Membrane Repair Mechanisms against Permeabilization by Pore-Forming Toxins


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
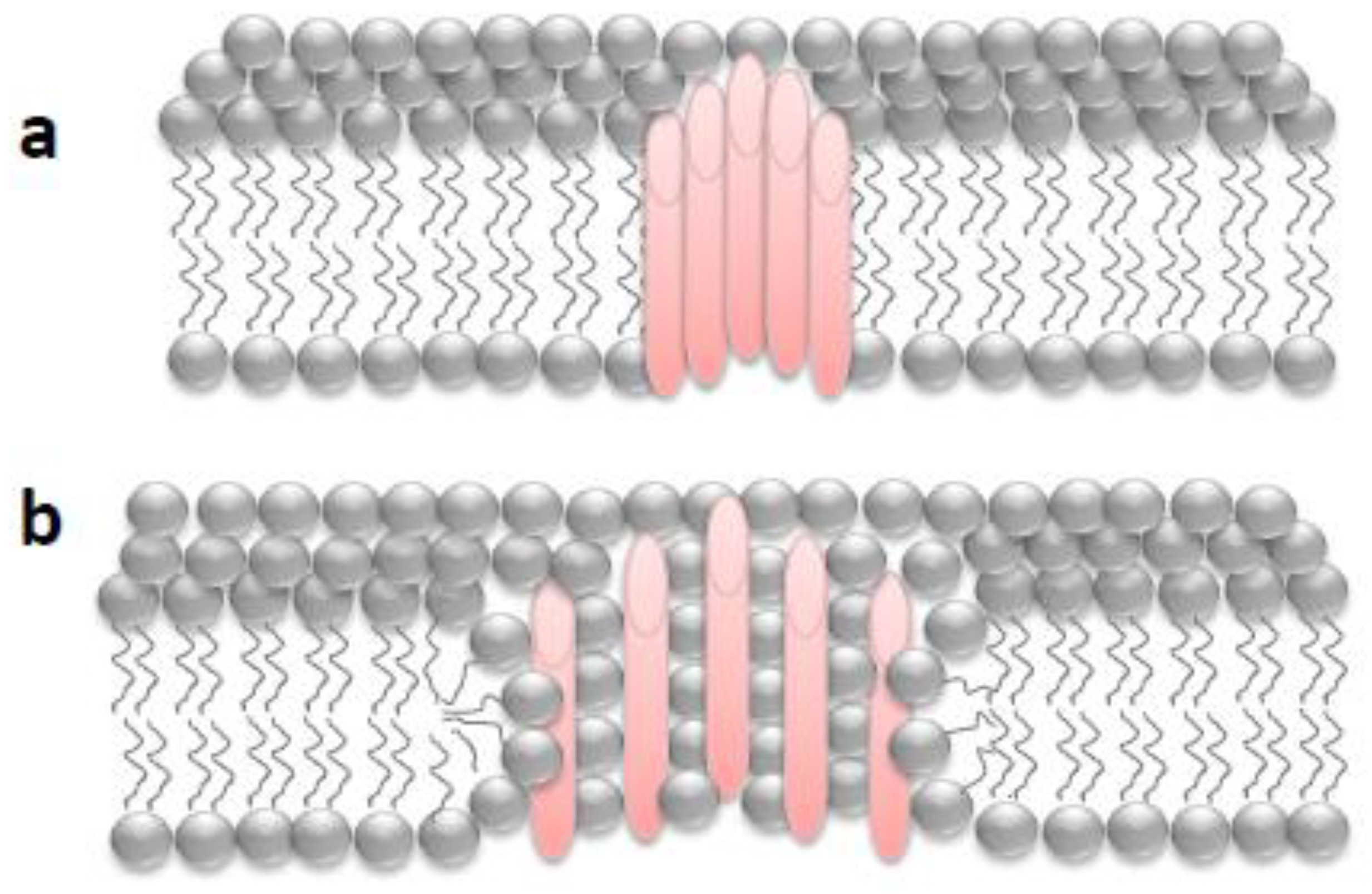
2. “Holes” in the Membrane: Some General Aspects
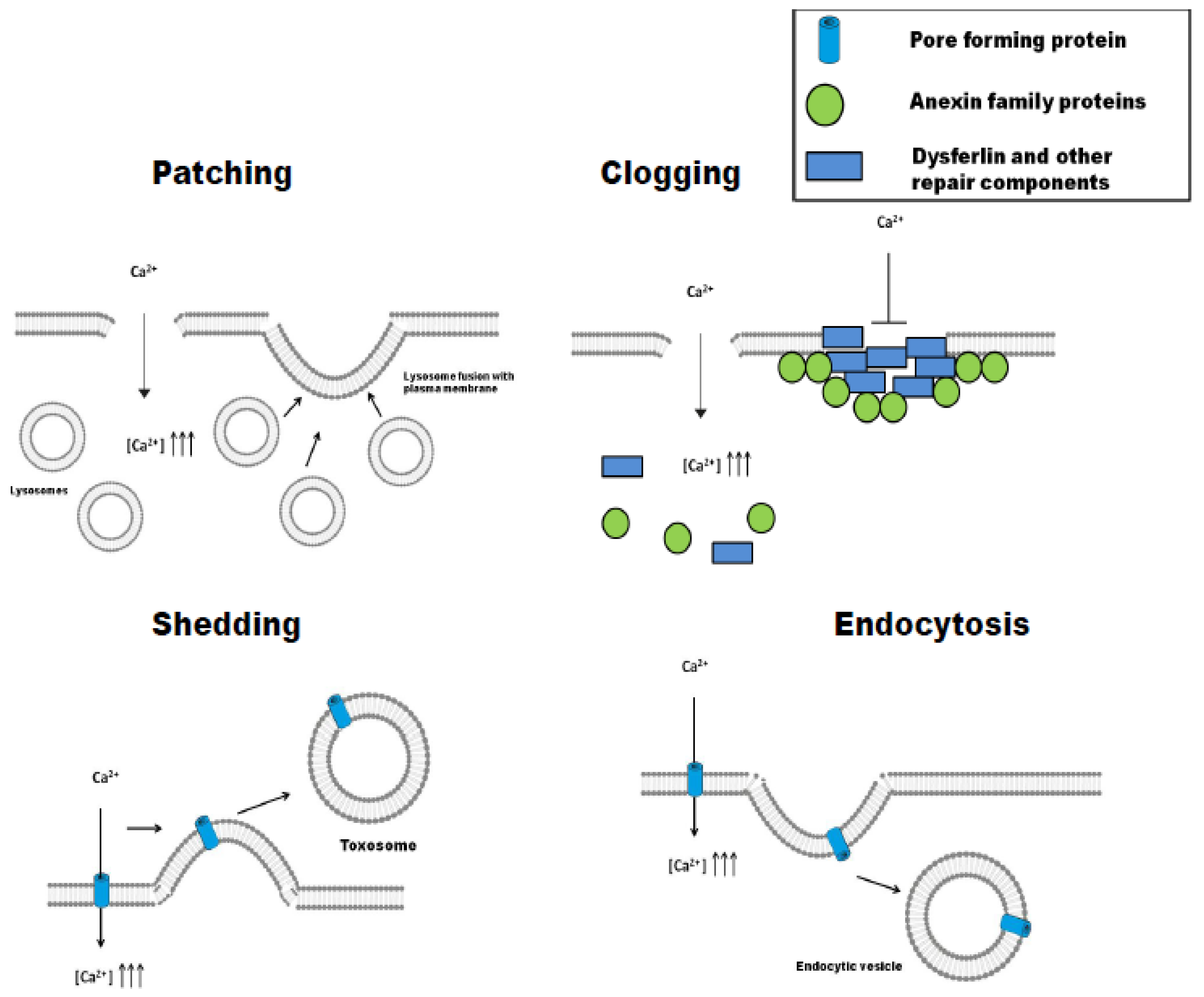
3. Different Strategies to the Same Problem: Patching, Clogging, Shedding, or Endocytosis
3.1. Patching
3.2. Clogging
3.3. Shedding (Ectocytosis)
3.4. Endocytosis
4. Repair Mechanisms Activated by Pore-Forming Toxins
4.1. Repair of Large Pores
4.2. Repair of Small Pores
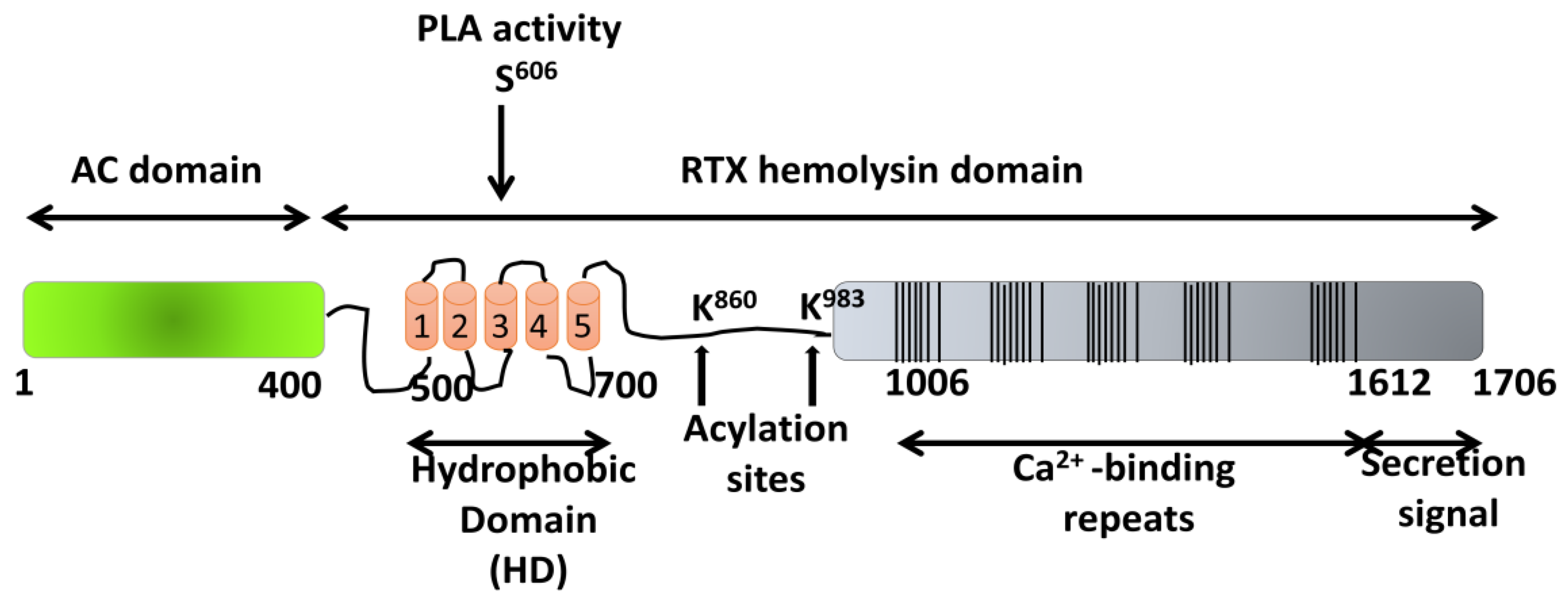
5. ACT Toxin: An Adenylate Cyclase Enzyme Fused to an RTX Hemolysin
5.1. Pore-Forming Activity of ACT
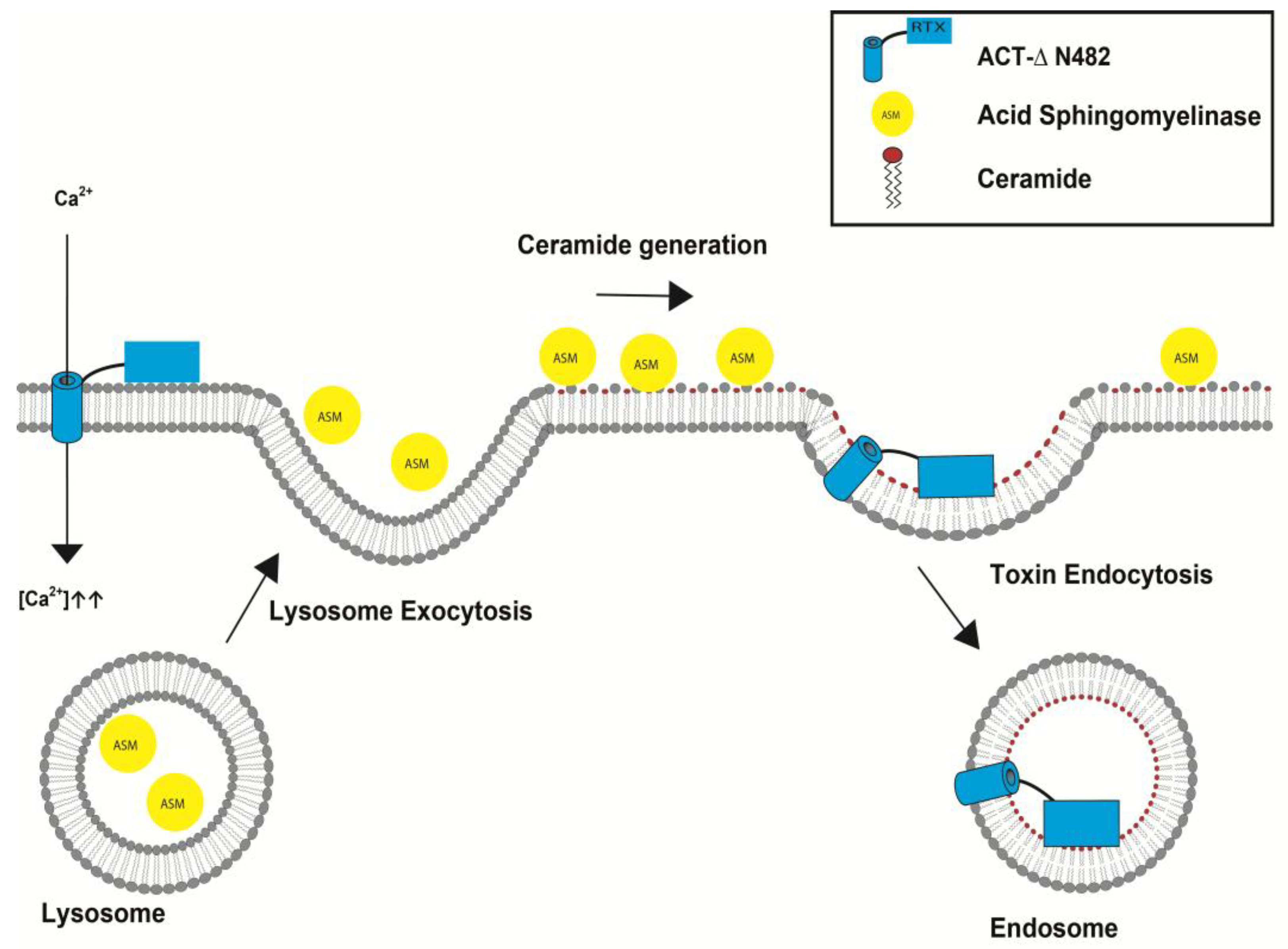
