The Peptide PnPP-19, a Spider Toxin Derivative, Activates μ-Opioid Receptors and Modulates Calcium Channels

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
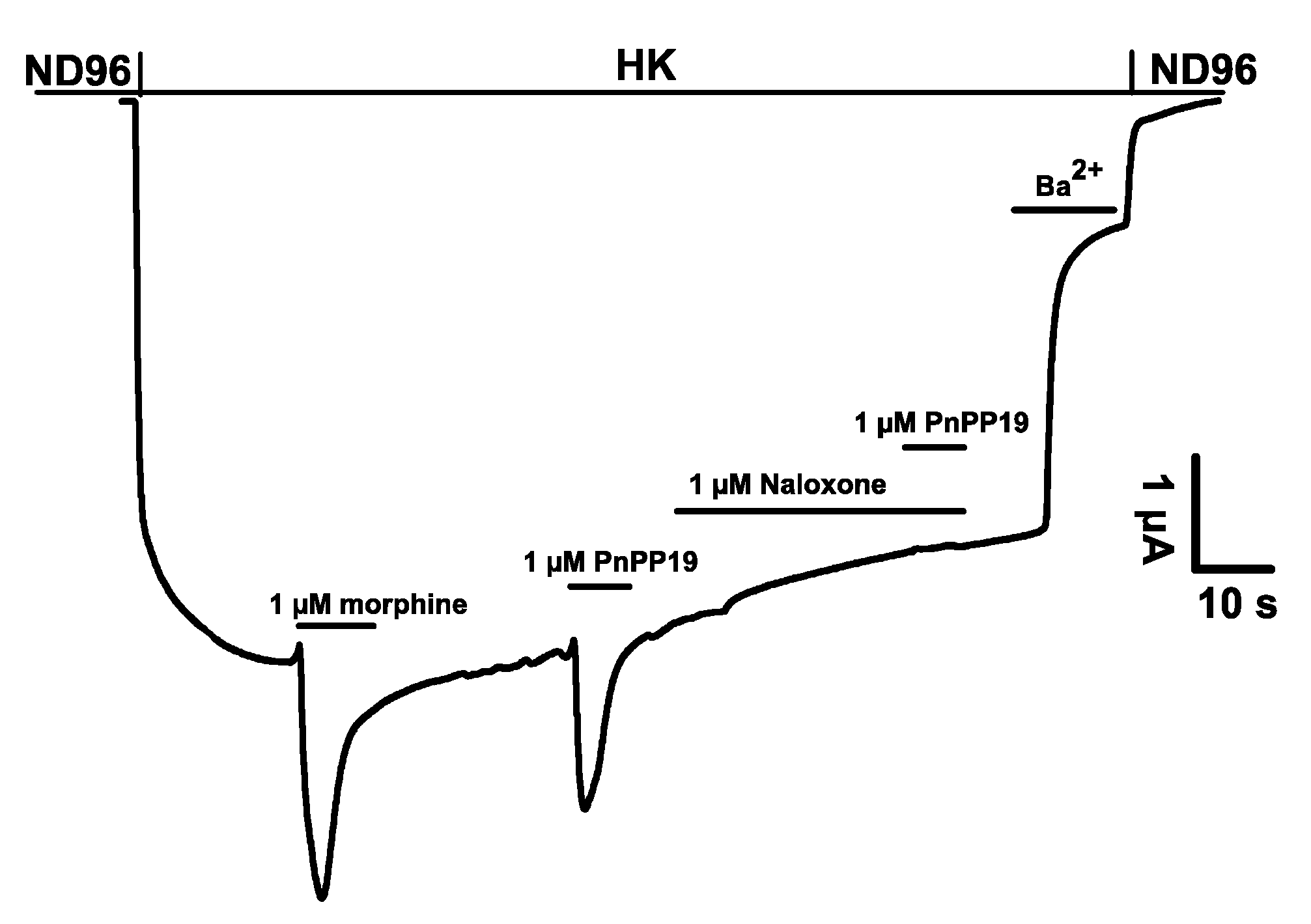
2.1. Electrophysiological Characterization of Direct Activation of Opioid Receptors Induced by PnPP-19 Using Two-Electrode Voltage-Clamp
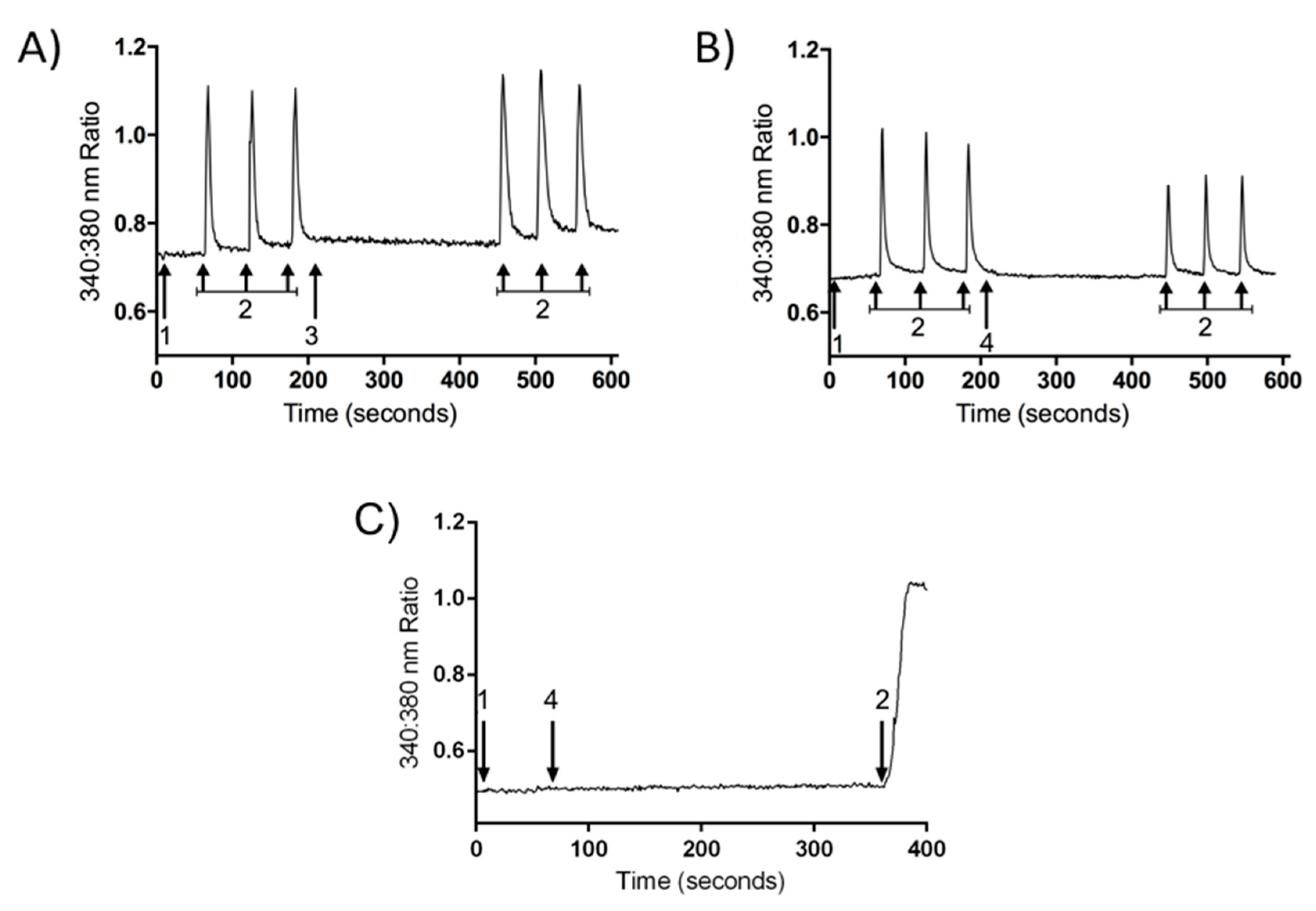
2.2. Inhibition of Calcium Current Induced by PnPP-19
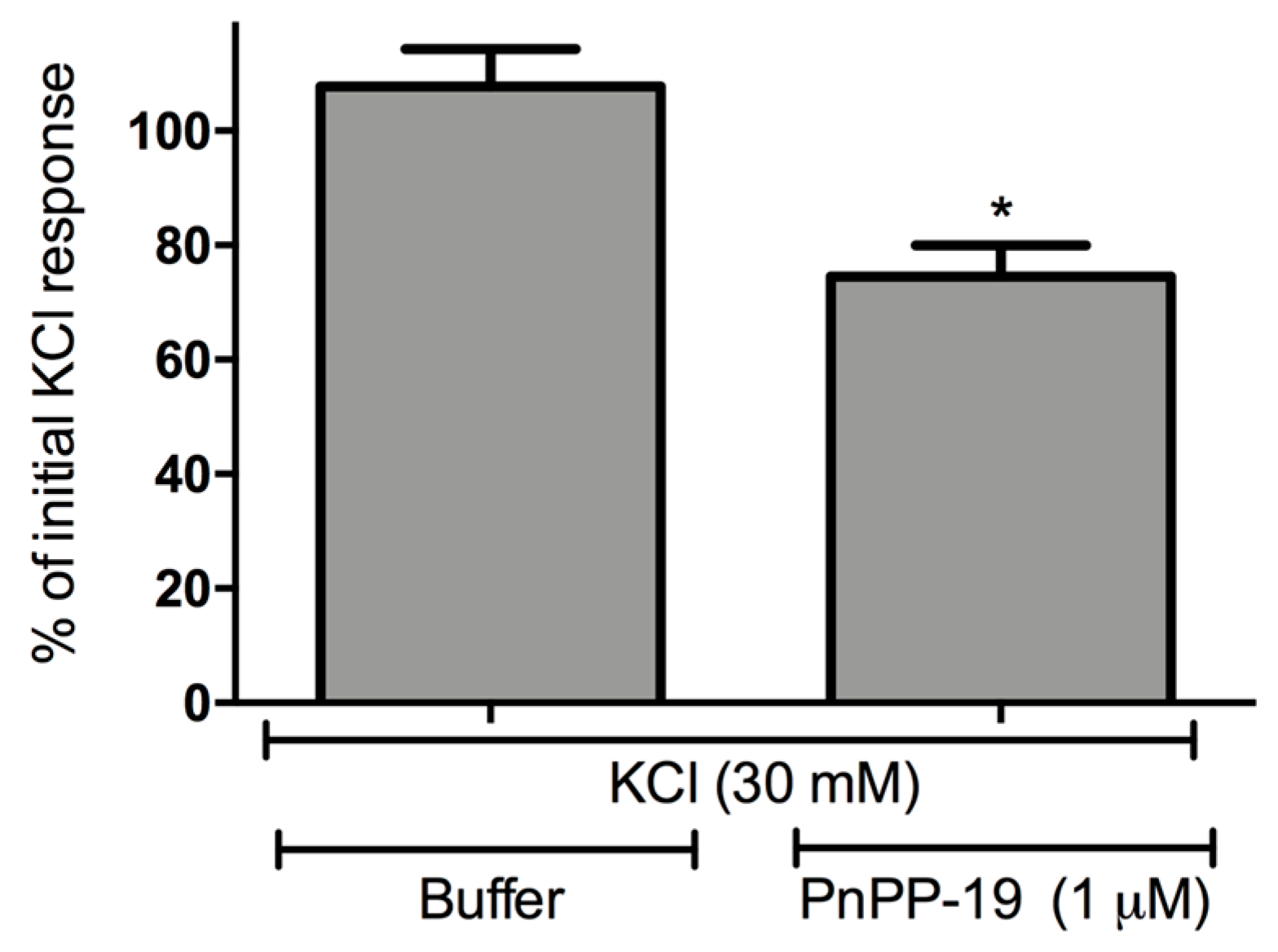
2.3. Inhibition of Calcium Influx Induced by PnPP-19
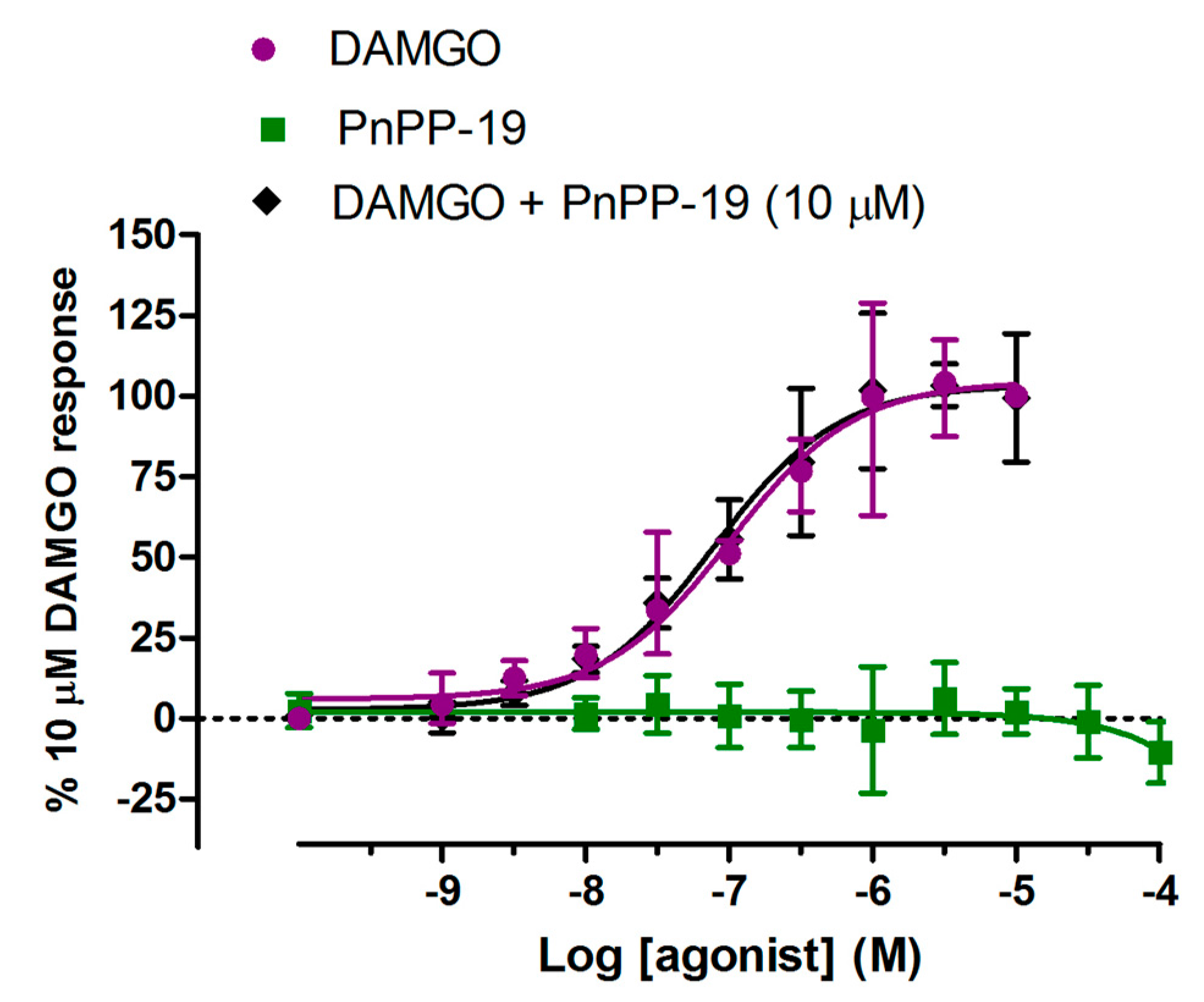
2.4. β-Arrestin2 Recruitment Induced by DAMGO and PnPP-19
3. Discussion
4. Materials and Methods
4.1. Expression of Voltage-Gated Potassium Channels in Xenopus laevis Oocytes
4.2. Electrophysiological Recordings: Xenopus laevis Oocytes
4.3. Data Analysis of Two-Microelectrode Voltage-Clamp
4.4. DRG Culture
4.5. Whole-Cell Voltage-Clamp
4.6. Calcium Imaging
4.7. Beta-Arrestin2 Recruitment
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Lima, M.E.; Figueiredo, S.G.; Matavel, A.; Nunes, K.P.; da Silva, C.N.; de Marco Almeida, F.; Ribeiro, M.; Diniz, V.; do Cordeiro, M.N.; Stankiewicz, M.; et al. Phoneutria nigriventer Venom and Toxins: A Review; Springer: Amsterdam, The Netherlands, 2015; pp. 1–24. [Google Scholar]
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, K.P.; Costa-Goncalves, A.; Lanza, L.F.; Côrtes, S.D.F.; Cordeiro, M.D.N.; Richardson, M.; Pimentad, A.M.C.; Webbe, R.C.; Leite, R.; De Lima, M.E. Tx2-6 toxin of the Phoneutria nigriventer