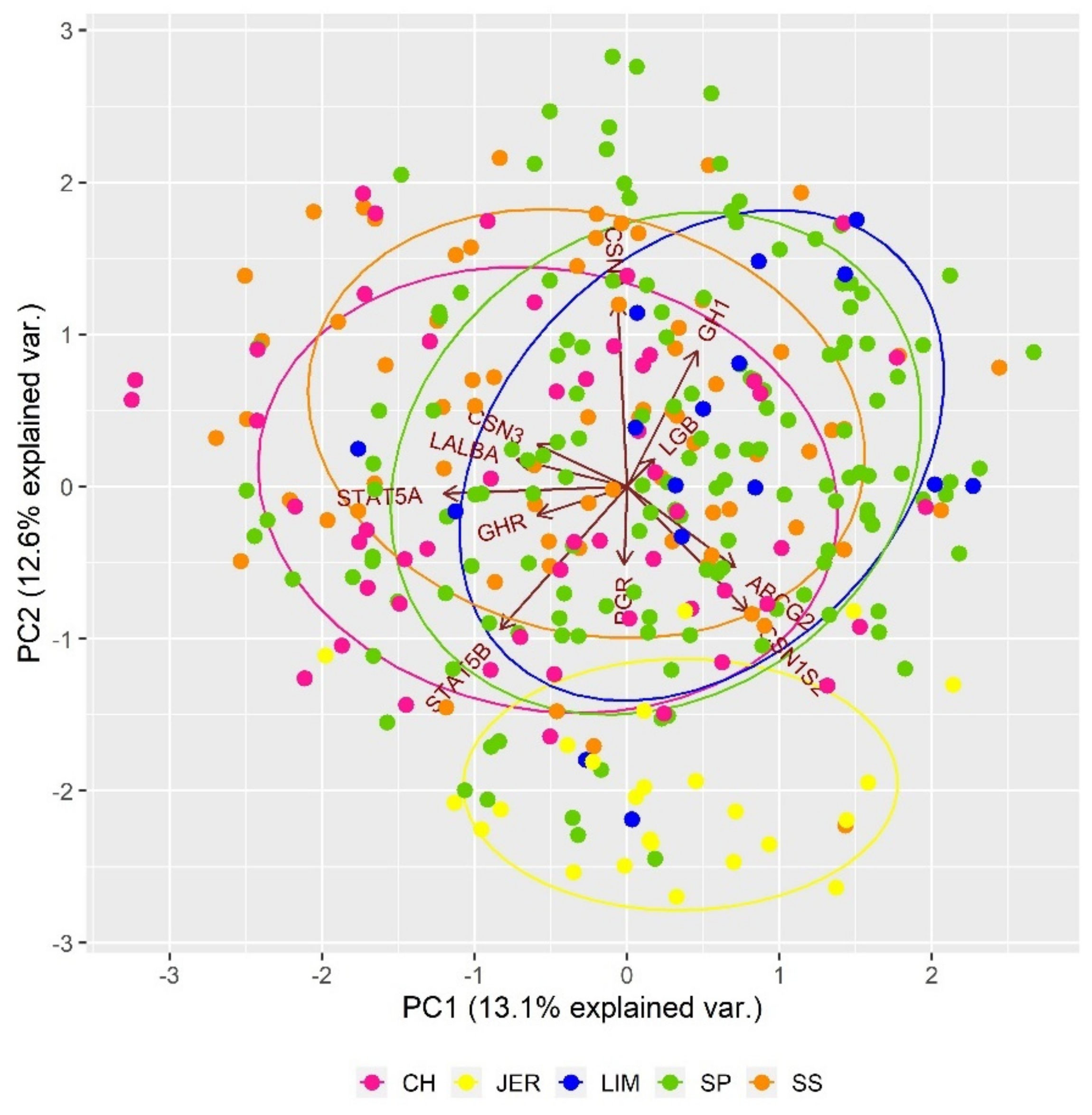
3.2. State of Inter-Population Genetic Diversity
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Date Availability Statement
Acknowledgments
Conflicts of Interest
References
- Féron, E.M. New food sources, conservation of biodiversity and sustainable development: Can unconventional animal species contribute to feeding the world? Biodivers. Conserv. 1995, 4, 233–240. [Google Scholar] [CrossRef]
- Thrupp, L.A. Linking Agricultural Biodiversity and Food Security: The Valuable Role of Sustainable Agriculture. Int. Aff. 2000, 2, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organization of the United Nations. Available online: http://www.fao.org/news/story/en/item/1180463/icode/ (accessed on 5 October 2020).
