Comparative Genomics and Identification of an Enterotoxin-Bearing Pathogenicity Island, SEPI-1/SECI-1, in Staphylococcus epidermidis Pathogenic Strains

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. SE90 and SE95 Whole Genome Assembly and Annotation
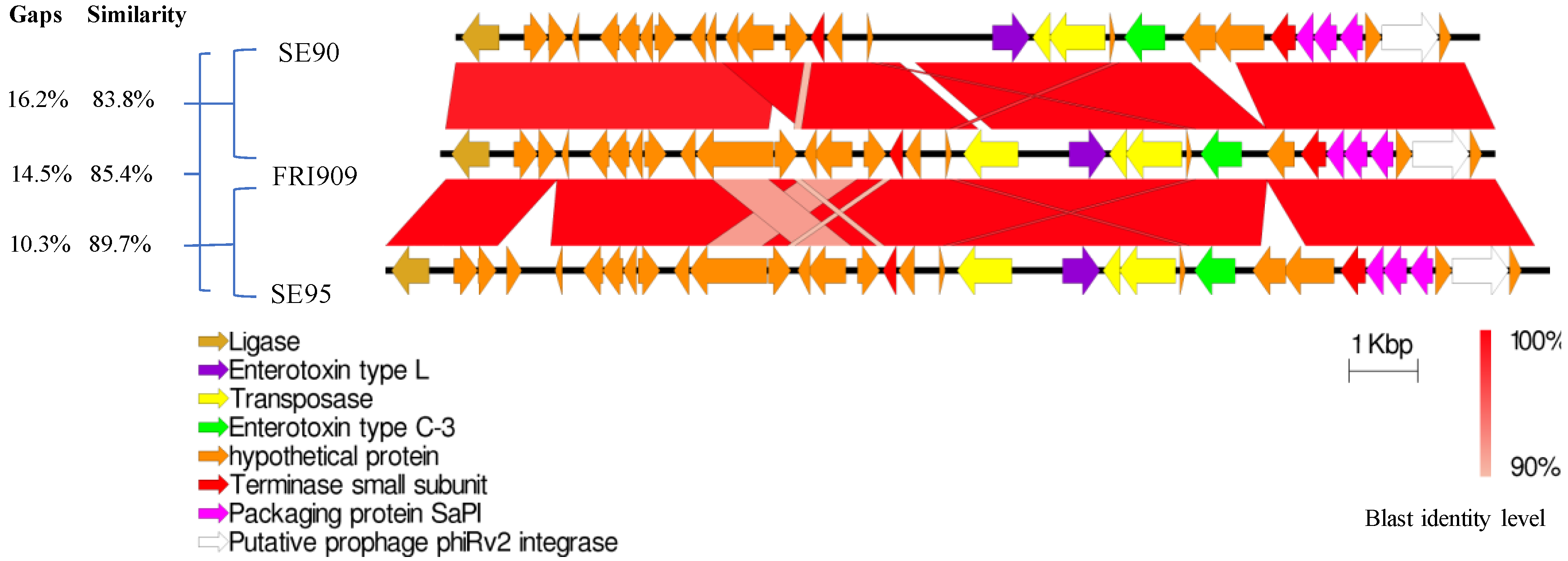
2.2. Identification of a Sec3-Containing Pathogenicity Island
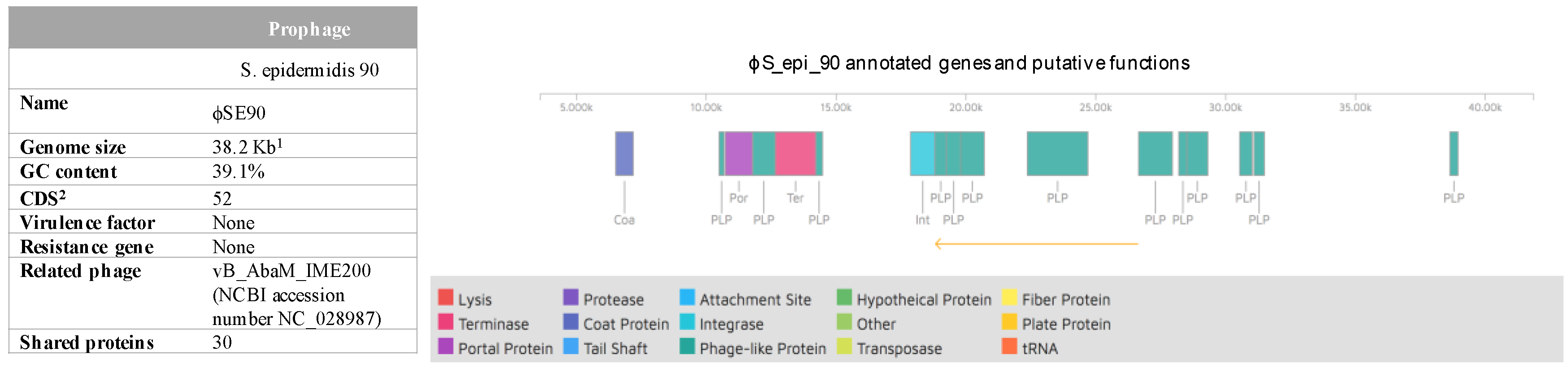
2.3. Identification of Plasmids and Prophages
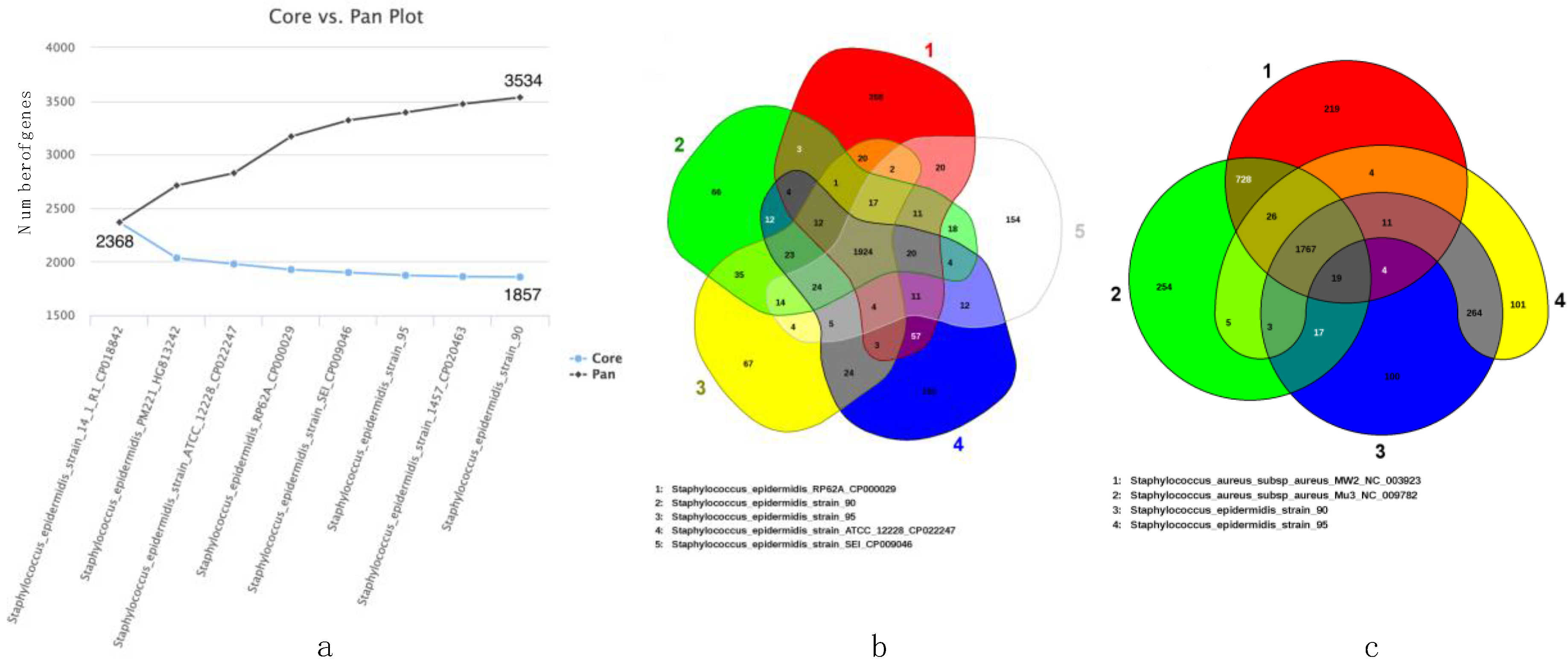
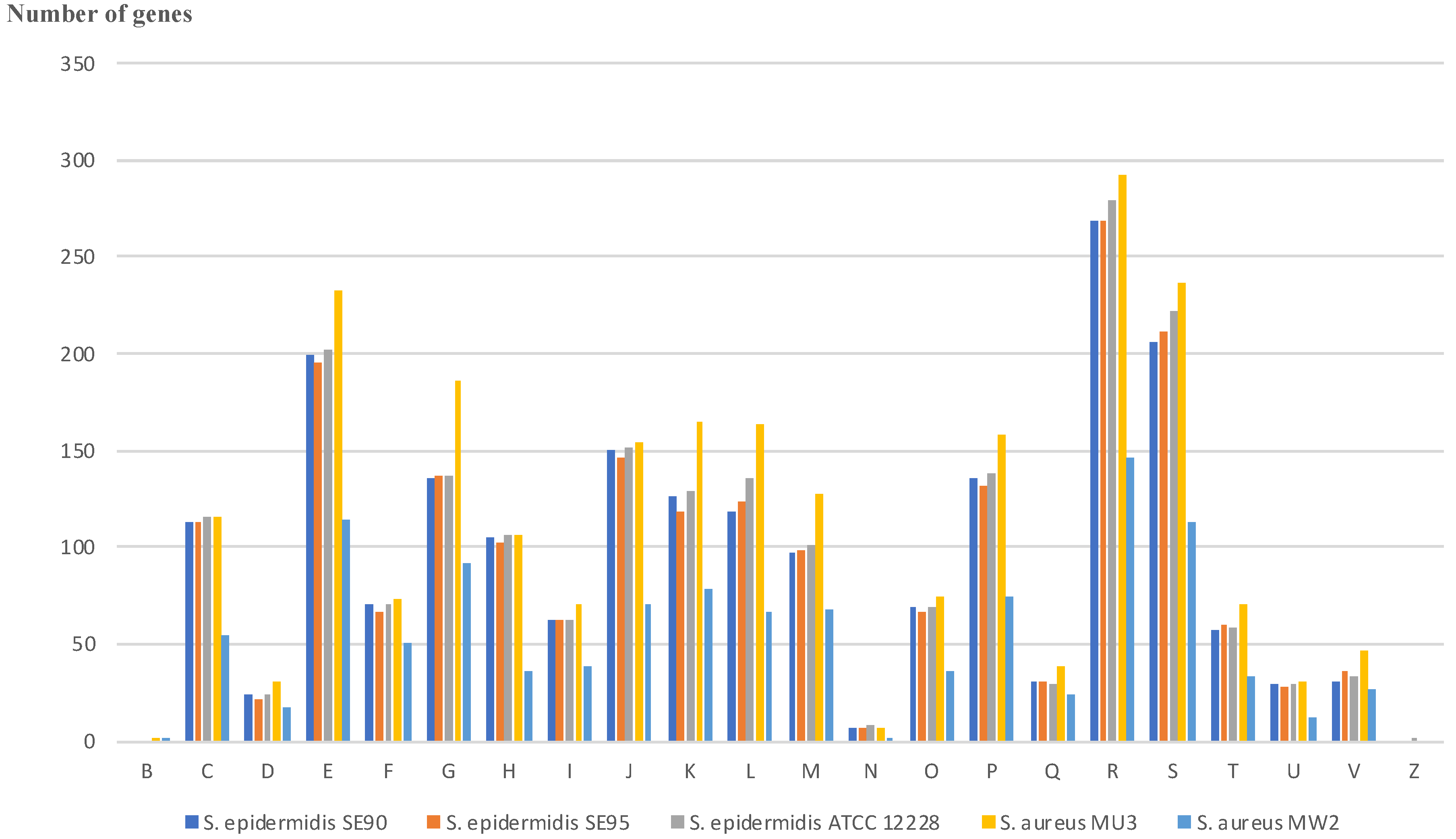
2.4. Whole Genome Comparisons between S. epidermidis and S. aureus
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Bacterial Strains, Culture, and DNA Extraction
5.2. Genome Sequencing, Assembly, and Annotations
5.3. Identification of Pathogenicity Islands, Plasmids Coding Sequences, and Prophages
5.4. Identification and Comparative Genomic of the SEC3-Coding Sequence
5.5. Whole-Genome Sequence Analysis
Supplementary Materials
Acknowledgments
Authors Contribution
Conflicts of Interest
Appendix
- -
- SE90: CP024408
- -
- Plasmid p1_90: CP024409
- -
- Plasmid p2_90: CP024410
- -
- SE95: CP024437
- -
- Plasmid p1_95: CP024438
- -
- Plasmid p2_95: CP024439
- -
- Plasmid p3_95: CP024440
- -
- Plasmid p4_95: CP024441
References
- Méric, G.; Miragaia, M.; de Been, M.; Yahara, K.; Pascoe, B.; Mageiros, L.; Mikhail, J.; Harris, L.G.; Wilkinson, T.S.; Rolo, J.; et al. Ecological Overlap and Horizontal Gene Transfer in Staphylococcus aureus and Staphylococcus epidermidis. Genome Biol. Evol. 2015, 7, 1313–1328. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Coagulase-negative staphylococci as reservoirs of genes facilitating MRSA infection. BioEssays News Rev. Mol. Cell. Dev. Biol. 2013, 35, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Novick, R.P. Phage-mediated intergeneric transfer of toxin genes. Science 2009, 323, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Carpena, N.; Quiles-Puchalt, N.; Ram, G.; Novick, R.P.; Penadés, J.R. Intra- and inter-generic transfer of pathogenicity island-encoded virulence genes by cos phages. ISME J. 2015, 9, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Madhusoodanan, J.; Seo, K.S.; Remortel, B.; Park, J.Y.; Hwang, S.Y.; Fox, L.K.; Park, Y.H.; Deobald, C.F.; Wang, D.; Liu, S.; et al. An Enterotoxin-Bearing Pathogenicity Island in Staphylococcus epidermidis. J. Bacteriol. 2011, 193, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Nanoukon, C.; Argemi, X.; Sogbo, F.; Orekan, J.; Keller, D.; Affolabi, D.; Schramm, F.; Riegel, P.; Baba-Moussa, L.; Prévost, G. Pathogenic features of clinically significant coagulase-negative staphylococci in hospital and community infections in Benin. Int. J. Med. Microbiol. 2017, 307, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. (Clifton NJ) 2014, 1079, 105–116. [Google Scholar]
- Kwan, T.; Liu, J.; DuBow, M.; Gros, P.; Pelletier, J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. USA 2005, 102, 5174–5179. [Google Scholar] [CrossRef] [PubMed]
- Ramia, N.F.; Tang, L.; Cocozaki, A.I.; Li, H. Staphylococcus epidermidis Csm1 is a 3′–5′ exonuclease. Nucleic Acids Res. 2014, 42, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, N.; Rajan, R.; Sontheimer, E.J. Primary processing of CRISPR RNA by the endonuclease Cas6 in Staphylococcus epidermidis. FEBS Lett. 2015, 589, 3197–3204. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, A.R.; Salgado-Pabón, W.; Kohler, P.L.; Horswill, A.R.; Leung, D.Y.M.; Schlievert, P.M. Staphylococcal and streptococcal superantigen exotoxins. Clin. Microbiol. Rev. 2013, 26, 422–447. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.Y.C.; Joo, H.-S.; Chatterjee, S.S.; Otto, M. Phenol-soluble modulins—Critical determinants of staphylococcal virulence. FEMS Microbiol. Rev. 2014, 38, 698–719. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Molecular basis of Staphylococcus epidermidis infections. Semin. Immunopathol. 2011, 34, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Staphylococcus epidermidis—The “accidental” pathogen. Nat. Rev. Microbiol. 2009, 7, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Sadykov, M.R. Restriction-Modification Systems as a Barrier for Genetic Manipulation of Staphylococcus aureus. Methods Mol. Biol. (Clifton NJ) 2016, 1373, 9–23. [Google Scholar]
- Conlan, S.; Mijares, L.A.; NISC Comparative Sequencing Program; Becker, J.; Blakesley, R.W.; Bouffard, G.G.; Brooks, S.; Coleman, H.; Gupta, J.; Gurson, N.; et al. Staphylococcus epidermidis pan-genome sequence analysis reveals diversity of skin commensal and hospital infection-associated isolates. Genome Biol. 2012, 13, R64. [Google Scholar] [CrossRef] [PubMed]
- Post, V.; Harris, L.G.; Morgenstern, M.; Mageiros, L.; Hitchings, M.D.; Méric, G.; Pascoe, B.; Sheppard, S.K.; Richards, R.G.; Moriarty, T.F. A comparative genomics study of Staphylococcus epidermidis from orthopedic device-related infections correlated with patient outcome. J. Clin. Microbiol. 2017, 55, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, L.C.; Florczak-Wyspianska, J.; de Jesus, L.B.; Viana, M.V.C.; Silva, A.; Ramos, R.T.J.; de Castro Soares, S. Inside the Pan-genome—Methods and Software Overview. Curr. Genom. 2015, 16, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Argemi, X.; Martin, V.; Loux, V.; Dahyot, S.; Lebeurre, J.; Guffroy, A.; Martin, M.; Velay, A.; Keller, D.; Riegel, P.; et al. Whole-Genome Sequencing of Seven Strains of Staphylococcus lugdunensis Allows Identification of Mobile Genetic Elements. Genome Biol. Evol. 2017, 9. [Google Scholar] [CrossRef]