spider potentiates rat erectile function. Toxicon 2008, 51, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.R.; Choi, Y.S.; Piao, S.; Park, Y.H.; Shrestha, K.R.; Jeon, S.H.; Hong, S.H.; Kim, S.W.; Hwang, T.K.; Kim, K.H.; et al. The effect of PnTx2-6 protein from Phoneutria nigriventer spider toxin on improvement of erectile dysfunction in a rat model of cavernous nerve injury. Urology 2014, 84, 730. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.N.; Nunes, K.P.; Torres, F.S.; Cassoli, J.S.; Santos, D.M.; Almeida, F.D.M.; Matavel, A.; Cruza, J.S.; Santos-Miranda, A.; Nunes, A.D.C.; et al. PnPP-19, a synthetic and non toxic peptide designed from a Phoneutria nigriventer toxin, potentiates erectile function via NO/cGMP. J. Urol. 2015, 194, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.C.; Freitas, A.C.N.; Pacheco, D.F.; Machado, M.F.M.; Carmona, A.K.; Duarte, I.D.G.; Lima, M.E. PnPP-19, a spider toxin peptide, induces peripheral antinociception through opioid and cannabinoid receptors and inhibition of neutral endopeptidase. Br. J. Pharmacol. 2016, 173, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.C.; Silva, G.C.; Pacheco, D.F.; Pimenta, A.M.C.; Lemos, V.S.; Duarte, I.D.G.; de Lima, M.E. The synthetic peptide PnPP-19 induces peripheral antinociception via activation of NO/cGMP/KATP pathway: Role of eNOS and nNOS. Nitric Oxide 2017, 64, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Da Fonseca Pacheco, D.; Freitas, A.C.N.; Pimenta, A.M.C.; Duarte, I.D.G.; de Lima, M.E. A spider derived peptide, PnPP-19, induces central antinociception mediated by opioid and cannabinoid systems. J. Venom. Anim. Toxins Incl. Trop. Dis. 2016, 22, 34. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.J. The Cost and Burden of Chronic Pain. Rev. Pain 2009, 3, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Law, P.Y.; Wong, Y.H.; Loh, H.H. Molecular mechanisms and regulation of opioid receptor signaling. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 389–430. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.H.; Ferreira, J.; do Nascimento Cordeiro, M.; Vieira, L.B.; De Castro, C.J.; Trevisan, G.; Reis, H.; Souza, I.A.; Richardson, M.; Prado, M.A.M.; et al. Analgesic effect in rodents of native and recombinant Phα1β toxin, a high-voltage-activated calcium channel blocker isolated from armed spider venom. Pain 2008, 140, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Dalmolin, G.D.; Silva, C.R.; Rigo, F.K.; Gomes, G.M.; do Nascimento Cordeiro, M.; Richardson, M.; Silva, M.A.R.; Prado, A.M.; Gomez, M.V.; Ferreira, J. Antinociceptive effect of Brazilian armed spider venom toxin Tx3-3 in animal models of neuropathic pain. Pain 2011, 152, 2224–2232. [Google Scholar] [CrossRef] [PubMed]
- McGivern, J.G. Ziconotide: A review of its pharmacology and use in the treatment of pain. Neuropsychiatr. Dis. Treat. 2007, 3, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Emerich, B.L.; Ferreira, R.; Cordeiro, M.N.; Borges, M.H.; Pimenta, A.; Figueiredo, S.G.; Duarte, I.D.G.; de Lima, M.E. δ-Ctenitoxin-Pn1a, a Peptide from Phoneutria nigriventer Spider Venom, Shows Antinociceptive Effect Involving Opioid and Cannabinoid Systems, in Rats. Toxins 2016, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Benyamin, R.; Rajive Adlaka, M.; Nalini Sehgal, M. Opioid complications and side effects. Pain Phys. 2008, 11, S105–S120. [Google Scholar]
- Wilson, K.C.; Saukkonen, J.J. Acute respiratory failure from abused substances. J. Intensiv. Care Med. 2004, 19, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M.; Walker, J.K.; Bohn, L.M. Morphine side effects in beta-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther. 2005, 314, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Lefkowitz, R.J.; Caron, M.G. Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J. Neurosci. 2002, 22, 10494–10500. [Google Scholar] [PubMed]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Gainetdinov, R.R.; Lin, F.T.; Lefkowitz, R.J.; Caron, M.G. μ-opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature 2000, 408, 720–723. [Google Scholar] [PubMed]
- DeWire, S.M.; Yamashita, D.S.; Rominger, D.H.; Liu, G.; Cowan, C.L.; Graczyk, T.M.; Chen, X.; Pitis, P.M.; Gotchev, D.; Yuan, C.; et al. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 2013, 344, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Soergel, D.G.; Subach, R.A.; Burnham, N.; Lark, M.W.; James, I.E.; Sadler, B.M.; Skobieranda, F.; Violin, J.D.; Webster, L.R. Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 2014, 155, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Piros, E.T.; Prather, P.L.; Law, P.Y.; Evans, C.J.; Hales, T.G. Voltage-dependent inhibition of Ca2+ channels in GH3 cells by cloned μ- and δ-opioid receptors. Mol. Pharmacol. 1996, 50, 947–956. [Google Scholar] [PubMed]