- Wang, J.; Santiago, E.; Caballero, A. Prediction and estimation of effective population size. Heredity 2016, 117, 193–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baes, C.F.; Makanjuola, B.O.; Miglior, F.; Marras, G.; Howard, J.T.; Fleming, A.; Maltecca, C. Symposium review: The genomic architecture of inbreeding: How homozygosity affects health and performance. J. Dairy Sci. 2019, 102, 2807–2817. [Google Scholar] [CrossRef] [PubMed]
- Kukučková, V.; Moravčíková, N.; Ferenčaković, M.; Simčič, M.; Mészáros, G.; Sölkner, J.; Trakovická, A.; Kadlečík, O.; Curik, I.; Kasarda, R. Genomic characterisation of Pinzgau cattle: Genetic conservation and breeding perspectives. Conserv. Genet. 2017, 18, 893–910. [Google Scholar] [CrossRef]
- Hoffmann, I. Livestock biodiversity and sustainability. Livest. Sci. 2011, 139, 1–2. [Google Scholar] [CrossRef]
- Kasarda, R.; Jamborová, L.; Moravčíková, N. Genetic diversity and production potential of animal food resources. Acta Fytotech. Zootech. 2020, 23, 102–108. [Google Scholar] [CrossRef]
- Toro, M.A.; Villanueva, B.; Fernández, J. Genomics applied to management strategies in conservation programmes. Livest. Sci. 2014, 166, 48–53. [Google Scholar] [CrossRef]
- Weir, B.S.; Anderson, A.D.; Hepler, A.B. Genetic relatedness analysis: Modern data and new challenges. Nat. Rev. Genet. 2006, 7, 771–780. [Google Scholar] [CrossRef]
- Doekes, H.P.; Veerkamp, R.F.; Bijma, P.; Hiemstra, S.J.; Windig, J.J. Trends in genome-wide and region-specific genetic diversity in the Dutch-Flemish Holstein–Friesian breeding program from 1986 to 2015. Genet. Sel. Evol. 2018, 50, 15. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, J.P.; Goyache, F. A note on ENDOG: A computer program for analysing pedigree information. J. Anim. Breed. Genet. 2005, 122, 172–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferenčaković, M.; Sölkner, J.; Curik, I. Estimating autozygosity from high-throughput information: Effects of SNP density and genotyping errors. Genet. Sel. Evol. 2013, 45, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livestock Science 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; McKeigue, P.M.; Wilson, J.F. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 2010, 5, e13996. [Google Scholar] [CrossRef] [Green Version]
- Bjelland, D.W.; Weigel, K.A.; Vukasinovic, N.; Nkrumah, J.D. Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding. J. Dairy Sci. 2013, 96, 4697–4706. [Google Scholar] [CrossRef]
- Charlesworth, D.; Willis, J.H. The genetics of inbreeding depression. Nat. Rev. Genet. 2009, 10, 783–796. [Google Scholar] [CrossRef]
- Qanbari, S.; Pimentel, E.C.G.; Tetens, J.; Thaller, G.; Lichtner, P.; Sharifi, A.R.; Simianer, H. The pattern of linkage disequilibrium in German Holstein cattle. Anim. Genet. 2010, 41, 346–356. [Google Scholar] [CrossRef]
- Selkoe, K.A.; Toonen, R.J. Microsatellites for ecologists: A practical guide to using and evaluating microsatellites markers. Ecol. Lett. 2006, 9, 615–629. [Google Scholar] [CrossRef]