5.2. Irreversible Membrane Permeabilization by ACT
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Heilbrunn, L. The Dynamics of Living Protoplasm; Academic Press: New York, NY, USA, 1956; p. 634. [Google Scholar]
- Chambers, R.; Chambers, E. Explorations into the Nature of the Living Cell; Harvard University Press: Cambridge, MA, USA, 1961; p. 352. [Google Scholar]
- Andrews, N.W.; Almeida, P.E.; Corrotte, M. Damage Control: Cellular Mechanisms of Plasma Membrane Repair. Trends Cell Biol. 2014, 24, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Bi, G.Q.; Alderton, J.M.; Steinhardt, R.A. Calcium-Regulated Exocytosis Is Required for Cell Membrane Resealing. J. Cell Biol. 1995, 131, 1747–1758. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; McNeil, P.L. Vesicle Accumulation and Exocytosis at Sites of Plasma Membrane Disruption. J. Cell Biol. 1995, 131, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Steinhardt, R.A.; Bi, G.; Alderton, J.M. Cell Membrane Resealing by a Vesicular Mechanism Similar to Neurotransmitter Release. Science 1994, 263, 390–393. [Google Scholar] [CrossRef] [PubMed]
- McNeil, P.L.; Vogel, S.S.; Miyake, K.; Terasaki, M. Patching Plasma Membrane Disruptions with Cytoplasmic Membrane. J. Cell Sci. 2000, 113, 1891–1902. [Google Scholar] [PubMed]
- McNeil, P.L.; Khakee, R. Disruptions of Muscle Fiber Plasma Membranes: Role in Exercise-Induced Damage. Am. J. Pathol. 1992, 140, 1097–1109. [Google Scholar] [PubMed]
- Los, F.C.O.; Kao, C.-Y.; Smitham, J.; McDonald, K.L.; Ha, C.; Peixoto, C.A.; Aroian, R.V. RAB-5-and RAB-11-Dependent Vesicle-Trafficking Pathways are Required for Plasma Membrane Repair after Attack by Bacterial Pore-Forming Toxin. Cell Host Microbe 2011, 9, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Los, F.C.O.; Huffman, D.L.; Wachi, S.; Kloft, N.; Husmann, M.; Karabrahimi, V.; Schwartz, J.-L.; Bellier, A.; Ha, C.; et al. Global Functional Analyses of Cellular Responses to Pore-Forming Toxins. PLoS Pathog. 2011, 7, e1001314. [Google Scholar] [CrossRef] [PubMed]
- Bischofberger, M.; Gonzalez, M.R.; van der Goot, F.G. