Pain-Causing Venom Peptides: Insights into Sensory Neuron Pharmacology




Abstract
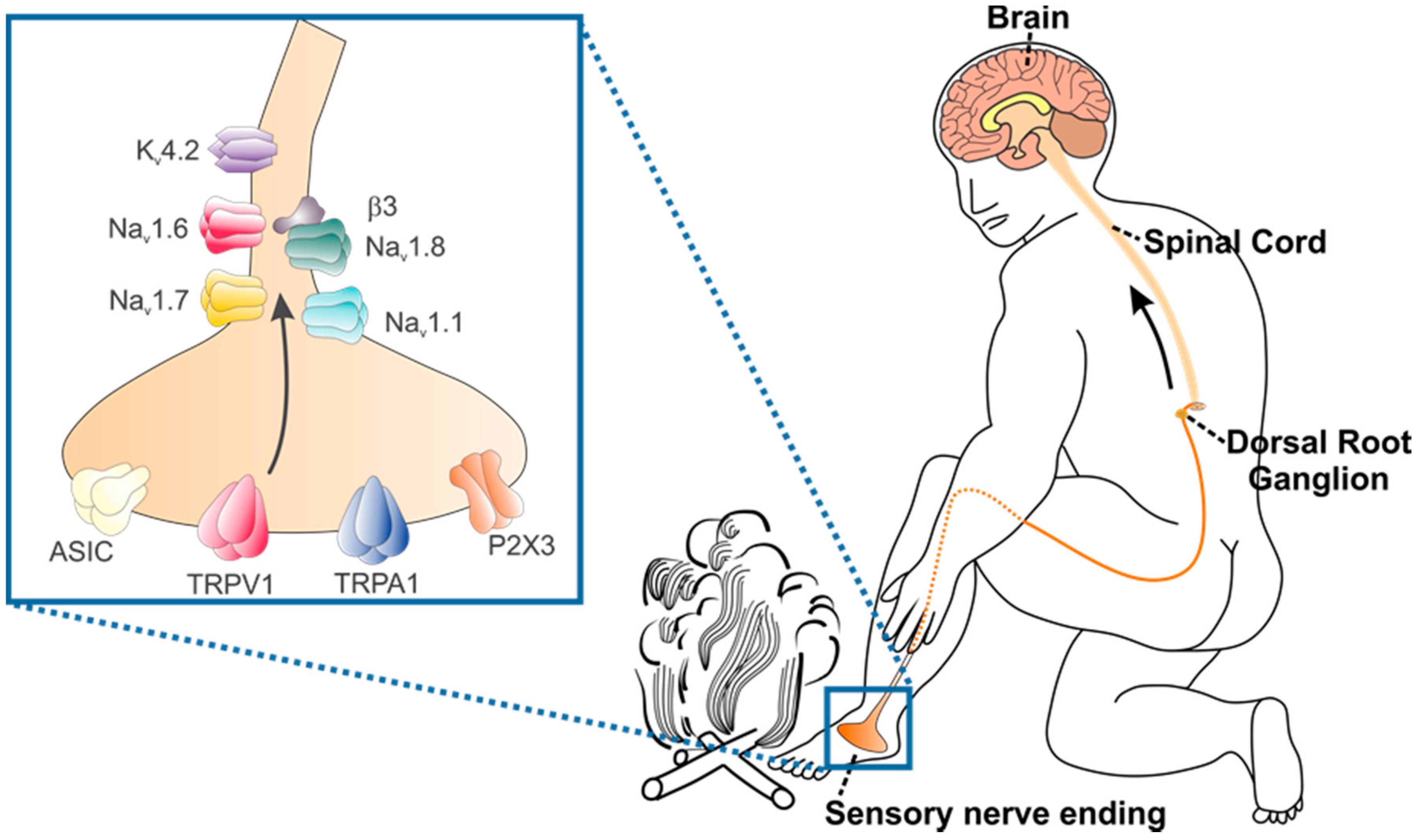
:1. Venoms and Their Pharmacological Effects
2. The Neurobiology of Peripheral Pain Detection and Processing
3. Voltage-Gated Sodium Channels
4. Transient Receptor Potential Channels
4.1. TRPV1
4.2. TRPA1
5. Voltage-Gated Potassium Channels
6. Acid-Sensing Ion Channels
7. Phospholipase A2
8. Pore-Forming Venom Peptides
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- SuppLewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar]
- Palagi, A.; Koh, J.M.; Leblanc, M.; Wilson, D.; Dutertre, S.; King, G.F.; Nicholson, G.M.; Escoubas, P. Unravelling the complex venom landscapes of lethal Australian funnel-web spiders (Hexathelidae: Atracinae) using LC-MALDI-TOF mass spectrometry. J. Proteom. 2013, 80, 292–310. [Google Scholar] [CrossRef] [PubMed]
- Phuong, M.A.; Mahardika, G.N.; Alfaro, M.E. Dietary breadth is positively correlated with venom complexity in cone snails. BMC Genom. 2016, 17, 401. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.; Davis, J.L.; Rash, L.D.; Anangi, R.; Mobli, M.; Alewood, P.F.; Lewis, R.J.; King, G.F. Venomics: A new paradigm for natural products-based drug discovery. Amino Acids 2011, 40, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Panagides, N.; Jackson, T.N.; Ikonomopoulou, M.P.; Arbuckle, K.; Pretzler, R.; Yang, D.C.; Ali, S.A.; Koludarov, I.; Dobson, J.; Sanker, B.; et al. How the Cobra Got Its Flesh-Eating Venom: Cytotoxicity as a Defensive Innovation and Its Co-Evolution with Hooding, Aposematic Marking, and Spitting. Toxins 2017, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J.; Callaghan, B.; Berecki, G. Analgesic contoxins: Block and G-protein-coupled receptor modulation of the N-type (CaV2.2) calcium channels. Br. J. Pharmacol. 2012, 166, 486–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetter, I.; Deuis, J.R.; Mueller, A.; Israel, M.R.; Starobova, H.; Zhang, A.; Rash, L.D.; Mobli, M. NaV1.7 as a pain target—From gene to pharmacology. Pharmacol. Ther. 2017, 172, 73–100. [Google Scholar]
- Vetter, I.; Lewis, R.J. Therapeutic potential of cone snail venom peptides (conopeptides). Curr. Top. Med. Chem. 2012, 12, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Jin, A.H.; Vetter, I.; Hamilton, B.; Sunagar, K.; Lavergne, V.; Dutertre, V.; Fry, B.G.; Antunes, A.; Venter, D.J.; et al. Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails. Nat. Commun. 2014, 5, 3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgenstern, D.; King, G.F. The venom optimization hypothesis revisited. Toxicon 2013, 63, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Boeve, J.L.; Kuhn-Nentwig, L.; Keller, S.; Nentwig, W. Quantity and quality of venom released by a spider (Cupiennius salei, Ctenidae). Toxicon 1995, 33, 1347–1357. [Google Scholar] [CrossRef]
- Carr, D.B.; Goudas, L.C. Acute pain. Lancet 1999, 353, 2051–2058. [Google Scholar] [CrossRef]