- Flury, C.; Tapio, M.; Sonstegard, C.; Drogemuller, C.; Leeb, T.; Simianer, H.; Hanotte, O.; Rieder, S. Effective population size of an indigenous Swiss cattle breed estimated from linkage disequilibrium. J. Anim. Breed. Genet. 2010, 127, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Coissac, E.; Pansu, J.; Pompanon, F. Conservation genetics of cattle, sheep and goats. Comptes Rendus Biol. 2011, 334, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Vellend, M.; Geber, M.A. Connections between species diversity and genetic diversity. Ecol. Lett. 2005, 8, 767–781. [Google Scholar] [CrossRef]
- Wennersten, L.; Forsman, A. Population-level consequences of polymorphism, plasticity and randomised phenotype switching: A review of predictions. Biol. Rev. Camb. Philos. Soc. 2012, 87, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Eding, H. Conservation of Genetic Diversity: Assessing Genetic Variation Using Marker Estimated Kinships. Ph.D. Thesis, Wageningen University, Wagening, Netherlands, 2008; 120p. [Google Scholar]
- Hall, S.J.G.; Lenstra, J.A.; Deeming, D.C. The European Cattle Genetic Diversity Consortium. Prioritisation based on neutral genetic diversity may fail to conserve important characteristics in cattle breeds. J. Anim. Breed. Genet. 2012, 129, 218–225. [Google Scholar] [CrossRef]
- Gauch, H.G.; Qian, S.; Piepho, H.P.; Zhou, L.; Chen, R. Consequences of PCA graphs, SNP codings, and PCA variants for elucidating population structure. PloS ONE 2019, 14, e0218306. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef] [Green Version]
- Mastrangelo, S.; Tolone, M.; Sardina, M.T.; Sottile, G.; Sutera, A.M.; Di Gerlando, R.; Portolano, B. Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep. Genet. Sel. Evol. 2017, 49, 84. [Google Scholar] [CrossRef] [Green Version]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, S. Genetics of populations. Encycl. Br. 1948, 10, 111–112. [Google Scholar]
- Makina, S.O.; Muchadeyi, F.C.; van Marle-Köster, E.; Taylor, J.F.; Makgahlela, M.L.; Maiwashe, A. Genome-wide scan for selection signatures in six cattle breeds in South Africa. Genet. Sel. Evol. 2015, 47, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef] [Green Version]
- Hayes, B.J.; Visscher, P.M.; McPartlan, H.C.; Goddard, M.E. Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res. 2003, 13, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Nei, M. Genetic distance between populations. Am. Nat. 1972, 106, 283–285. [Google Scholar] [CrossRef]
- Pembleton, L.W.; Cogan, N.O.; Forster, J.W. StAMPP: An R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Res. 2013, 13, 946–952. [Google Scholar] [CrossRef]
- Rainer, J.; Gatto, L.; Weichenberger, C.X. ensembldb: An R package to create and use Ensembl-based annotation resources. Bioinformatics 2019, 35, 3151–3153. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Herrero-Medrano, J.M.; Megens, H.J.; Groenen, M.A.M.; Ramis, G.; Bosse, M.; Pérez-Enciso, M.; Crooijmans, R.P.M.A. Conservation genomic analysis of domestic and wild pig populations from the Iberian Peninsula. BMC Genetics 2013, 14, 106. [Google Scholar] [CrossRef] [Green Version]
- Franklin, I.R. Evolutionary change in small populations. In Conservation Biology: An Evolutionary-Ecological Perspective; Sinauer Associates: Sunderland, UK, 1980; pp. 135–140. [Google Scholar]
- Meuwissen, T.H.E. Genetic management of small populations: A review. Acta. Agric. Scand. Sect. Anim. Sci. 2009, 59, 71–79. [Google Scholar] [CrossRef]
- Koohmaraie, M. Biochemical factors regulating the tougheningand tenderisation processes of meat. Meat Sci. 1996, 43, 193–201. [Google Scholar] [CrossRef]
