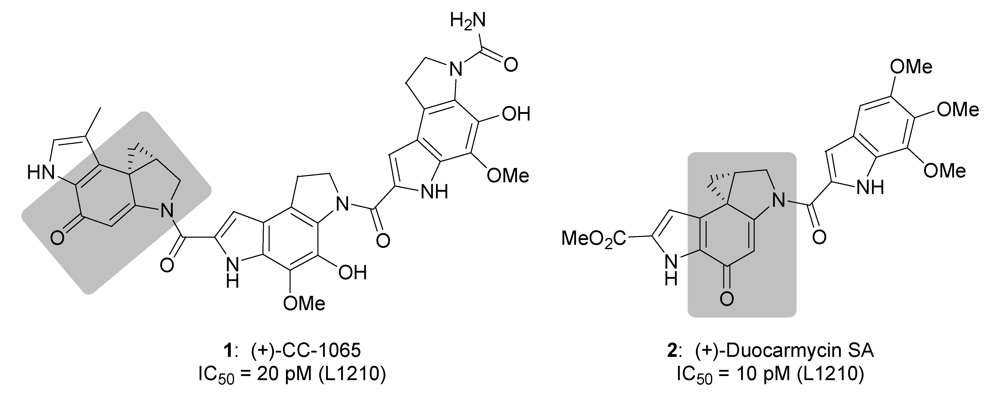
Determination of the Biological Activity and Structure Activity Relationships of Drugs Based on the Highly Cytotoxic Duocarmycins and CC-1065

Abstract
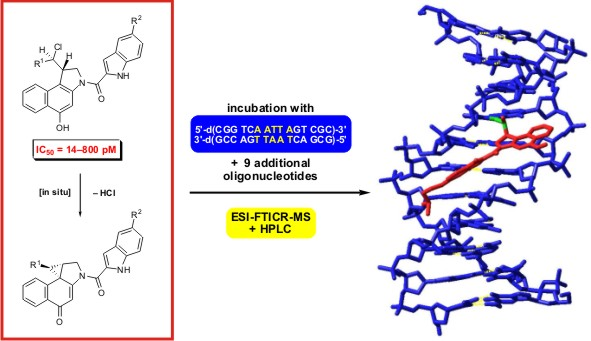
:
1. Introduction


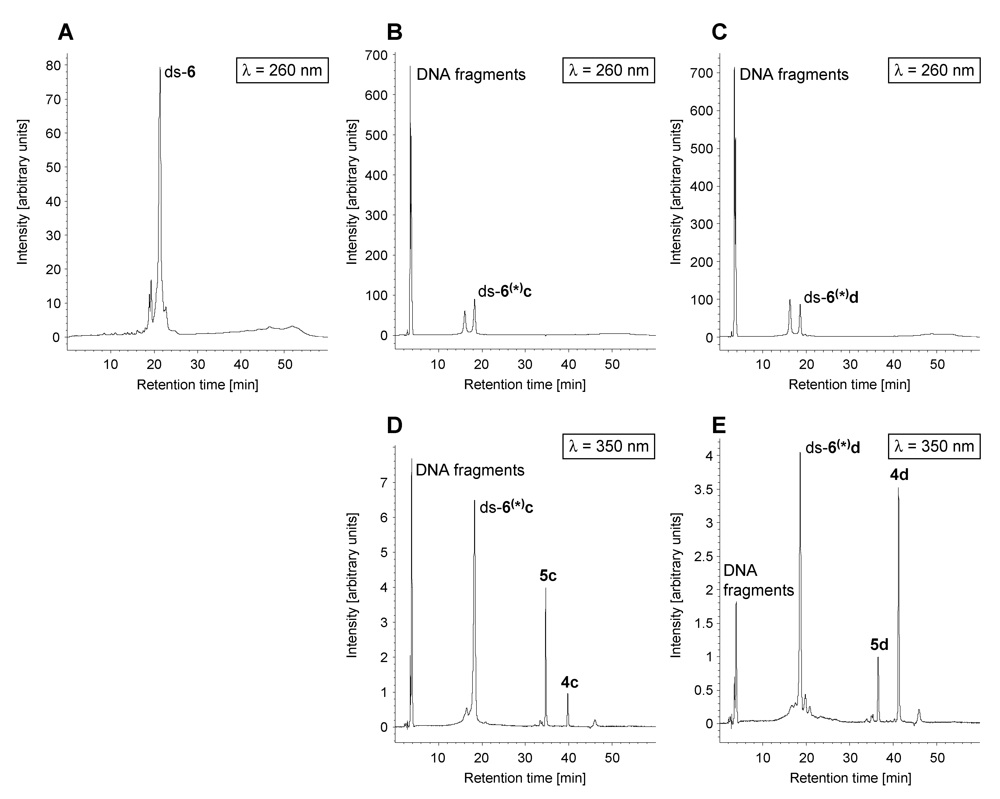
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
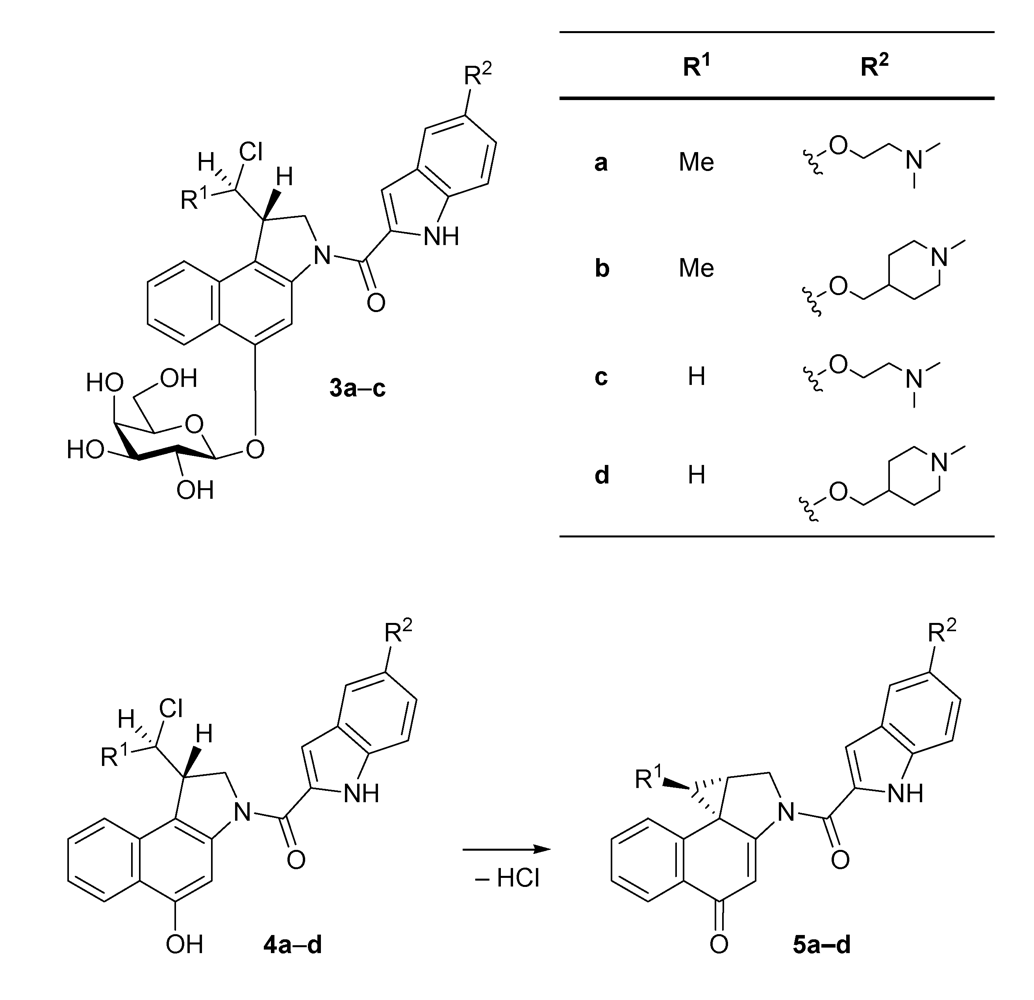
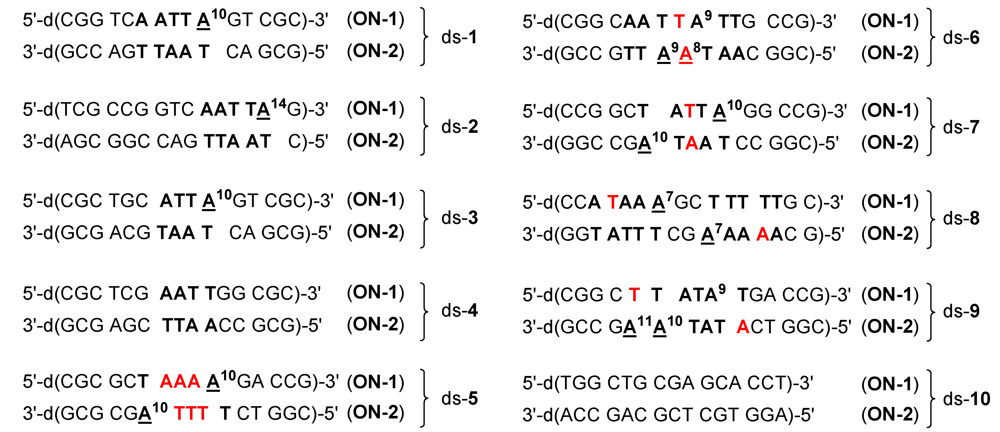
| seco-drug | IC50 (pm) |
|---|---|
| 4a·HCl | 750 |
| 4b·HCl | 800 |
| 4c·HCl | 26 |
| 4d·HCl | 14 |
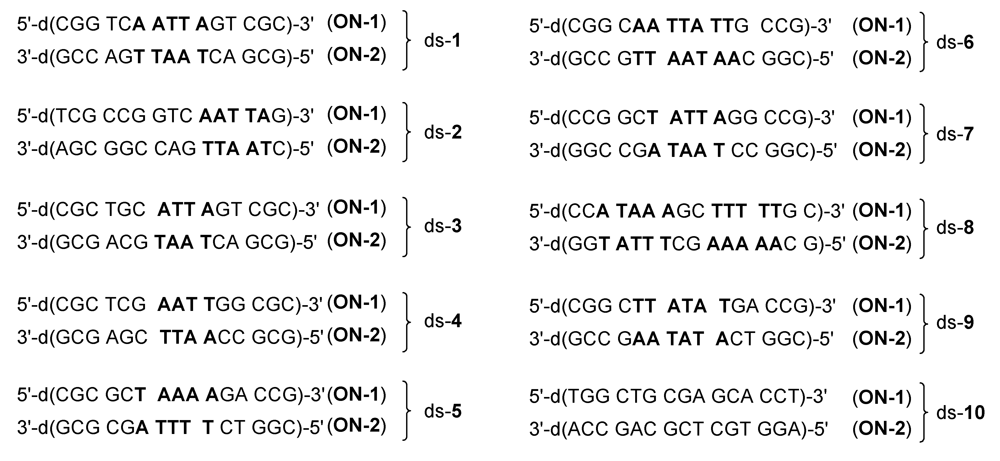
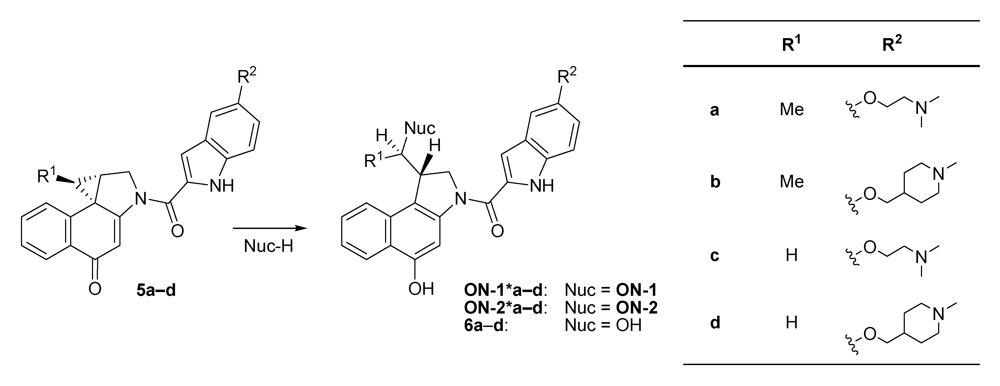
2.1. Investigations on the Reactivity and Sequence Selectivity of 4a-d against DNA Oligomers



| Alkylation [%] | Alkylation position | |||||||
|---|---|---|---|---|---|---|---|---|
| DNA | 4a | 4b | 4c | 4d | 4a | 4b | 4c | 4d |
| ds-1 | 75 | 53 | 46 | 28 | A10 (ON-1) | |||
| ds-2 | 60 | 66 | 9 | 31 | A14 (ON-1) | |||
| ds-3 | 55 | 67 | 29 | 32 | A10 (ON-1) | |||
| ds-4 | - a | - a | - a | 32 | - b | ON-1 a | ||
| ds-5 | ON-1 and ON-2 c | 6 | A10 (ON-1) ≥ A10 (ON-2) | A10 (ON-1) | ||||
| ds-6 | 30 | 20 | 17 | 13 | A8 (ON-2) ≥ A9 (ON-2) | |||
| ds-7 | ON-1 and ON-2 c | A10 (ON-2) ≥ A10 (ON-1) | ||||||
| ds-8 | ON-1 and ON-2 c | A7 (ON-2) ≥ A7 (ON-1) | ||||||
| ds-9 | 77 | 82 | 21 | 30 | A10 (ON-2) ≥ A11 (ON-2) | |||