- Swain, M.T.; Tsai, I.J.; Assefa, S.A.; Newbold, C.; Berriman, M.; Otto, T.D. A Post-assembly genome-improvement toolkit (PAGIT) to obtain annotated genomes from contigs. Nat. Protoc. 2012, 7, 1260–1284. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, B.K.; Laird, M.R.; Shay, J.A.; Winsor, G.L.; Lo, R.; Nizam, F.; Pereira, S.K.; Waglechner, N.; McArthur, A.G.; Langille, M.G.I.; et al. IslandViewer 3: More flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 2015, 43, W104–W108. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Smillie, C.; Garcillán-Barcia, M.P.; Francia, M.V.; Rocha, E.P.C.; de la Cruz, F. Mobility of Plasmids. Microbiol. Mol. Biol. Rev. 2010, 74, 434–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thammavongsa, V.; Kim, H.K.; Missiakas, D.; Schneewind, O. Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol. 2015, 13, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Staphylococcus aureus toxins. Curr. Opin. Microbiol. 2014, 17, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Höök, M. Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Blom, J.; Kreis, J.; Spänig, S.; Juhre, T.; Bertelli, C.; Ernst, C.; Goesmann, A. EDGAR 2.0: An enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 2016, 44, W22–W28. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhu, Z.; Fu, L.; Niu, B.; Li, W. WebMGA: A customizable web server for fast metagenomic sequence analysis. BMC Genom. 2011, 12, 444. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. epidermidis Strain | ATCC 12228 2 | S. epidermidis SE90 | S. epidermidis SE95 |
