Development of a Deimmunized Bispecific Immunotoxin dDT2219 against B-Cell Malignancies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
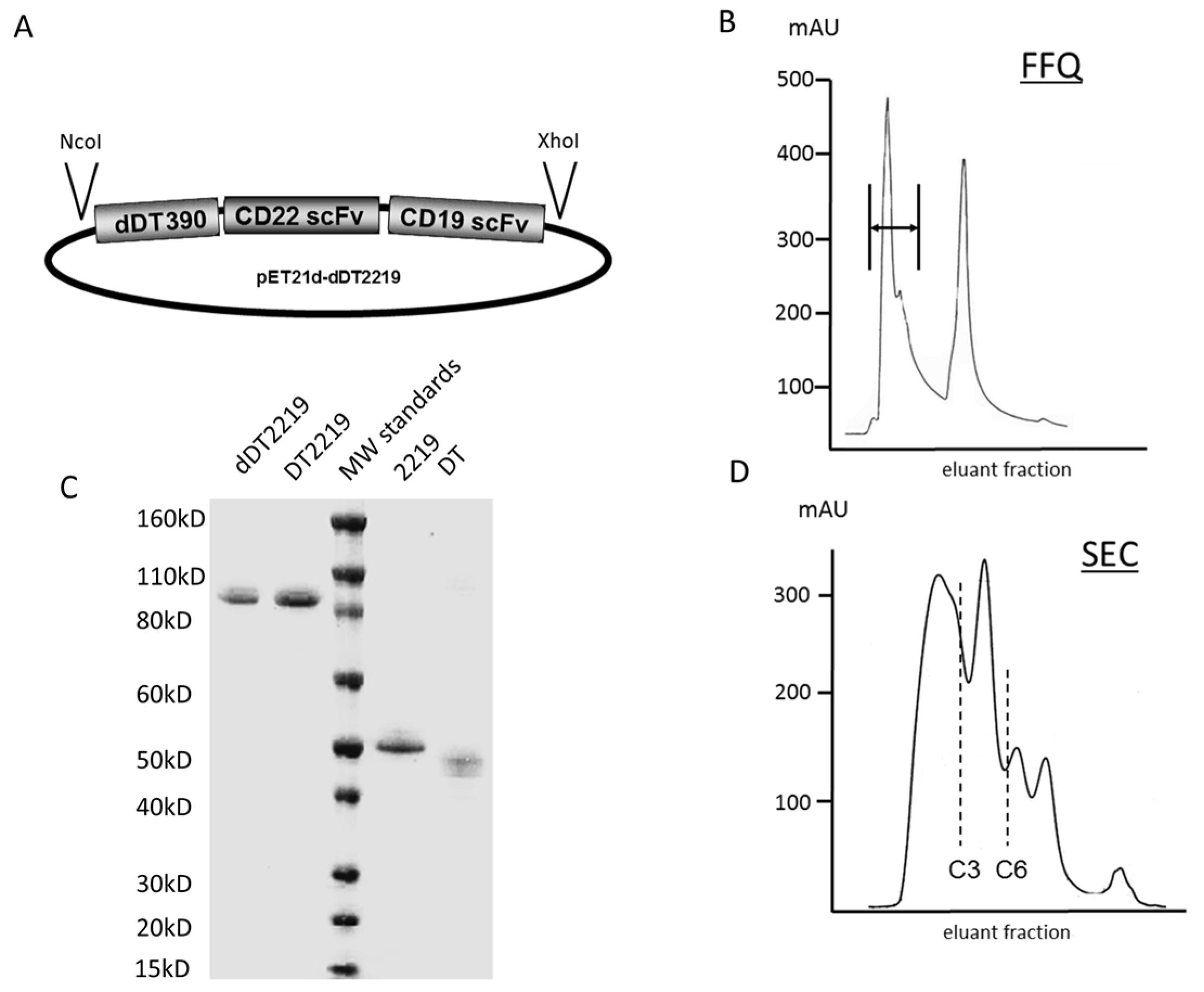
2.1. Construction of dDT2219
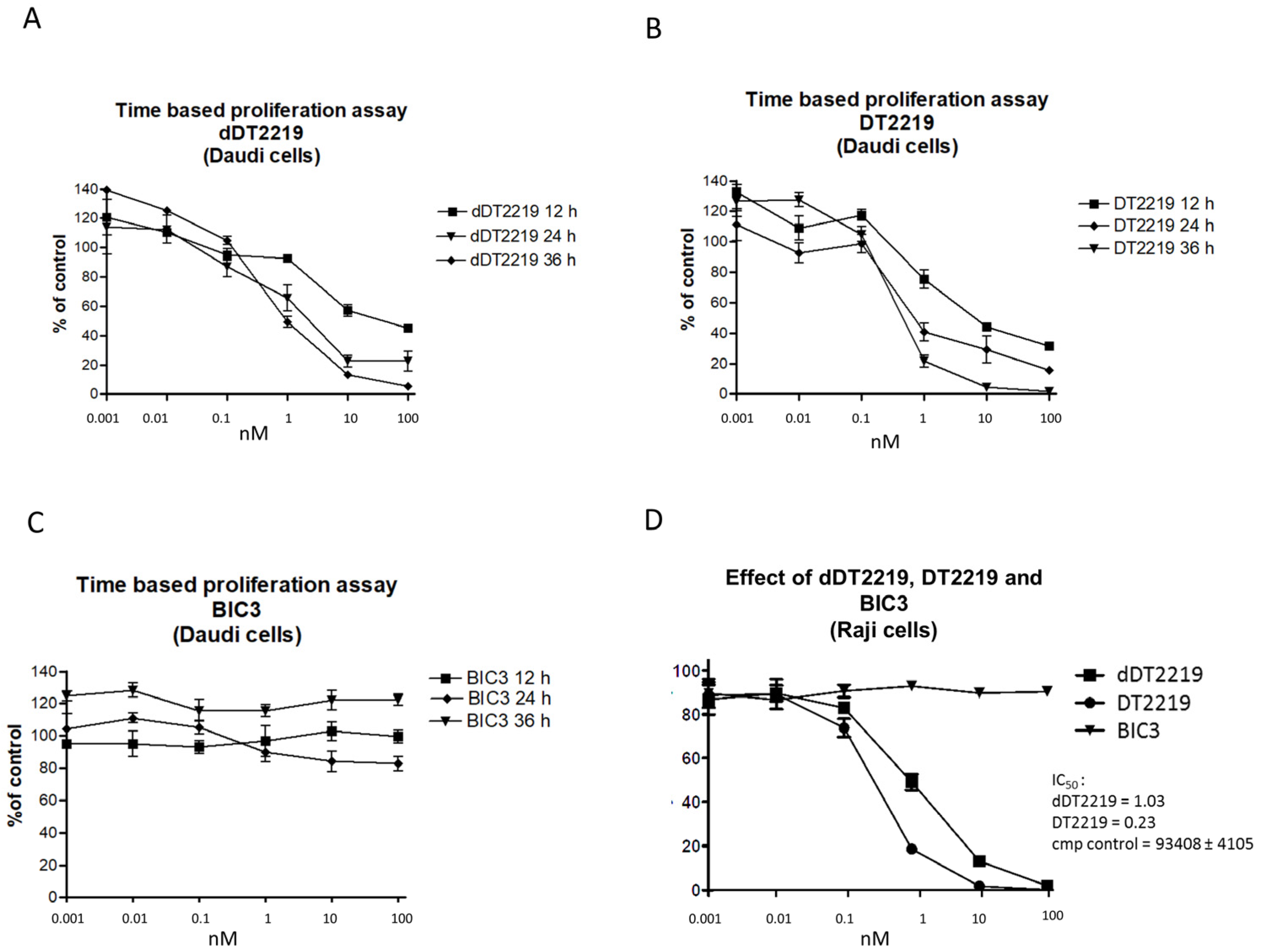
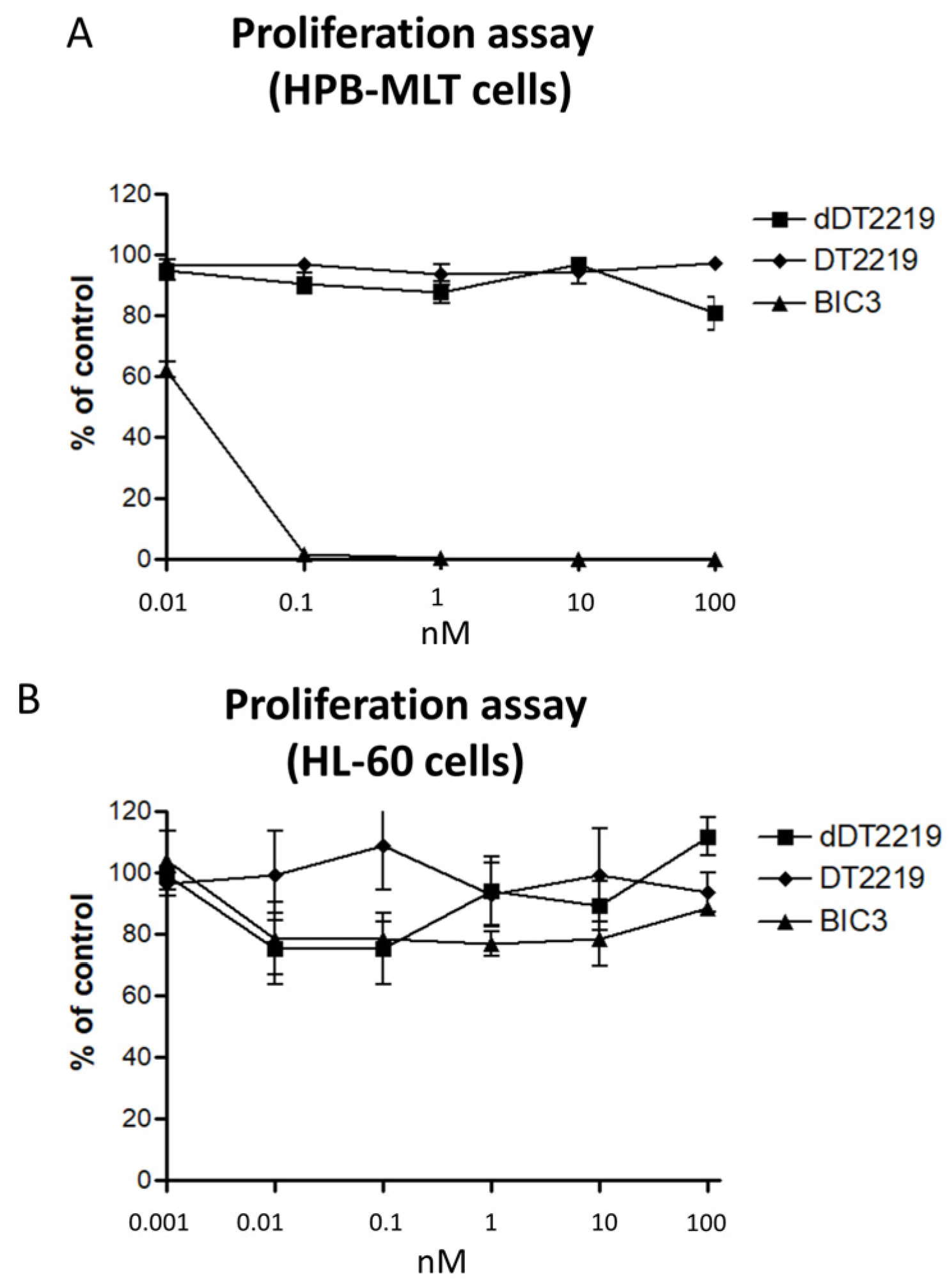
2.2. Activity/Specificity of the Mutated dDT2219
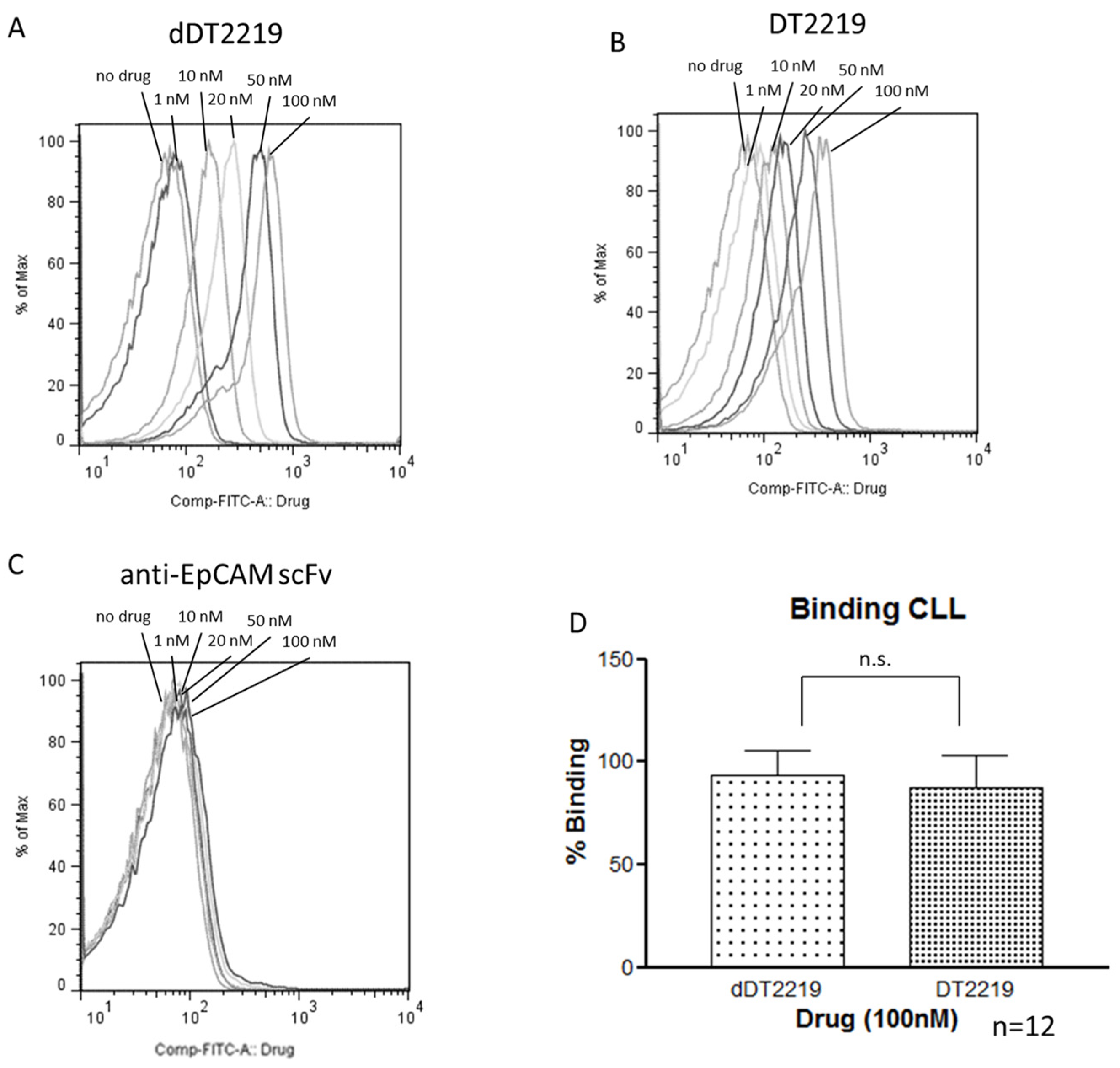
2.3. Binding Characteristics of dDT2219
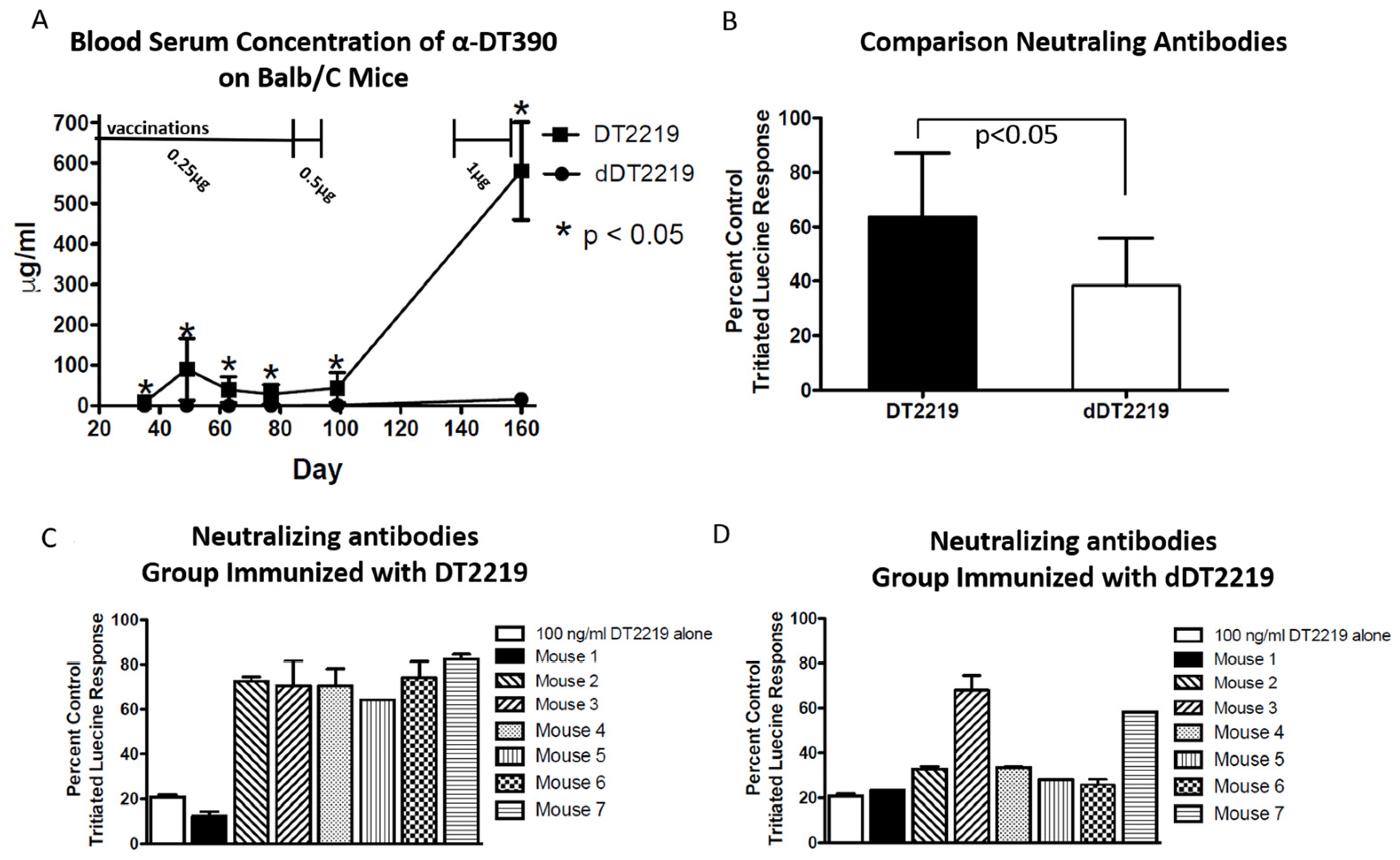
2.4. Immunogenicity in Mice
3. Methods
3.1. Construction of dDT2219
3.2. Inclusion Body Isolation, Refolding, and Purification
3.3. Tissue Cultures
3.4. Proliferation Assay/Blocking Assay
3.5. Patient Samples and Binding Assay
3.6. Detecting Anti-Toxin Antibodies in Mice
3.7. Detecting Neutralizing Antibodies in Mice
3.8. Statistical Analyses
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kreitman, R.J. Recombinant immunotoxins for the treatment of chemoresistant hematologic malignancies. Curr. Pharm. Des. 2009, 15, 2652–2664. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Vallera, D.A.; Hall, W.A. Diphtheria toxin-based targeted toxin therapy for brain tumors. J. Neurooncol. 2013, 114, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Bachanova, V.; Frankel, A.E.; Cao, Q.; Lewis, D.; Grzywacz, B.; Verneris, M.R.; Ustun, C.; Lazaryan, A.; McClune, B.; Warlick, E.D.; et al. Phase i study of a bispecific ligand-directed toxin targeting cd22 and cd19 (dt2219) for refractory b-cell malignancies. Clin. Cancer Res. 2015, 21, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.J. Diphtheria toxin: Mode of action and structure. Bacteriol. Rev. 1975, 39, 54–85. [Google Scholar] [PubMed]
- Tamilvanan, S.; Raja, N.L.; Sa, B.; Basu, S.K. Clinical concerns of immunogenicity produced at cellular levels by biopharmaceuticals following their parenteral administration into human body. J. Drug Target. 2010, 18, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Zuckero, S.L.; Mankin, A.A.; Grable, M.; Mitchell, K.; Lee, Y.J.; Neville, D.M.; Woo, J.H. Anti-cd3 recombinant diphtheria immunotoxin therapy of cutaneous t cell lymphoma. Curr. Drug Targets 2009, 10, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Fleming, D.R.; Powell, B.L.; Gartenhaus, R. Dab389il2 (ontak) fusion protein therapy of chronic lymphocytic leukaemia. Expert Opin. Biol. Ther. 2003, 3, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. Who classification of tumours of haematopoietic and lymphoid tissues; World Cancer Report; IARC: Lyon, France, 2008. [Google Scholar]
- Scheuermann, R.H.; Racila, E. Cd19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk. Lymphoma 1995, 18, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Daridon, C.; Blassfeld, D.; Reiter, K.; Mei, H.E.; Giesecke, C.; Goldenberg, D.M.; Hansen, A.; Hostmann, A.; Frolich, D.; Dorner, T. Epratuzumab targeting of cd22 affects adhesion molecule expression and migration of b-cells in systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12, R204. [Google Scholar] [CrossRef] [PubMed]