| ds-10 | - a | - a | - a | - a | - b | |||
| ON-1 of ds-1 | 20 | 25 | - a | - a | - b | |||
| 5' | 4 | 3 | 2 | 1 | 0 | -1 | 3' | |
|---|---|---|---|---|---|---|---|---|
| 4a, 4c, 4b, 4d | A/T > G/C, G/C > A/T | A > T | A > T | A > T | A | A/G > C/T |
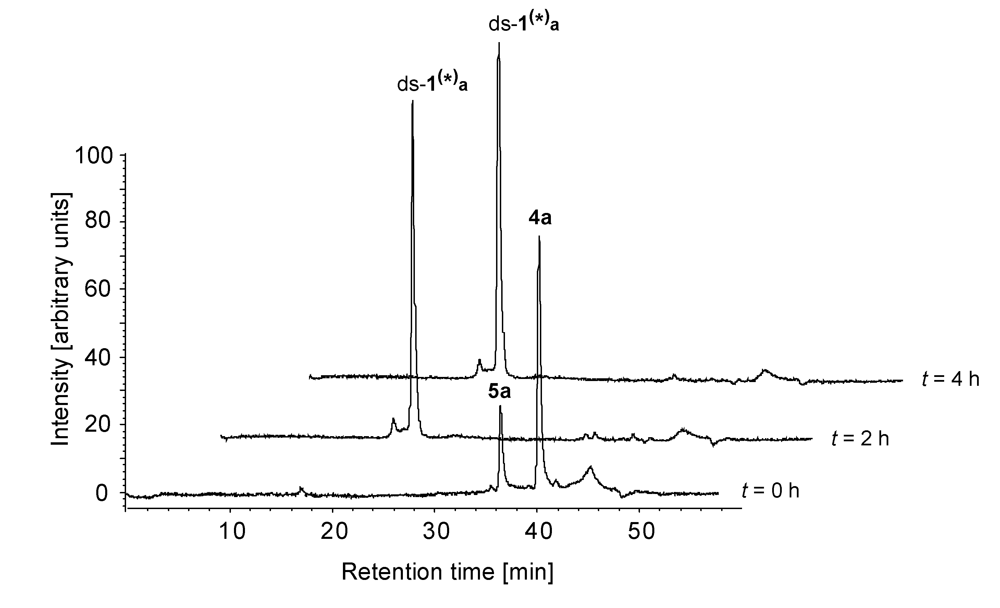
2.2. Investigations on the Reaction Kinetics of the Seco-Drugs

| AuC [%] after the indicated time of incubation | |||||
|---|---|---|---|---|---|
| Reaction mixture | Species | 0 h | 2 h | 4 h | 6 h |
| ds-1/4a | 4a | 78 | - | - | - |
| 5a | 22 | - | - | - | |
| ds-1(*)a | - | 100 | 100 | 100 | |
| ds-1/4b | 4b | 89 | - | - | - |
| 5b | 11 | - | - | - | |
| ds-1(*)b | - | 100 | 100 | 100 | |
| ds-1/4c | 4c | 74 | 45 | 19 | 12 |
| 5c | 26 | 20 | 14 | - | |
| ds-1(*)c | - | 35 | 67 | 88 | |
| ds-1/4d | 4d | 76 | 51 | 34 | 19 |
| 5d | 24 | 20 | 17 | 19 | |
| ds-1(*)d | - | 29 | 49 | 62 | |
| ds-x/4a | ds-x/4b | |||||
|---|---|---|---|---|---|---|
| ds-x | 4a [%] | 5a [%] | ds-x(*)a [%] | 4b [%] | 5b [%] | ds-x(*)b[%] |
| ds-1 | - | - | 100 | 2 | 1 | 97 |
| ds-2 | - | 2 | 98 | 9 | 2 | 89 |
| ds-3 | - | 1 | 99 | - | 4 | 96 |
| ds-4 | - | 64 | 36 | - | 62 | 38 |
| ds-5 | - | 1 | 99 | 3 | 1 | 96 |
| ds-6 | - | 27 | 73 | - | 40 | 60 |
| ds-7 | - | 3 | 97 | - | 3 | 97 |
| ds-8 | - | - | 100 | 2 | 1 | 97 |