- Nagasako, E.M.; Oaklander, A.L.; Dworkin, R.H. Congenital insensitivity to pain: An update. Pain 2003, 101, 213–219. [Google Scholar] [CrossRef]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Schaible, H.G. Peripheral and central mechanisms of pain generation. Handb. Exp. Pharmacol. 2007, 177, 3–28. [Google Scholar]
- Du, X.; Hao, H.; Gigout, S.; Huang, D.; Yang, Y.; Li, L.; Wang, C.; Sundt, D.; Jaffe, D.B.; Zhang, H.; et al. Control of somatic membrane potential in nociceptive neurons and its implications for peripheral nociceptive transmission. Pain 2014, 155, 2306–2322. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Leipold, E.; Liebmann, L.; Korenke, G.C.; Heinrich, T.; Giesselmann, S.; Baets, J.; Ebbinghaus, M.; Goral, R.O.; Stodberg, T.; Hennings, J.C.; et al. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat. Genet. 2013, 45, 1399–1404. [Google Scholar]
- Zhang, X.Y.; Wen, J.; Yang, W.; Wang, C.; Gao, L.; Zheng, L.H.; Wang, T.; Ran, K.; Li, Y.; Li, X.; et al. Gain-of-function mutations in SCN11A cause familial episodic pain. Am. J. Hum. Genet. 2013, 93, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Inserra, M.C.; Israel, M.R.; Caldwell, A.; Castro, J.; Deuis, J.R.; Harrington, A.M.; Keramidas, A.; Garcia-Caraballo, S.; Maddern, J.; Erickson, A.; et al. Multiple sodium channel isoforms mediate the pathological effects of Pacific ciguatoxin-1. Sci. Rep. 2017, 7, 42810. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.; Touska, F.; Hess, A.; Hinsbey, R.; Sattler, S.; Lampert, A.; Sergejeva, M.; Sharov, A.; Collins, L.S.; Eberhardt, M.; et al. Ciguatoxins activate specific cold pain pathways to elicit burning pain from cooling. EMBO J. 2012, 31, 3795–3808. [Google Scholar] [CrossRef] [PubMed]
- Deuis, J.R.; Zimmermann, K.; Romanovsky, A.A.; Possani, L.D.; Cabot, P.J.; Lewis, R.J.; Vetter, I. An animal model of oxaliplatin-induced cold allodynia reveals a crucial role for Nav1.6 in peripheral pain pathways. Pain 2013, 154, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Rowe, A.H.; Xiao, Y.; Scales, J.; Linse, K.D.; Rowe, M.P.; Cummins, T.R.; Zakon, H.H. Isolation and Characterization of CvIV4: A Pain Inducing α—Scorpion Toxin. PLoS ONE 2011, 6, e23520. [Google Scholar] [CrossRef] [PubMed]
- Deuis, J.R.; Wingerd, J.S.; Winter, Z.; Durek, T.; Dekan, Z.; Sousa, S.R.; Zimmermann, K.; Hoffmann, T.; Weidner, C.; Nassar, M.A.; et al. Analgesic Effects of GpTx-1, PF-04856264 and CNV1014802 in a Mouse Model of NaV1.7-Mediated Pain. Toxins 2016, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Durek, T.; Vetter, I.; Wang, C.I.; Motin, L.; Knapp, O.; Adams, D.J.; Lewis, R.J.; Alewood, P.F. Chemical engineering and structural and pharmacological characterization of the alpha-scorpion toxin OD1. ACS Chem. Biol. 2013, 8, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Osteen, J.D.; Herzig, V.; Gilchrist, J.; Emrick, J.J.; Zhang, C.; Wang, X.; Castro, J.; Garcia-Caraballo, S.; Grundy, L.; Rychkov, G.Y.; et al. Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature 2016, 534, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Sahara, Y.; Gotoh, M.; Konno, K.; Miwa, A.; Tsubokawa, H.; Robinson, H.P.; Kawai, N. A new class of neurotoxin from wasp venom slows inactivation of sodium current. Eur. J. Neurosci. 