- Rhim, H.; Miller, R.J. Opioid receptors modulate diverse types of calcium channels in the nucleus tractus solitarius of the rat. J. Neurosci. 1994, 14, 7608–7615. [Google Scholar] [PubMed]
- North, R.A.; Williams, J.T.; Surprenant, A.; Christie, M.J. μ and δ receptors belong to a family of receptors that are coupled to potassium channels. Proc. Natl. Acad. Sci. USA 1987, 84, 5487–5491. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.P.; Eckert, W.A.; Light, A.R. Opioid-activated postsynaptic, inward rectifying potassium currents in whole cell recordings in substantia gelatinosa neurons. J. Neurophysiol. 1998, 80, 2954–2962. [Google Scholar] [CrossRef] [PubMed]
- Marker, C.L.; Luján, R.; Loh, H.H.; Wickman, K. Spinal G-protein-gated potassium channels contribute in a dose-dependent manner to the analgesic effect of μ- and δ- but not kappa-opioids. J. Neurosci. 2005, 25, 3551–3559. [Google Scholar] [CrossRef] [PubMed]
- Berecki, G.; Motin, L.; Adams, D.J. Voltage-Gated R-Type Calcium Channel Inhibition via Human μ-, δ-, and κ-opioid Receptors Is Voltage-Independently Mediated by Gβγ Protein Subunits. Mol. Pharmacol. 2016, 89, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Seseña, E.; Vega, R.; Soto, E. Activation of μ-opioid receptors inhibits calcium-currents in the vestibular afferent neurons of the rat through a cAMP dependent mechanism. Front. Cell. Neurosci. 2014, 8, 90. [Google Scholar] [PubMed]