| ds-9 | - | 1 | 99 | - | 1 | 99 |
| ds-10 | - | 63 | 37 | - | 40 | 60 |
| ds-x/4c | ds-x/4d | |||||||
|---|---|---|---|---|---|---|---|---|
| ds-x | 4c [%] | 5c [%] | ds-x(*)c [%] | DNA fragments [%] | 4d [%] | 5d [%] | ds-x(*)d [%] | DNA fragments [%] |
| ds-1 | 1 | - | 99 | - | 45 | 3 | 52 | - |
| ds-2 | 13 | - | 87 | - | 17 | 2 | 81 | - |
| ds-3 | 8 | - | 92 | - | 19 | 3 | 78 | - |
| ds-4 | 15 | 68 | 17 | - | 19 | 68 | 13 | - |
| ds-5 | 59 | - | 41 | - | 63 | 3 | 34 | - |
| ds-6 | 4 | 13 | 53 | 30 | 27 | 8 | 48 | 17 |
| ds-7 | 3 | 11 | 73 | 13 | 7 | 2 | 84 | 7 |
| ds-8 | 5 | - | 95 | - | 30 | 3 | 67 | - |
| ds-9 | 2 | - | 83 | 15 | 21 | 3 | 70 | 6 |
| ds-10 | 29 | 53 | 18 | - | 33 | 41 | 26 | - |

3. Experimental Section
3.1. Materials
3.2. Incubation of 4a-d with Synthetic DNA Oligonucleotides
3.3. Electrospray Ionisation Fourier Transform Ion Cyclotron Resonance Mass Spectrometry (ESI-FTICR MS)
3.4. High Performance Liquid Chromatography (HPLC)
4. Conclusions
Acknowledgements
References and Notes
- Martin, D.G.; Biles, C.; Gerpheide, S.A.; Hanka, L.J.; Krueger, W.C.; McGovren, J.P.; Mizsak, S.A.; Neil, G.L.; Stewart, J.C.; Visser, J. CC-1065 (NSC 298223), a Potent New Antitumor Agent. Improved Production and Isolation, Characterization and Antitumor Activity. J. Antibiot. 1981, 34, 1119–1125. [Google Scholar] [PubMed]