2000, 12, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.C.; Deuis, J.R.; Dashevsky, D.; Dobson, J.; Jackson, T.N.; Brust, A.; Xie, B.; Koludarov, I.; Debono, J.; Hendrikx, I.; et al. The Snake with the Scorpion’s Sting: Novel Three-Finger Toxin Sodium Channel Activators from the Venom of the Long-Glanded Blue Coral Snake (Calliophis bivirgatus). Toxins 2016, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- Klinger, A.B.; Eberhardt, M.; Link, A.S.; Namer, B.; Kutsche, L.K.; Schuy, E.T.; Sittl, R.; Hoffmann, T.; Alzheimer, C.; Huth, T.; et al. Sea-anemone toxin ATX-II elicits A-fiber-dependent pain and enhances resurgent and persistent sodium currents in large sensory neurons. Mol. Pain 2012, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.H.; Israel, M.R.; Inserra, M.C.; Smith, J.J.; Lewis, R.J.; Alewood, P.F.; Vetter, I.; Dutertre, S. Delta-Conotoxin SuVIA suggests an evolutionary link between ancestral predator defence and the origin of fish-hunting behaviour in carnivorous cone snails. Proc. Biol. Sci. 2015, 282, 1811. [Google Scholar] [CrossRef] [PubMed]
- Deuis, J.R.; Mueller, A.; Israel, M.R.; Vetter, I. The pharmacology of voltage-gated sodium channel activators. Neuropharmacology 2017, 127, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.R.; Tay, B.; Deuis, J.R.; Vetter, I. Sodium Channels and Venom Peptide Pharmacology. Adv. Pharmacol. 2017, 79, 67–116. [Google Scholar] [PubMed]
- Kuiken, S.D.; Lindeboom, R.; Tytgat, G.N.; Boeckxstaens, G.E. Relationship between symptoms and hypersensitivity to rectal distension in patients with irritable bowel syndrome. Aliment. Pharmacol. Ther. 2005, 22, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Verne, G.N. New insights into visceral hypersensitivity—Clinical implications in IBS. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Rowe, A.H.; Xiao, Y.; Rowe, M.P.; Cummins, T.R.; Zakon, H.H. Voltage-Gated Sodium Channel in Grasshopper Mice Defends Against Bark Scorpion Toxin. Science 2013, 342, 441–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopp, B.H.; Arvidson, R.S.; Adams, M.E.; Razak, K.A. Arizona bark scorpion venom resistance in the pallid bat, Antrozous pallidus. PLoS ONE 2017, 12, e0183215. [Google Scholar] [CrossRef] [PubMed]
- Klint, J.K.; Senff, S.; Rupasinghe, D.B.; Er, S.Y.; Herzig, V.; Nicholson, G.M.; King, G.F. Spider-venom peptides that target voltage-gated sodium channels: Pharmacological tools and potential therapeutic leads. Toxicon 2012, 60, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Montell, C. TRP Channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Owsianik, G. The transient receptor potential family of ion channels. Genom. Biol. 2011, 12, 218. [Google Scholar] [CrossRef] [PubMed]