- Casas, E.; Shackelford, S.D.; Keele, J.W.; Koohmaraie, M.; Smith, T.P.; Stone, R.T. Detection of quantitative trait loci for growth and carcass composition in cattle. J. Anim. Sci. 2003, 81, 2976–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Ekerljung, M.; Lundström, K.; Lundén, A. Associationof polymorphisms at DGAT1, leptin, SCD1, CAPN1 and CAST genes with color, marbling and water holding capacity in meatfrom beef cattle populations in Sweden. Meat Sci. 2013, 94, 153–158. [Google Scholar] [CrossRef]
- Cheong, H.; Yoon, D.H.; Park, B.; Kim, L.; Bae, J.; Namgoong, S.; Lee, H.; Han, C.; Kim, J.; Cheong, I.C.; et al. A single nucleotide polymorphism in CAPN1 associated with marbling score in Korean cattle. BMC Genetics 2008, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Choi, B.H.; Lim, D.; Gondro, C.; Cho, Y.M.; Dang, C.G.; Sharma, A.; Jang, G.W.; Lee, K.T.; Yoon, D.; et al. Genome-wide association study identifies major loci for carcass weight on BTA14 in Hanwoo (Korean cattle). PLoS ONE 2013, 8, e74677. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Xu, L.; Chen, Y.; Zhang, L.; Gao, H.; Zhu, B.; Niu, H.; Zhang, W.; Xia, J.; Gao, X.; et al. Genome-Wide Association Study Reveals the PLAG1 Gene for Knuckle, Biceps and Shank Weight in Simmental Beef Cattle. PLoS ONE 2016, 11, e0168316. [Google Scholar] [CrossRef] [Green Version]
- Purfield, D.C.; Evans, R.D.; Berry, D.P. Reaffirmation of known major genes and the identification of novel candidate genes associated with carcass-related metrics based on whole genome sequence within a large multi-breed cattle population. BMC Genom. 2019, 20, 720. [Google Scholar] [CrossRef]
- Gamarra, D.; Aldai, N.; Arakawa, A.; de Pancorbo, M.M.; Taniguchi, M. Association of a polymorphism in SREBP1 with fatty acid composition and lipogenic gene expression in beef cattle breeds. Res. Sq. 2019. [Google Scholar] [CrossRef]
- Ujan, J.A.; Zan, L.S.; Shengjuan, W.; Adoligbe, C.; Wang, H.B. Meat tenderness and water holding capacity are associated with a 959 A G mutation in the MYOG gene of Chinese indigenous cattle. Afr. J. Biotechnol. 2011, 10, 5654–5660. [Google Scholar]
- Sarti, F.M.; Lasagna, E.; Ceccobelli, S.; Di Lorenzo, P.; Filippini, F.; Sbarra, F.; Giontella, A.; Pieramati, C.; Panella, F. Influence of single nucleotide polymorphisms in the myostatin and myogenic factor 5 muscle growth-related genes on the performance traits of Marchigiana beef cattle. J. Anim. Sci. 2014, 92, 3804–3810. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Raza, S.H.A.; Khan, R.; Sabek, A.; Khan, S.; Ullah, I.; Memon, S.; Abd El-Aziz, A.H.; Shah, M.A.; Shijun, L.; et al. Genetic variants in MYF5 affected growth traits and beef quality traits in Chinese Qinchuan cattle. Genomics 2020, 112, 2804–2812. [Google Scholar] [CrossRef] [PubMed]
- Du, X.H.; Gan, Q.F.; Yuan, Z.R.; Gao, X.; Zhang, L.P.; Gao, H.J.; Li, J.Y.; Xu, S.Z. Polymorphism of MyoD1 and Myf6 genes and associations with carcass and meat quality traits in beef cattle. Genet. Mol. Res. 2013, 12, 6708–6717. [Google Scholar] [CrossRef] [PubMed]
- Fiems, L.O. Double Muscling in Cattle: Genes, Husbandry, Carcasses and Meat. Animals 2012, 2, 472–506. [Google Scholar] [CrossRef] [PubMed]