- Ichimura, M.; Ogawa, T.; Takahashi, K.; Kobayashi, E.; Kawamoto, I.; Yasuzawa, T.; Takahashi, I.; Nakano, H. Duocarmycin SA, a New Antitumor Antibiotic from Streptomyces sp. J. Antibiot. 1990, 43, 1037–1038. [Google Scholar] [PubMed]
- Tietze, L.F.; Krewer, B. Novel Analogues of CC-1065 and the Duocarmycins for the Use in Targeted Tumour Therapies. Anti-Cancer Agents Med. Chem. 2009, 9, 304–325. [Google Scholar]
- Baraldi, P.G.; Bovero, A.; Fruttarolo, F.; Preti, D.; Tabrizi, M.A.; Pavani, M.G.; Romagnoli, R. DNA Minor Groove Binders as Potential Antitumor and Antimicrobial Agents. Med. Res. Rev. 2004, 24, 475–528. [Google Scholar] [PubMed]
- Boger, D.L.; Johnson, D.S. CC-1065 and the Duocarmycins: Understanding Their Biological Function through Mechanistic Studies. Angew. Chem. Int. Ed. Eng. 1996, 35, 1438–1474. [Google Scholar]
- Tietze, L.F.; Major, F.; Schuberth, I. Antitumor Agents: Development of Highly Potent Glycosidic Duocarmycin Analogues for Selective Cancer Therapy. Angew. Chem. Int. Ed. 2006, 45, 6574–6577. [Google Scholar]
- Tietze, L.F.; Major, F.; Schuberth, I.; Spiegl, D.A.; Krewer, B.; Maksimenka, K.; Bringmann, G.; Magull, J. Selective treatment of Cancer: Synthesis, Biological Evaluation and Structural Elucidation of Novel Analogues of the Antibiotic CC-1065 and the Duocarmycins. Chem. Eur. J. 2007, 13, 4396–4409. [Google Scholar]