- Togashi, K.; Hara, Y.; Tominaga, T.; Higashi, T.; Konishi, Y.; Mori, Y.; Tominaga, M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006, 25, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, K.; Lennerz, J.K.; Hein, A.; Link, A.S.; Kaczmarek, J.S.; Delling, M.; Uysal, S.; Pfeifer, J.D.; Riccio, A.; Clapham, D.E. Transient receptor potential cation channel, subfamily C, member 5 (TRPC5) is a cold-transducer in the peripheral nervous system. Proc. Natl. Acad. Sci. USA 2011, 108, 18114–18119. [Google Scholar] [CrossRef] [PubMed]
- Story, G.M.; Peier, A.M.; Reeve, A.J.; Eid, S.R.; Mosbacher, J.; Hricik, T.R.; Earley, T.J.; Hergarden, A.C.; Andersson, D.A.; Hwang, S.W.; et al. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 2003, 112, 819–829. [Google Scholar] [CrossRef]
- Vriens, J.; Owsianik, G.; Hofmann, T.; Philipp, S.E.; Stab, J.; Chen, X.; Benoit, M.; Xue, F.; Janssens, A.; Kerselaers, S.; et al. TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 2011, 70, 482–494. [Google Scholar] [CrossRef] [PubMed]
- McKemy, D.D.; Neuhausser, W.M.; Julius, D. Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 2002, 416, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Talavera, K.; Yasumatsu, K.; Voets, T.; Droogmans, G.; Shigemura, N.; Ninomiya, Y.; Margolskee, R.F.; Nilius, B. Heat activation of TRPM5 underlies thermal sensitivity of sweet taste. Nature 2005, 438, 1022–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, S.; Tominaga, M. Functional diversity and evolutionary dynamics of thermoTRP channels. Cell Calcium 2015, 57, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; McNaughton, P.A. The TRPM2 ion channel is required for sensitivity to warmth. Nature 2016, 536, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Patapoutian, A.; Peier, A.M.; Story, G.M.; Viswanath, V. ThermoTRP channels and beyond: Mechanisms of temperature sensation. Nat. Rev. Neurosci. 2003, 4, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Szallasi, A.; Cortright, D.N.; Blum, C.A.; Eid, S.R. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat. Rev. Drug Discov. 2007, 6, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [PubMed]
- Zhang, N.; Inan, S.; Cowan, A.; Sun, R.; Wang, J.M.; Rogers, T.J.; Caterina, M.; Oppenheim, J.J. A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc. Natl. Acad. Sci. USA 2005, 102, 4536–4541. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yang, F.; Wei, N.; Hong, J.; Li, B.; Luo, L.; Rong, M.; Yarov-Yarovoy, V.; Zheng, J.; Wang, K.; et al. A pain-inducing centipede toxin targets the heat activation machinery of nociceptor TRPV1. Nat. Commun. 2015, 6, 8297. [Google Scholar] [CrossRef] [PubMed]
- Hakim, M.A.; Jiang, W.; Luo, L.; Li, B.; Yang, S.; Song, Y.; Lai, R. Scorpion Toxin, BmP01, Induces Pain by Targeting TRPV1 Channel. Toxins 2015, 7, 3671–3687. [Google Scholar] [CrossRef] [PubMed]