- He, M.L.; Stanford, K.; Dugan, M.E.R.; Marquess, L.; McAllister, T.A. Association of leptin genotype with growth performance, adipocyte cellularity, meat quality, and fatty acid profile in beef steers fed flaxseed or high-oleate sunflower seed diets with or without triticale dried distiller’s grains. Meat Sci. 2020, 98, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, P.D.S.; de Souza, F.R.P.; de Camargo, G.M.F.; Gil, F.M.M.; Cardoso, D.F.; Zetouni, L.-; Braz, C.U.; Boligon, A.A.; Branco, R.H.; de Albuquerque, L.G.; et al. Association of ADIPOQ, OLR1 and PPARGC1A gene polymorphisms with growth and carcass traits in Nelore cattle. Meta Gene 2015, 4, 1–7. [Google Scholar] [CrossRef]
- Zhang, L.; Ren, H.; Yang, J.; Gan, Q.; Thao, F.; Gao, H.; Li, J. Effect of thyroglobulin gene polymorphisms on growth, carcass composition and meat quality traits in Chinese beef cattle. Mol. Biol. Rep. 2015, 42, 1403–1407. [Google Scholar] [CrossRef]
- Blecha, I.M.Z.; Siqueira, F.; Ferreira, A.B.R.; Feijó, G.L.D.; Torres Junior, R.A.A.; Medeiros, S.R.; Sousa, I.I.; Santiago, G.G.; Ferraz, A.L.J. Identification and evaluation of polymorphisms in FABP3 and FABP4 in beef cattle. Genet. Mol. Res. 2015, 14, 16353–16363. [Google Scholar] [CrossRef]
- Nafikov, R.A.; Schoonmaker, J.P.; Korn, K.T.; Noack, K.; Garrick, D.J.; Koehler, K.J.; Minick-Bormann, J.; Reecy, J.M.; Sputlock, D.E.; Beitz, D.C. Association of polymorphisms in solute carrier family 27, isoform A6 (SLC27A6) and fatty acid-binding protein-3 and fatty acid-binding protein-4 (FABP3 and FABP4) with fatty acid composition of bovine milk. J. Dairy Sci. 2013, 96, 6007–6021. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.; Lee, Y.; La, B.; Yeo, E.; Kim, Y.; Lee, C. Fatty acid composition of beef is associated with exonic nucleotide variants of the gene encoding FASN. Mol. Biol. Rep. 2012, 39, 4083–4090. [Google Scholar] [CrossRef]
- Rincon, G.; Islas-Trejo, A.; Castillo, A.R.; Bauman, D.E.; German, B.J.; Medrano, J.F. Polymorphisms in genes in the SREBP1 signalling pathway and SCD are associated with milk fatty acid composition in Holstein cattle. J. Dairy Sci. 2012, 79, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamarra, D.; Aldai, N.; Arakawwa, A.; Barron, L.J.R.; López-Oceja, A.; de Pancorbo, M.M.; Taniguchi, M. Distinct correlations between lipogenic gene expression and fatty acid composition of subcutaneous fat among cattle breeds. BMC Vet. Res. 2018, 14, 167. [Google Scholar] [CrossRef] [PubMed]
- Kučerová, J.; Matějíček, A.; Jandurová, O.M.; Sørensen, P.; Němcová, E.; Štípková, M.; Kott, T.; Bouška, J.; Frelich, J. Milk protein genes CSN1S1, CSN2, CSN3, LGB and their relation to genetic values of milk production parameters in Czech Fleckvieh. Czech J. Anim. Sci. 2006, 51, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Caroli, A.M.; Chessa, S.; Erhardt, G.J. Invited review: Milk protein polymorphisms in cattle: Effect on animal breeding and human nutrition. J. Dairy Sci. 2009, 92, 5335–5352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, L.F.; Milazzotto, M.P.; Feitosa, W.B.; Coutinho, A.R.S.; Simoes, R.; Marques, M.G.; Assumpcao, M.E.O.A.; Visintin, J.A. Sequence variation of the alpha-lactalbumin gene in Holstein and Nellore cows. Anim. Biotechnol. 2008, 19, 194–198. [Google Scholar] [CrossRef]