- Schmohl, J.U.; Todhunter, D.; Oh, S.; Vallera, D.A. Mutagenic deimmunization of diphtheria toxin for use in biologic drug development. Toxins (Basel) 2015, 7, 4067–4082. [Google Scholar] [CrossRef] [PubMed]
- Schmohl, J.U.; Gleason, M.K.; Dougherty, P.R.; Miller, J.S.; Vallera, D.A. Heterodimeric bispecific single chain variable fragments (scfv) killer engagers (bikes) enhance nk-cell activity against cd133+ colorectal cancer cells. Target. Oncol. 2015, 11, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Schmohl, J.U.; Felices, M.; Taras, E.; Miller, J.S.; Vallera, D.A. Enhanced adcc and nk cell activation of an anti-carcinoma bispecific antibody by genetic insertion of a modified il-15 cross-linker. Mol. Ther. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.; Klein, G.; Nadkarni, J.S.; Nadkarni, J.J.; Wigzell, H.; Clifford, P. Surface igm-kappa specificity on a burkitt lymphoma cell in vivo and in derived culture lines. Cancer Res. 1968, 28, 1300–1310. [Google Scholar] [PubMed]
- Vallera, D.A.; Todhunter, D.; Kuroki, D.W.; Shu, Y.; Sicheneder, A.; Panoskaltsis-Mortari, A.; Vallera, V.D.; Chen, H. Molecular modification of a recombinant, bivalent anti-human cd3 immunotoxin (bic3) results in reduced in vivo toxicity in mice. Leuk. Res. 2005, 29, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Todhunter, D.A.; Panoskaltsis-Mortari, A.; Buchsbaum, D.J.; Toma, S.; Vallera, D.A. A deimmunized bispecific ligand-directed toxin that shows an impressive anti–pancreatic cancer effect in a systemic nude mouse orthotopic model. Pancreas 2012, 41, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Hayes, G.M.; Busch, R.; Voogt, J.; Siah, I.M.; Gee, T.A.; Hellerstein, M.K.; Chiorazzi, N.; Rai, K.R.; Murphy, E.J. Isolation of malignant b cells from patients with chronic lymphocytic leukemia (cll) for analysis of cell proliferation: Validation of a simplified method suitable for multi-center clinical studies. Leuk. Res. 2010, 34, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Olsen, E.; Duvic, M.; Frankel, A.; Kim, Y.; Martin, A.; Vonderheid, E.; Jegasothy, B.; Wood, G.; Gordon, M.; Heald, P.; et al. Pivotal phase iii trial of two dose levels of denileukin diftitox for the treatment of cutaneous t-cell lymphoma. J. Clin. Oncol. 2001, 19, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase i study of ss1p, a recombinant anti-mesothelin immunotoxin given as a bolus i.V. Infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Cancer Res. 2007, 13, 5144–5149. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Wilson, W.H.; White, J.D.; Stetler-Stevenson, M.; Jaffe, E.S.; Giardina, S.; Waldmann, T.A.; Pastan, I. Phase i trial of recombinant immunotoxin anti-tac(fv)-pe38 (lmb-2) in patients with hematologic malignancies. J. Clin. Oncol. 2000, 18, 1622–1636. [Google Scholar] [CrossRef] [PubMed]
- Roscoe, D.M.; Pai, L.H.; Pastan, I. Identification of epitopes on a mutant form of pseudomonas exotoxin using serum from humans treated with pseudomonas exotoxin containing immunotoxins. Eur. J. Immunol. 1997, 27, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Molineux, G. Pegylation: Engineering improved biopharmaceuticals for oncology. Pharmacotherapy 2003, 23, 3S–8S. [Google Scholar] [CrossRef] [PubMed]
- Schirrmann, T.; Krauss, J.; Arndt, M.A.; Rybak, S.M.; Dubel, S. Targeted therapeutic rnases (immunornases). Expert Opin. Biol. Ther. 2009, 9, 79–95. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Williams-Gould, J.; Watson, T.; Pai-Scherf, L.; Pastan, I. Pretreatment with rituximab does not inhibit the human immune response against the immunogenic protein lmb-1. Clin. Cancer Res. 2004, 10, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Onda, M.; Nagata, S.; FitzGerald, D.J.; Beers, R.; Fisher, R.J.; Vincent, J.J.; Lee, B.; Nakamura, M.; Hwang, J.; Kreitman, R.J.; et al. Characterization of the b cell epitopes associated with a truncated form of pseudomonas exotoxin (pe38) used to make immunotoxins for the treatment of cancer patients. J. Immunol. 2006, 177, 8822–8834. [Google Scholar] [CrossRef] [PubMed]