- Bohlen, C.J.; Priel, A.; Zhou, S.; King, D.; Siemens, J.; Julius, D. A bivalent tarantula toxin activates the capsaicin receptor, TRPV1, by targeting the outer pore domain. Cell 2010, 141, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Geron, M.; Kumar, R.; Matzner, H.; Lahiani, A.; Gincberg, G.; Cohen, G.; Lazarovici, P.; Priel, A. Protein toxins of the Echis coloratus viper venom directly activate TRPV1. Biochim. Biophys. Acta 2017, 1861, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Siemens, J.; Zhou, S.; Piskorowski, R.; Nikai, T.; Lumpkin, E.A.; Basbaum, A.I.; King, D.; Julius, D. Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature 2006, 444, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yang, F.; Zhang, B.; Lee, B.H.; Li, B.; Luo, L.; Zheng, J.; Lai, R. A bimodal activation mechanism underlies scorpion toxin–induced pain. Sci. Adv. 2017, 3, e1700810. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Cao, E.; Julius, D.; Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 2016, 534, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Single particle electron cryo-microscopy of a mammalian ion channel. Curr. Opin. Struct. Biol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cao, E.; Liao, M.; Cheng, Y.; Julius, D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 2013, 504, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.; Liu, B.; Cao, G.; Lipchik, A.M.; Perez, M.; Dekan, Z.; Mobli, M.; Daly, N.L.; Alewood, P.F.; Parker, L.L.; et al. A tarantula-venom peptide antagonizes the TRPA1 nociceptor ion channel by binding to the S1–S4 gating domain. Curr. Biol. 2014, 24, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Bressan, E.; Touska, F.; Vetter, I.; Kistner, K.; Kichko, T.I.; Teixeira, N.B.; Picolo, G.; Cury, Y.; Lewis, R.J.; Fischer, M.J.; et al. Crotalphine desensitizes TRPA1 ion channels to alleviate inflammatory hyperalgesia. Pain 2016, 157, 2504–2516. [Google Scholar] [CrossRef] [PubMed]
- Tonello, R.; Fusi, C.; Materazzi, S.; Marone, I.M.; de Logu, F.; Benemei, S.; Goncalves, M.C.; Coppi, E.; Castro-Junior, C.J.; Gomez, M.V.; et al. The peptide Phalpha1beta, from spider venom, acts as a TRPA1 channel antagonist with antinociceptive effects in mice. Br. J. Pharmacol. 2017, 174, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Logashina, Y.A.; Mosharova, I.V.; Korolkova, Y.V.; Shelukhina, I.V.; Dyachenko, I.A.; Palikov, V.A.; Palikova, Y.A.; Murashev, A.N.; Kozlov, S.A.; Stensvag, K.; et al. Peptide from Sea Anemone Metridium senile Affects Transient Receptor Potential Ankyrin-repeat 1 (TRPA1) Function and Produces Analgesic Effect. J. Biol. Chem. 2017, 292, 2992–3004. [Google Scholar] [CrossRef] [PubMed]
- Konno, K.; Picolo, G.; Gutierrez, V.P.; Brigatte, P.; Zambelli, V.O.; Camargo, A.C.M.; Cury, Y. Crotalphine, a novel potent analgesic peptide from the venom of the South American rattlesnake Crotalus durissus terrificus. Peptides 2008, 29, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Zhang, J.R.; Wang, Y.; Li, C.L.; Lu, D.; Guan, S.M.; Chen, J. Effects of a non-selective TRPC channel blocker, SKF-96365, on melittin-induced spontaneous persistent nociception and inflammatory pain hypersensitivity. Neurosci. Bull. 2012, 28, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Grizel, A.V.; Glukhov, G.S.; Sokolova, O.S. Mechanisms of activation of voltage-gated potassium channels. Acta Nat. 2014, 6, 10–26. [Google Scholar]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science 2005, 309, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; McKinnon, D.; Pardo, L.A.