- McClure, M.C.; McCarthy, J.; Flynn, P.; McClure, J.C.; Dair, E.; O’Connel, D.K.; Kearney, J.F. SNP Data Quality Control in a National Beef and Dairy Cattle System and Highly Accurate SNP Based Parentage Verification and Identification. Front Genet. 2018, 9, 84. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S.; Yoshinari, K.; Uchiyama, K.; Takeda, M.; Kojima, T. Relationship between call rate per individual and genotyping accuracy of bovine single-nucleotide polymorphism array using deoxyribonucleic acid of various qualities. J. Anim. Sci. 2018, 89, 1533–1539. [Google Scholar] [CrossRef]
- Kharzinova, V.; Sermyagin, A.A.; Gladyr, E.A.; Okhlopkov, I.M.; Brem, G.; Zinovieva, N.A. A Study of Applicability of SNP Chips Developed for Bovine and Ovine Species to Whole-Genome Analysis of Reindeer Rangifer tarandus. J. Hered. 2015, 106, 758–761. [Google Scholar]
- Moravčíková, N.; Kirchner, R.; Šidlová, V.; Kasarda, R.; Trakovická, A. Estimation of genomic variation in cervids using cross-species application of SNP arrays. Poljoprivreda 2015, 21, 33–36. [Google Scholar] [CrossRef]
- More, M.; Gutiérrez, G.; Rothschild, M.; Bertolini, F.; Abel Ponce de León, F. Evaluation of SNP genotyping in alpacas using the bovine HD genotyping beadchip. Front. Genet. 2019, 10, 361. [Google Scholar] [CrossRef]
- Matukumalli, L.K.; Lawley, C.T.; Schnabel, R.D.; Taylor, J.F.; Allan, M.F.; Heaton, M.P.; O’Connell, J.; Moore, S.S.; Smith, T.P.L.; Sonstegard, T.S.; et al. Development and characterisation of a high density SNP genotyping assay for cattle. PloS ONE 2009, 4, e5350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sermyagin, A.A.; Dotsev, A.V.; Gladyr, E.A.; Traspov, A.A.; Deniskova, T.E.; Kostyunina, O.V.; Reyer, H.; Wimmers, K.; Barbato, M.; Paronyan, I.A.; et al. Whole-genome SNP analysis elucidates the genetic structure of Russian cattle and its relationship with Eurasian taurine breeds. Genet. Sel. Evol. 2018, 50, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilie, D.E.; Gao, Y.; Nicolae, I.; Dun, D.; Li, J.; Han, B.; Gavojdian, D. Evaluation of single nucleotide polymorphisms identified through the use of SNP assay in Romanian and Chinese Holstein and Simmental cattle breeds. Acta Biochim. Pol. 2020, 67, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Makanjuola, B.O.; Miglior, F.; Abdalla, E.A.; Maltecca, C.; Schenkel, F.S.; Baes, C.F. Effect of genomic selection on rate of inbreeding and coancestry and effective population size of Holstein and Jersey cattle populations. J. Dairy Sci. 2020, 103, 5183–5199. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Ciani, E.; Marsan, P.A.; Bagnato, A.; Battaglini, L.; Bozzi, R.; Carta, A.; Catillo, G.; Cassandro, M.; Casu, S.; et al. Conservation status and historical relatedness of Italian cattle breeds. Genet. Sel. Evol. 2018, 50, 35. [Google Scholar] [CrossRef] [Green Version]
- Kadlečík, O.; Hazuchová, E.; Moravčíková, N.; Kukučková, V.; Kasarda, R. Genetic Diversity in Slovak Spotted breed. AGROFOR Int. J. 2017, 2, 124–131. [Google Scholar] [CrossRef]
- Kadlečík, O.; Moravčíková, N.; Kukučková, V.; Kasarda, R. Inbreeding and Genetic Diversity Loss in Slovak Pinzgau Breed. Agric. Conspec. Sci. 2017, 82, 259–262. [Google Scholar]