- Tietze, L.F.; von Hof, J.M.; Krewer, B.; Müller, M.; Major, F.; Schuster, H.J.; Schuberth, I.; Alves, F. Asymmetric Synthesis and Biological Evaluation of New Glycosidic Prodrugs for a Selective Cancer Therapy. ChemMedChem 2008, 3, 1946–1955. [Google Scholar] [PubMed]
- Tietze, L.F.; Krewer, B.; Major, F.; Schuberth, I. CD-Spectroscopy as a Powerful Tool for Investigating the Mode of Action of Unmodified Drugs in Living Cells. J. Am. Chem. Soc. 2009, 131, 13031–13036. [Google Scholar] [PubMed]
- Tietze, L.F.; Feuerstein, T.; Fecher, A.; Haunert, F.; Panknin, O.; Borchers, U.; Schuberth, I.; Alves, F. Proof of Principle in the Selective Treatment of Cancer by Antibody Directed Enzyme Prodrug Therapy. The Development of a Highly Potent Prodrug. Angew. Chem. Int. Ed. 2002, 41, 759–761. [Google Scholar] [CrossRef]
- Tietze, L.F.; Feuerstein, T. Highly Selective Compounds for the Antibody-Directed Enzyme Prodrug Therapy of Cancer. Aust. J. Chem. 2003, 56, 841–854. [Google Scholar]
- Boger, D.L.; Ishizaki, T.; Wysocki, R.J., Jr.; Munk, S.A.; Kitos, P.A.; Suntornwat, O. Total Synthesis and Evaluation of (±)-N-(tert-Butyloxycarbonyl)-CBI, (±)-CBI-CDPI1, and (±)-CBI-CDPI2: CC-1065 Functional Agents Incorporating the Equivalent 1,2,9,9a-Tetrahydrocycloprop[1,2-cbenz[1,2-e]indol-4-one (CBI) Left-Hand Subunit. J. Am. Chem. Soc. 1989, 111, 6461–6463. [Google Scholar]