- Schroeder, J.E.; Fischbach, P.S.; Zheng, D.; McCleskey, E.W. Activation of mu opioid receptors inhibits transient high- and low-threshold Ca2+ currents, but spares a sustained current. Neuron 1991, 6, 13–20. [Google Scholar] [CrossRef]
- Rusin, K.I.; Moises, H.C. μ-Opioid receptor activation reduces multiple components of high-threshold calcium current in rat sensory neurons. J. Neurosci. 1995, 15, 4315–4327. [Google Scholar] [PubMed]
- Liman, E.R.; Tytgat, J.; Hess, P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 1992, 9, 861–871. [Google Scholar] [CrossRef]
- Ulens, C.; Daenens, P.; Tytgat, J. Changes in GIRK1/GIRK2 deactivation kinetics and basal activity in the presence and absence of RGS4. Life Sci. 2000, 67, 2305–2317. [Google Scholar] [CrossRef]
- Lindsay, R.M. Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J. Neurosci. 1988, 8, 2394–2405. [Google Scholar] [PubMed]
- Bezanilla, F.; Armstrong, C.M. A low-cost signal averager and data-acquisition device. Am. J. Physiol. 1977, 232, C211–C215. [Google Scholar] [CrossRef] [PubMed]
- Bundey, R.A.; Kendall, D.A. Inhibition of receptor-mediated calcium responses by corticotrophin-releasing hormone in the CATH.a cell line. Neuropharmacology 1999, 38, 39–47. [Google Scholar] [CrossRef]