- Zhang, Q.; Calus, M.P.L.; Guldbrandtsen, B.; Lund, M.S.; Sahana, G. Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. Bmc Genet. 2015, 16, 88. [Google Scholar] [CrossRef] [Green Version]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2017, 46, 110–121. [Google Scholar] [CrossRef]
- Jemaa, S.B.; Thamri, N.; Mnara, S.; Rebours, E.; Rocha, D.; Boussaha, M. Linkage disequilibrium and past effective population size in native Tunisian cattle. Genet. Mol. Biol. 2019, 42, 52–56. [Google Scholar] [CrossRef]
- Porto-Neto, L.R.; Lee, S.H.; Sonstegard, T.S.; Van Tassell, C.P.; Lee, H.K.; Gibson, J.P.; Gondro, C. Genome-wide detection of signatures of selection in Korean Hanwoo cattle. Anim. Genet. 2014, 2, 2–190. [Google Scholar] [CrossRef] [PubMed]
- Al-Mamun, H.A.; Clark, S.A.; Kwan, P.; Gondro, C. Genome-wide linkage disequilibrium and genetic diversity in five populations of Australian domestic sheep. Genet. Sel. Evol. 2015, 47, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEvoy, B.P.; Powell, J.E.; Goddard, M.E.; Visscher, P.M. Human population dispersal "Out of Africa" estimated from linkage disequilibrium and allele frequencies of SNPs. Genome Res. 2011, 21, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moravčíková, N.; Kasarda, R.; Vostrý, L.; Krupová, Z.; Krupa, E.; Lehocká, K.; Olšanská, B.; Trakovická, A.; Nádaský, R.; Židek, R.; et al. Analysis of selection signatures in the beef cattle genome. Czech J. Anim. Sci. 2019, 64, 491–503. [Google Scholar] [CrossRef] [Green Version]
- Jahuey-Martínez, F.J.; Parra-Bracamonte, G.M.; Sifuentes-Rincón, A.M.; Moreno-Medina, V.R. Signatures of selection in Charolais beef cattle identified by genome-wide analysis. J. Anim. Breed. Genet. 2019, 136, 378–389. [Google Scholar] [CrossRef]
- Paim, T.D.P.; Hay, E.H.A.; Wilson, C.; Thomas, M.G.; Kuehn, L.A.; Paiva, S.R.; McManus, C.; Blackburn, H. Genomic Breed Composition of Selection Signatures in Brangus Beef Cattle. Front. Genet. 2020, 11, 710. [Google Scholar] [CrossRef]
- Hayes, B.J.; Lien, S.; Nilsen, H.; Olsen, H.G.; Berg, P.; Maceachern, S.; Potter, S.; Meuwissen, T.H.E. The origin of selection signatures on bovine chromosome 6. Anim. Genet. 2008, 39, 105–111. [Google Scholar] [CrossRef]
- Naderi, S.; Moradi, M.H.; Farhadian, M.; Yin, T.; Jaeger, M.; Scheper, C.; Korkuc, P.; Brockmann, G.A.; König, S.; May, K. Assessing selection signatures within and between selected lines of dual-purpose black and white and German Holstein cattle. Anim. Genet. 2020, 51, 391–408. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed | Abbreviation | Production Type | Sample Size | Genotyping Microarray |
|---|---|---|---|---|
| Jersey | JER | Dairy | 29 | GeneSeek GGP Bovine 150 k |
| Charolais | CHAR | Beef | 71 | International Dairy and Beef Chip |
| Limousin | LIM | Beef | 17 | International Dairy and Beef Chip |
| Slovak Pinzgau | SP | Dual-purpose | 152 | BovineSNP50v1 BeadChip |
| Slovak Spotted | SS | Dual-purpose | 87 | BovineSNP50v1 BeadChip International Dairy and Beef Chip |
| Trait | Gene Name | Chr | Start Position (bp) | End Position (bp) | No of SNPs in Region |
|---|---|---|---|---|---|
| Carcass | MSTN | 2 | 6278630 | 6285486 | 2 |
| LEP | 4 | 92436922 | 92453653 | 2 | |
| MYF6 | 5 | 10275115 | 10277301 | 2 | |
| MYF5 | 5 | 10284434 | 10287669 | 2 | |
| OLR1 | 5 | 99803497 | 99815138 | 2 | |
| LCORL | 6 | 37380296 | 37557106 | 6 | |
| CAST | 7 | 96033978 | 96167151 | 1 | |
| CAPN3 | 10 | 37711578 | 37766813 | 1 | |