- Boger, D.L.; Ishizaki, T. Resolution of a CBI Precursor and Incorporation into the Synthesis of (+)-CBI, (+)-CBI-CDPI1, (+)-CBI-CDPI2: Enhanced Functional Analogs of (+)-CC-1065. A Critical Appraisal of a Proposed Relationship Between Electrophile Reactivity, DNA Binding Properties,and Cytotoxic Potency. Tetrahedron Lett. 1990, 31, 793–794. [Google Scholar] [CrossRef]
- Boger, D.L.; Ishizaki, T. Synthesis of N-(tert-Butyloxycarbonyl)-CBI, CBI, CBI-CDPI1, and CBI-CDPI2: Enhanced Functional Analogues of CC-1065 Incorporating the 1,2,9,9a-Tetrahydrocyclopropa[1,2-c]benz[1,2-e]indol-4-one (CBI) Left-Hand Subunit. J. Org. Chem. 1990, 55, 5823–5832. [Google Scholar]
- Jin, W.; Trzupek, J.D.; Rayl, T.J.; Broward, M.A.; Vielhauer, G.A.; Weir, S.J.; Hwang, I.; Boger, D.L. A Unique Class of Duocarmycin and CC-1065 Analogues Subject to Reductive Activation. J. Am. Chem. Soc. 2007, 129, 15391–15397. [Google Scholar] [PubMed]
- Tietze, L.F.; Krewer, B.; Frauendorf, H. Investigation of the transformations of a novel anticancer agent combining HPLC, HPLC-MS and direct ESI-HRMS analyses. Anal. Bioanal. Chem. 2009, 395, 437–448. [Google Scholar] [PubMed]
- Tietze, L.F.; von Hof, J.M.; Krewer, B. Unpublished Results.
- Tietze, L.F.; Krewer, B.; Frauendorf, H.; Major, F.; Schuberth, I. Investigation of Reactivity and Selectivity of DNA-Alkylating Duocarmycin Analogues by High-Resolution Mass Spectrometry. Angew. Chem. Int. Ed. 2006, 45, 6570–6574. [Google Scholar]
- Tietze, L.F.; Krewer, B.; Frauendorf, H. Probing the mechanism of action of potential anticancer agents at a molecular level using electrospray Fourier transform ion cyclotron resonance mass spectrometry. Eur. J. Mass. Spectrom. 2009, 15, 661–672. [Google Scholar]
- Shinohara, K.-I.; Bando, T.; Sasaki, S.; Sakakibara, Y.; Minoshima, M.; Sugiyama, H. Antitumor activity of sequence-specific alkylating agents: Pyrrole-imidazole CBI conjugates with indole linker. Cancer Sci. 2006, 97, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.M.; Ferguson, L.R.; Denny, W.A. DNA and the chromosome-varied targets for chemotherapy. Cell Chromosome 2004, 3:2. [Google Scholar]
- Tichenor, M.S.; MacMillen, K.S.; Stover, J.S.; Wolkenberg, S.E.; Pavani, M.G.; Zanella, L.; Zaid, A.N.; Spalluto, G.; Rayl, T.J.; Hwang, I.; Baraldi, P.G.; Boger, D.L. Rational Design, Synthesis, and Evaluation of Key Analogues of CC-1065 and the Duocarmycins. J. Am Chem. Soc. 2007, 129, 14092–14099. [Google Scholar] [PubMed]
- Parrish, J.P.; Hughes, T.V.; Hwang, I.; Boger, D.L. Establishing the Parabolic Relationship between Reactivity and Activity for Derivatives and Analogues of the Duocarmycins and CC‑1065 Alkylation Subunits. J. Am. Chem. Soc. 2004, 126, 80–81. [Google Scholar] [PubMed]
- Tietze, L.F.; Hannemann, R.; Buhr, W.; Lögers, M.; Menningen, P.; Lieb, M.; Starck, D.; Grote, T.; Döring, A.; Schuberth, I. Prodrugs of the Cytostatic CC-1065 That Can Be Activated in a Tumor-Selective Manner. Angew. Chem. Int. Ed. Engl. 1996, 35, 2674–2677. [Google Scholar]
- Milbank, J.J.B.; Tercel, M.; Atwell, G.J.; Wilson, W.R.; Hogg, A.; Denny, W.A. Synthesis of 1-Substituted 3-(Chloromethyl)-6-aminoindoline (6-Amino-seco-CI) DNA Minor Groove Alkylating Agents and Structure-Activity Relationships for Their Cytotoxicity. J. Med. Chem. 1999, 42, 649–658. [Google Scholar] [PubMed]
- Atwell, G.J.; Milbank, J.J.B.; Wilson, W.R.; Hogg, A.; Denny, W.A. 5-Amino-1-(chloromethyl)-1,2-dihydro-3H-benz[e]indoles: Relationships between Structure and Cytotoxicity for Analogues Bearing Different DNA Minor Groove Binding Subunits. J. Med. Chem. 1999, 42, 3400–3411. [Google Scholar] [PubMed]
- Parrish, J.P.; Kastrinsky, D.B.; Stauffer, F.; Hedrick, M.P.; Hwang, I.; Boger, D.L. Establishment of Substituent Effects in the DNA Binding Subunit of CBI Analogues of the Duocarmycins and CC-1065. Bioorg. Med. Chem. 2003, 11, 3815–3838. [Google Scholar] [PubMed]
- Boger, D.L.; Bollinger, B.; Johnson, D.S. Examination of the role of the duocarmycin SA methoxy substituents: identification of the minimum, fully potent DNA binding subunit. Bioorg. Med. Chem. Lett. 1996, 6, 2207–2210. [Google Scholar]
- Hofstadler, S.A.; Sannes-Lowery, K.A.; Hannis, J.C. Analysis of Nucleic Acids by FTICR MS. Mass Spectrom. Rev. 2005, 24, 265–285. [Google Scholar] [CrossRef] [PubMed]
- Banoub, J.H.; Newton, R.P.; Esmans, E.; Ewing, D.F.; Mackenzie, G. Recent Developments in Mass Spectrometry for the Characterization of Nucleosides, Nucleotides, Oligonucleotides, and Nucleic Acids. Chem. Rev. 2005, 105, 1869–1915. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.L.; Colgrave, M.L.; Stephen, F.R.; Sheil, M.M. Electrospray Ionization Mass Spectrometry of Oligonucleotide Complexes with Drugs, Metals, and Proteins. Mass Spectrom. Rev. 2001, 20, 61–87. [Google Scholar] [CrossRef] [PubMed]
- McLuckey, S.; Habibi-Goudarzi, S. Decompositions of Multiply Charged Oligonucleotide Anions. J. Am. Chem. Soc. 1993, 115, 12085–12095. [Google Scholar] [CrossRef]
- Cao, P.-R.; McHugh, M.M.; Melendy, T.; Beerman, T. The DNA Minor Groove-alkylating Cyclopopylpyrroloindole Drugs Adozelesin and Bizelesin Induce Different DNA Damage Response Pathways in Human Colon Carcinoma HCT116 Cells. Mol. Canc. Ther. 2003, 2, 651–659. [Google Scholar]
- Schärer, O.D. Chemistry and Biology of DNA Repair. Angew. Chem. Int. Ed. 2003, 42, 2946–2974. [Google Scholar] [CrossRef]
- Parrish, J.P.; Hughes, T.V.; Hwang, I.; Boger, D.L. Establishing the Parabolic Relationship between Reactivity and Activity for Derivatives and Analogues of Duocarmycin and CC-1065 Alkylation Subunits. J. Am. Chem. Soc. 2004, 126, 80–81. [Google Scholar] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tietze, L.F.; Krewer, B.; Von Hof, J.M.; Frauendorf, H.; Schuberth, I. Determination of the Biological Activity and Structure Activity Relationships of Drugs Based on the Highly Cytotoxic Duocarmycins and CC-1065. Toxins 2009, 1, 134-150. https://doi.org/10.3390/toxins1020134
Tietze LF, Krewer B, Von Hof JM, Frauendorf H, Schuberth I. Determination of the Biological Activity and Structure Activity Relationships of Drugs Based on the Highly Cytotoxic Duocarmycins and CC-1065. Toxins. 2009; 1(2):134-150. https://doi.org/10.3390/toxins1020134
Chicago/Turabian StyleTietze, Lutz F., Birgit Krewer, J. Marian Von Hof, Holm Frauendorf, and Ingrid Schuberth. 2009. "Determination of the Biological Activity and Structure Activity Relationships of Drugs Based on the Highly Cytotoxic Duocarmycins and CC-1065" Toxins 1, no. 2: 134-150. https://doi.org/10.3390/toxins1020134
APA StyleTietze, L. F., Krewer, B., Von Hof, J. M., Frauendorf, H., & Schuberth, I. (2009). Determination of the Biological Activity and Structure Activity Relationships of Drugs Based on the Highly Cytotoxic Duocarmycins and CC-1065. Toxins, 1(2), 134-150. https://doi.org/10.3390/toxins1020134