- Liu, M.; Richardson, R.R.; Mountford, S.J.; Zhang, L.; Tempone, M.H.; Herzog, H.; Holliday, N.D.; Thompson, P.E. Identification of a Cyanine-Dye Labeled Peptidic Ligand for Y1R and Y4R, Based upon the Neuropeptide Y C-Terminal Analogue, BVD-15. Bioconjug. Chem. 2016, 27, 2166–2175. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freitas, A.C.N.; Peigneur, S.; Macedo, F.H.P.; Menezes-Filho, J.E.; Millns, P.; Medeiros, L.F.; Arruda, M.A.; Cruz, J.; Holliday, N.D.; Tytgat, J.; et al. The Peptide PnPP-19, a Spider Toxin Derivative, Activates μ-Opioid Receptors and Modulates Calcium Channels. Toxins 2018, 10, 43. https://doi.org/10.3390/toxins10010043
Freitas ACN, Peigneur S, Macedo FHP, Menezes-Filho JE, Millns P, Medeiros LF, Arruda MA, Cruz J, Holliday ND, Tytgat J, et al. The Peptide PnPP-19, a Spider Toxin Derivative, Activates μ-Opioid Receptors and Modulates Calcium Channels. Toxins. 2018; 10(1):43. https://doi.org/10.3390/toxins10010043
Chicago/Turabian StyleFreitas, Ana C. N., Steve Peigneur, Flávio H. P. Macedo, José E. Menezes-Filho, Paul Millns, Liciane F. Medeiros, Maria A. Arruda, Jader Cruz, Nicholas D. Holliday, Jan Tytgat, and et al. 2018. "The Peptide PnPP-19, a Spider Toxin Derivative, Activates μ-Opioid Receptors and Modulates Calcium Channels" Toxins 10, no. 1: 43. https://doi.org/10.3390/toxins10010043
APA StyleFreitas, A. C. N., Peigneur, S., Macedo, F. H. P., Menezes-Filho, J. E., Millns, P., Medeiros, L. F., Arruda, M. A., Cruz, J., Holliday, N. D., Tytgat, J., Hathway, G., & De Lima, M. E. (2018). The Peptide PnPP-19, a Spider Toxin Derivative, Activates μ-Opioid Receptors and Modulates Calcium Channels. Toxins, 10(1), 43. https://doi.org/10.3390/toxins10010043