| TG | 14 | 8217490 | 8453614 | 5 | |
| PLAG1 | 14 | 23330541 | 23375751 | 1 | |
| MYOD1 | 15 | 34794122 | 34796767 | 2 | |
| MYOG | 16 | 797547 | 800444 | 2 | |
| CAPN2 | 16 | 27079890 | 27138327 | 2 | |
| SREBP1 | 19 | 34633133 | 34649213 | 2 | |
| CAPN1 | 29 | 43400333 | 43427397 | 2 | |
| Fatty acid composition | FABP3 | 2 | 122285620 | 122294666 | 2 |
| SLC27A6 | 7 | 25037025 | 25117897 | 2 | |
| FABP1 | 11 | 47917375 | 47923252 | 2 | |
| FABP4 | 14 | 44676542 | 44681059 | 2 | |
| SREBP1 | 19 | 34633133 | 34649213 | 2 | |
| FASN | 19 | 50775674 | 50796012 | 2 | |
| SCD | 26 | 21263727 | 21279185 | 2 | |
| Milk production | LALBA | 5 | 31183432 | 31213145 | 2 |
| ABCG2 | 6 | 36475377 | 36603209 | 1 | |
| CSN1S1 | 6 | 85411118 | 85429268 | 1 | |
| CSN2 | 6 | 85449164 | 85457744 | 1 | |
| CSN1S2 | 6 | 85529905 | 85548556 | 1 | |
| CSN3 | 6 | 85645854 | 85658926 | 1 | |
| PAEP | 11 | 103255824 | 103264276 | 2 | |
| PGR | 15 | 7854787 | 7966985 | 1 | |
| STAT5B | 19 | 42319170 | 42357910 | 1 | |
| STAT5A | 19 | 42395221 | 42416545 | 2 | |
| GH1 | 19 | 48117957 | 48119752 | 2 | |
| GHR | 20 | 31868624 | 32178311 | 2 |
| Breed | Ho ± SD | He ± SD | MAF ± SD |
|---|---|---|---|
| Jersey | 0.31 ± 0.20 | 0.30 ± 0.18 | 0.23 ± 0.16 |
| Charolais | 0.35 ± 0.17 | 0.34 ± 0.15 | 0.27 ± 0.15 |
| Limousin | 0.34 ± 0.20 | 0.32 ± 0.16 | 0.26 ± 0.15 |
| Slovak Pinzgau | 0.36 ± 0.15 | 0.35 ± 0.14 | 0.27 ± 0.14 |
| Slovak Spotted | 0.34 ± 0.16 | 0.34 ± 0.15 | 0.26 ± 0.14 |
| Inbreeding Estimator | Population | ||||
|---|---|---|---|---|---|
| JER ± SD | CHAR ± SD | LIM ± SD | SP ± SD | SS ± SD | |
| FROH > 4 Mbp | 9.58 ± 2.97 | 2.20 ± 1.13 | 1.79 ± 0.87 | 2.12 ± 1.97 | 1.67 ± 1.44 |
| FROH > 8 Mbp | 5.95 ± 2.43 | 1.17 ± 0.95 | 0.74 ± 0.66 | 1.45 ± 1.72 | 0.86 ± 1.19 |
| FROH > 16 Mbp | 2.87 ± 2.05 | 0.58 ± 0.85 | 0.43 ± 0.65 | 0.79 ± 1.40 | 0.41 ± 1.08 |
| FGRM | −0.21 ± 0.07 | −0.05 ± 0.07 | −0.14 ± 0.09 | −0.01 ± 0.09 | −0.02 ± 0.07 |
| FHOM | −0.04 ± 0.04 | −0.02 ± 0.05 | −0.05 ± 0.04 | −0.01 ± 0.08 | −0.01 ± 0.06 |
| FUNI | −0.04 ± 0.03 | −0.02 ± 0.03 | −0.06 ± 0.03 | −0.01 ± 0.03 | −0.01 ± 0.02 |
| SS | SP | CHAR | LIM | JER | |
|---|---|---|---|---|---|
| SS | 0.02 | 0.03 | 0.04 | 0.13 | |
| SP | 0.04 | 0.03 | 0.05 | 0.12 | |
| CHAR | 0.04 | 0.05 | 0.05 | 0.13 | |
| LIM | 0.06 | 0.06 | 0.07 | 0.15 | |
| JER | 0.19 | 0.18 | 0.20 | 0.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasarda, R.; Vostrý, L.; Vostrá-Vydrová, H.; Candráková, K.; Moravčíková, N. Food Resources Biodiversity: The Case of Local Cattle in Slovakia. Sustainability 2021, 13, 1296. https://doi.org/10.3390/su13031296
Kasarda R, Vostrý L, Vostrá-Vydrová H, Candráková K, Moravčíková N. Food Resources Biodiversity: The Case of Local Cattle in Slovakia. Sustainability. 2021; 13(3):1296. https://doi.org/10.3390/su13031296
Chicago/Turabian StyleKasarda, Radovan, Luboš Vostrý, Hana Vostrá-Vydrová, Kristína Candráková, and Nina Moravčíková. 2021. "Food Resources Biodiversity: The Case of Local Cattle in Slovakia" Sustainability 13, no. 3: 1296. https://doi.org/10.3390/su13031296
APA StyleKasarda, R., Vostrý, L., Vostrá-Vydrová, H., Candráková, K., & Moravčíková, N. (2021). Food Resources Biodiversity: The Case of Local Cattle in Slovakia. Sustainability, 13(3), 1296. https://doi.org/10.3390/su13031296