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stuhmer, W.; et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 2005, 57, 473–508. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Gamper, N. Potassium channels in peripheral pain pathways: Expression, function and therapeutic potential. Curr. Neuropharmacol. 2013, 11, 621–640. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulas, C. Emerging potassium channel targets for the treatment of pain. Curr. Opin. Support. Palliat. Care 2015, 9, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 2009, 8, 982–1001. [Google Scholar] [CrossRef] [PubMed]
- Jacob, N.T. Drug targets: Ligand and voltage gated ion channels. Int. J. Basic Clin. Pharmacol. 2017, 6, 11. [Google Scholar] [CrossRef]
- Diochot, S.; Schweitz, H.; Beress, L.; Lazdunski, M. Sea anemone peptides with a specific blocking activity against the fast inactivating potassium channel Kv3.4. J. Biol. Chem. 1998, 273, 6744–6749. [Google Scholar] [CrossRef] [PubMed]
- Pucca, M.B.; Cerni, F.A.; Cordeiro, F.A.; Peigneur, S.; Cunha, T.M.; Tytgat, J.; Arantes, E.C. Ts8 scorpion toxin inhibits the Kv4.2 channel and produces nociception in vivo. Toxicon 2016, 119, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Swartz, K.J.; MacKinnon, R. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron 1995, 15, 941–949. [Google Scholar] [CrossRef]
- Tytgat, J.; Debont, T.; Carmeliet, E.; Daenens, P. The alpha-dendrotoxin footprint on a mammalian potassium channel. J. Biol. Chem. 1995, 270, 24776–24781. [Google Scholar] [CrossRef] [PubMed]
- Busserolles, J.; Tsantoulas, C.; Eschalier, A.; Garcia, J.A.L. Potassium channels in neuropathic pain: Advances, challenges, and emerging ideas. Pain 2016, 157 (Suppl. 1), S7–S14. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Toyoda, H. Role of leak potassium channels in pain signaling. Brain Res. Bull. 2015, 119, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lonnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggstrom, J.; Kharchenko, O.; Kharchenko, P.V.; et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015, 18, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Madrid, R.; de la Pena, E.; Donovan-Rodriguez, T.; Belmonte, C.; Viana, F. Variable threshold of trigeminal cold-thermosensitive neurons is determined by a balance between TRPM8 and Kv1 potassium channels. J. Neurosci. 2009, 29, 3120–3131. [Google Scholar] [CrossRef] [PubMed]
- Cristofori-Armstrong, B.; Rash, L.D. Acid-sensing ion channel (ASIC) structure and function: Insights from spider, snake and sea anemone venoms. Neuropharmacology 2017, 127, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Lee, L.Y. Acid-Sensing Ion Channels and Pain. Pharmaceuticals 2010, 3, 1411–1425. [Google Scholar] [CrossRef] [PubMed]
- Dube, G.R.; Elagoz, A.; Mangat, H. Acid sensing ion channels and acid nociception. Curr. Pharm. Des. 2009, 15, 1750–1766. [Google Scholar] [CrossRef] [PubMed]
- Hesselager, M.; Timmermann, D.B.; Ahring, P.K. pH Dependency and desensitization kinetics of heterologously expressed combinations of acid-sensing ion channel subunits. J. Biol. Chem. 2004, 279, 11006–11015. [Google Scholar] [CrossRef] [PubMed]
- Bohlen, C.J.; Chesler, A.T.; Sharif-Naeini, R.; Medzihradszky, K.F.; Zhou, S.; King, D.; Sanchez, E.E.; Burlingame, A.L.; Basbaum, A.I.; Julius, D. A heteromeric Texas coral snake toxin targets acid-sensing ion channels to produce pain. Nature 2011, 479, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.B.; Scott-Davey, T. Secreted phospholipases A2 of snake venoms: Effects on the peripheral neuromuscular system with comments on the role of phospholipases A2 in disorders of the CNS and their uses in industry. Toxins 2013, 5, 2533–2571. [Google Scholar] [CrossRef] [PubMed]
- Jouiaei, M.; Yanagihara, A.A.; Madio, B.; Nevalainen, T.J.; Alewood, P.F.; Fry, B.G. Ancient Venom Systems: A Review on Cnidaria Toxins. Toxins 2015, 7, 2251–2271. [Google Scholar] [CrossRef] [PubMed]
- Kini, R.M. Excitement ahead: Structure, function and mechanism of snake venom phospholipase A2 enzymes. Toxicon 2003, 42, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Caccin, P.; Rigoni, M.; Bisceglie, A.; Rossetto, O.; Montecucco, C. Reversible skeletal neuromuscular paralysis induced by different lysophospholipids. FEBS Lett. 2006, 580, 6317–6321. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Blumberg, P.M.; Szallasi, A. Endovanilloid signaling in pain. Curr. Opin. Neurobiol. 2002, 12, 372–379. [Google Scholar] [CrossRef]
- Chacur, M.; Longo, I.; Picolo, G.; Gutierrez, J.M.; Lomonte, B.; Guerra, J.L.; Teixeira, C.F.; Cury, Y. Hyperalgesia induced by Asp49 and Lys49 phospholipases A2 from Bothrops asper snake venom: Pharmacological mediation and molecular determinants. Toxicon 2003, 41, 667–678. [Google Scholar] [CrossRef]
- Zambelli, V.O.; Chioato, L.; Gutierrez, V.P.; Ward, R.J.; Cury, Y. Structural determinants of the hyperalgesic activity of myotoxic Lys49-phospholipase A2. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 7. [Google Scholar] [CrossRef] [PubMed]
- Belo, C.A.; Leite, G.B.; Toyama, M.H.; Marangoni, S.; Corrado, A.P.; Fontana, M.D.; Southan, A.; Rowan, E.G.; Hyslop, S.; Rodrigues-Simioni, L. Pharmacological and structural characterization of a novel phospholipase A2 from Micrurus dumerilii carinicauda venom. Toxicon 2005, 46, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Lambeau, G.; Lazdunski, M. Receptors for a growing family of secreted phospholipases A2. Trends Pharmacol. Sci. 1999, 20, 162–170. [Google Scholar] [CrossRef]
- Sharma, S.V. Melittin-induced hyperactivation of phospholipase A2 activity and calcium influx in ras-transformed cells. Oncogene 1993, 8, 939–947. [Google Scholar] [PubMed]
- Raghuraman, H.; Chattopadhyay, A. Melittin: A membrane-active peptide with diverse functions. Biosci. Rep. 2007, 27, 189–223. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guan, S.M.; Sun, W.; Fu, H. Melittin, the Major Pain-Producing Substance of Bee Venom. Neurosci. Bull. 2016, 32, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Villegas, E.; Corzo, G. Pore-Forming Peptides from Spiders. Toxin Rev. 2005, 24, 345–357. [Google Scholar] [CrossRef]