|---|---|---|---|
| NCBI accession number | CP022247.1 | CP024408 | CP024437 |
| Clinical origin | Not clinical | Bacteremia | Bacteremia |
| N50 after SPAdes assembly (kb) 1 | - | 179 | 195 |
| N90 after SPAdes assembly (kb) | - | 39 | 43 |
| Contigs number after SPAdes assembly 3 | - | 26 | 38 |
| Mean coverage after SPAdes assembly | - | 422× | 410× |
| Contigs number after PAGIT assembly termination | - | 7 | 10 |
| Full length of the scaffold (bp) 4 | 2,497,508 | 2,395,274 | 2,407,125 |
| GC content (%) | 32.03 | 32.47 | 31.95 |
| Coding sequences | 2545 | 2189 | 2189 |
| tRNA 5 | 60 | 57 | 59 |
| rRNA 6 | 19 | 9 | 7 |
| tmRNA 7 | 0 | 1 | 1 |
| Plasmids | ||||||
|---|---|---|---|---|---|---|
| S. epidermidis SE90 | S. epidermidis SE95 | |||||
| Name | p_1_90 | p_2_90 | p_1_95 | p_2_95 | p_3_95 | p_4_95 |
| Accession number | CP024409 | CP024410 | CP024438 | CP024439 | CP024440 | CP024441 |
| Genome size 1 | 18.6 kb | 6.6 kb | 12.1 kb | 9 kb | 4.5 kb | 3.3 kb |
| Contig coverage | 1968× | 2095× | 1212× | 1214× | 3040× | 1567× |
| GC content | 26.12% | 28.17% | 28.69% | 29.29% | 30.29% | 33.56% |
| CDS 2 | 20 | 7 | 14 | 10 | 4 | 3 |
| Virulence factor | None | None | None | None | None | None |
| Resistance gene | None | Beta-lactamase | Beta-lactamase | None | hsrA 4 | dfrG 5 |
| Replication gene | repA | None | None | repA | repN | None |
| Mobilization module | None | None | None | MobA | None | None |
| T4CP/T4SS genes | None | None | None | None | None | None |
| Plasmid category | Non-mobilizable | Doubtful | Doubtful | Mobilizable | Non-mobilizable | Doubtful |