- Atassi, M.Z.; Dolimbek, B.Z.; Hayakari, M.; Middlebrook, J.L.; Whitney, B.; Oshima, M. Mapping of the antibody-binding regions on botulinum neurotoxin h-chain domain 855-1296 with antitoxin antibodies from three host species. J. Protein Chem. 1996, 15, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Bugelski, P.J.; Treacy, G. Predictive power of preclinical studies in animals for the immunogenicity of recombinant therapeutic proteins in humans. Curr. Opin. Mol. Ther. 2004, 6, 10–16. [Google Scholar] [PubMed]
- Atassi, M.Z.; Dolimbek, B.Z. Mapping of the antibody-binding regions on the hn-domain (residues 449-859) of botulinum neurotoxin a with antitoxin antibodies from four host species. Full profile of the continuous antigenic regions of the h-chain of botulinum neurotoxin a. The Protein J. 2004, 23, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Vlasveld, L.T.; Hekman, A.; Vyth-Dreese, F.A.; Melief, C.J.; Sein, J.J.; Voordouw, A.C.; Dellemijn, T.A.; Rankin, E.M. Treatment of low-grade non-hodgkin's lymphoma with continuous infusion of low-dose recombinant interleukin-2 in combination with the b-cell-specific monoclonal antibody clb-cd19. Cancer Immunol. Immunother. 1995, 40, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Grossbard, M.L.; Multani, P.S.; Freedman, A.S.; O'Day, S.; Gribben, J.G.; Rhuda, C.; Neuberg, D.; Nadler, L.M. A phase ii study of adjuvant therapy with anti-b4-blocked ricin after autologous bone marrow transplantation for patients with relapsed b-cell non-hodgkin's lymphoma. Clin. Cancer Res. 1999, 5, 2392–2398. [Google Scholar] [PubMed]