- Sher, D.; Fishman, Y.; Zhang, M.; Lebendiker, M.; Gaathon, A.; Mancheno, J.M.; Zlotkin, E. Hydralysins, a new category of beta-pore-forming toxins in cnidaria. J. Biol. Chem. 2005, 280, 22847–22855. [Google Scholar] [CrossRef] [PubMed]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Dekan, Z.; Headey, S.J.; Scanlon, M.; Baldo, B.A.; Lee, T.H.; Aguilar, M.I.; Deuis, J.R.; Vetter, I.; Elliott, A.G.; Amado, M.; et al. Delta-Myrtoxin-Mp1a is a Helical Heterodimer from the Venom of the Jack Jumper Ant that has Antimicrobial, Membrane-Disrupting, and Nociceptive Activities. Angew.Chem. Int. Ed. Engl. 2017, 56, 8495–8499. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Heesters, B.A.; Ghasemlou, N.; von Hehn, C.A.; Zhao, F.; Tran, J.; Wainger, B.; Strominger, A.; Muralidharan, S.; Horswill, A.R.; et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 2013, 501, 52–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Venom Peptide | Species | Pharmacological Target(s) | Pain Phenotype (Route) | Reference |
|---|---|---|---|---|
| δ-theraphotoxin-Hm1a | Heteroscodra maculata | NaV1.1 | Spontaneous pain (i.pl.), mechanical allodynia (i.pl.) | [27] |
| OD1 | Odontobuthus doriae | NaV1.7 | Spontaneous pain (i.pl.) | [25] |
| Cn2 | Centruroides noxius | NaV1.6 | Spontaneous pain (i.pl.), mechanical allodynia (i.pl.) | [23] |
| δ-conotoxin SuVIA | Conus suturatus | NaV1.3, NaV1.4, NaV1.6, NaV1.7 | Spontaneous pain (i.pl.) | [31] |
| α-scorpion toxin CvIV4 | Centruroides vittatus | NaV1.2, NaV1.3, NaV1.4, NaV1.7 | Spontaneous pain (i.pl.) | [24] |
| Venom Peptide | Species | Pharmacological Target(s) | Reference |
|---|---|---|---|
| RhTx | Scolopendra subspinipes mutilans | TRPV1 | [53] |
| BmP01 | Mesobuthus martensii | TRPV1 | [54] |
| DkTx | Ornithoctonus huwena | TRPV1 | [55] |
| Echis coloratus venom | Echis coloratus | TRPV1 | [56] |
| VaTx1 (τ/κ-theraphotoxin-Pc1a) | Psalmopoeus cambridgei | TRPV1, KV2.1 | [57] |
| VaTx2 (τ-theraphotoxin-Pc1b) | Psalmopoeus cambridgei | TRPV1, KV2.1 | [57] |
| VaTx3 (τ-theraphotoxin-Pc1c) | Psalmopoeus cambridgei | TRPV1 | [57] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jami, S.; Erickson, A.; Brierley, S.M.; Vetter, I. Pain-Causing Venom Peptides: Insights into Sensory Neuron Pharmacology. Toxins 2018, 10, 15. https://doi.org/10.3390/toxins10010015
Jami S, Erickson A, Brierley SM, Vetter I. Pain-Causing Venom Peptides: Insights into Sensory Neuron Pharmacology. Toxins. 2018; 10(1):15. https://doi.org/10.3390/toxins10010015
Chicago/Turabian StyleJami, Sina, Andelain Erickson, Stuart M. Brierley, and Irina Vetter. 2018. "Pain-Causing Venom Peptides: Insights into Sensory Neuron Pharmacology" Toxins 10, no. 1: 15. https://doi.org/10.3390/toxins10010015
APA StyleJami, S., Erickson, A., Brierley, S. M., & Vetter, I. (2018). Pain-Causing Venom Peptides: Insights into Sensory Neuron Pharmacology. Toxins, 10(1), 15. https://doi.org/10.3390/toxins10010015