| Related plasmid (strain, nucleotide length, CDS) | p1457 (S. epidermidis, 17 CDS, 15 kb) | pSC-SNUDS-2-1 (S. cohnii, 30 CDS, 29.4 kb) | pETB DNA (S. aureus, 63 CDS, 60.5 kb) | pVISLISI_5 (S. lugdunensis, 13 CDS, 12.6 kb) | SAP085A (S. aureus, 3 CDS, 4.4 kb) | pUSA04-2-SUR11 (S. aureus, 28 CDS, 26 kb) |
| Nucleotide similarities: | ||||||
| - Sequence cov 3 | −42% | 93% | 75% | 52% | 100% | 88% |
| - Identities | 99% | 97% | 99% | 91% | 99% | 99% |
| - E-value | 0.0 | 0.0 | 0.0 | 0.0 | 0% | 0.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Argemi, X.; Nanoukon, C.; Affolabi, D.; Keller, D.; Hansmann, Y.; Riegel, P.; Baba-Moussa, L.; Prévost, G. Comparative Genomics and Identification of an Enterotoxin-Bearing Pathogenicity Island, SEPI-1/SECI-1, in Staphylococcus epidermidis Pathogenic Strains. Toxins 2018, 10, 93. https://doi.org/10.3390/toxins10030093
Argemi X, Nanoukon C, Affolabi D, Keller D, Hansmann Y, Riegel P, Baba-Moussa L, Prévost G. Comparative Genomics and Identification of an Enterotoxin-Bearing Pathogenicity Island, SEPI-1/SECI-1, in Staphylococcus epidermidis Pathogenic Strains. Toxins. 2018; 10(3):93. https://doi.org/10.3390/toxins10030093
Chicago/Turabian StyleArgemi, Xavier, Chimène Nanoukon, Dissou Affolabi, Daniel Keller, Yves Hansmann, Philippe Riegel, Lamine Baba-Moussa, and Gilles Prévost. 2018. "Comparative Genomics and Identification of an Enterotoxin-Bearing Pathogenicity Island, SEPI-1/SECI-1, in Staphylococcus epidermidis Pathogenic Strains" Toxins 10, no. 3: 93. https://doi.org/10.3390/toxins10030093
APA StyleArgemi, X., Nanoukon, C., Affolabi, D., Keller, D., Hansmann, Y., Riegel, P., Baba-Moussa, L., & Prévost, G. (2018). Comparative Genomics and Identification of an Enterotoxin-Bearing Pathogenicity Island, SEPI-1/SECI-1, in Staphylococcus epidermidis Pathogenic Strains. Toxins, 10(3), 93. https://doi.org/10.3390/toxins10030093