- Topp, M.S.; Gokbuget, N.; Zugmaier, G.; Klappers, P.; Stelljes, M.; Neumann, S.; Viardot, A.; Marks, R.; Diedrich, H.; Faul, C.; et al. Phase ii trial of the anti-cd19 bispecific t cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory b-precursor acute lymphoblastic leukemia. J. Clin. Oncol. 2014, 32, 4134–4140. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.P.; Coleman, M.; Ketas, J.C.; Chadburn, A.; Ely, S.; Furman, R.R.; Wegener, W.A.; Hansen, H.J.; Ziccardi, H.; Eschenberg, M.; et al. Phase i/ii trial of epratuzumab (humanized anti-cd22 antibody) in indolent non-hodgkin's lymphoma. J. Clin. Oncol. 2003, 21, 3051–3059. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Thomas, D.; Jorgensen, J.; Jabbour, E.; Kebriaei, P.; Rytting, M.; York, S.; Ravandi, F.; Kwari, M.; Faderl, S.; et al. Inotuzumab ozogamicin, an anti-cd22-calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: A phase 2 study. Lancet Oncol. 2012, 13, 403–411. [Google Scholar] [CrossRef]
- Vallera, D.A.; Todhunter, D.A.; Kuroki, D.W.; Shu, Y.; Sicheneder, A.; Chen, H. A bispecific recombinant immunotoxin, dt2219, targeting human cd19 and cd22 receptors in a mouse xenograft model of b-cell leukemia/lymphoma. Clin. Cancer Res. 2005, 11, 3879–3888. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Chen, H.; Sicheneder, A.R.; Panoskaltsis-Mortari, A.; Taras, E.P. Genetic alteration of a bispecific ligand-directed toxin targeting human cd19 and cd22 receptors resulting in improved efficacy against systemic b cell malignancy. Leuk. Res. 2009, 33, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Oh, S.; Chen, H.; Shu, Y.; Frankel, A.E. Bioengineering a unique deimmunized bispecific targeted toxin that simultaneously recognizes human cd22 and cd19 receptors in a mouse model of b-cell metastases. Mol. Cancer Ther. 2010, 9, 1872–1883. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmohl, J.U.; Todhunter, D.; Taras, E.; Bachanova, V.; Vallera, D.A. Development of a Deimmunized Bispecific Immunotoxin dDT2219 against B-Cell Malignancies. Toxins 2018, 10, 32. https://doi.org/10.3390/toxins10010032
Schmohl JU, Todhunter D, Taras E, Bachanova V, Vallera DA. Development of a Deimmunized Bispecific Immunotoxin dDT2219 against B-Cell Malignancies. Toxins. 2018; 10(1):32. https://doi.org/10.3390/toxins10010032
Chicago/Turabian StyleSchmohl, Joerg U., Deborah Todhunter, Elizabeth Taras, Veronika Bachanova, and Daniel A. Vallera. 2018. "Development of a Deimmunized Bispecific Immunotoxin dDT2219 against B-Cell Malignancies" Toxins 10, no. 1: 32. https://doi.org/10.3390/toxins10010032
APA StyleSchmohl, J. U., Todhunter, D., Taras, E., Bachanova, V., & Vallera, D. A. (2018). Development of a Deimmunized Bispecific Immunotoxin dDT2219 against B-Cell Malignancies. Toxins, 10(1), 32. https://doi.org/10.3390/toxins10010032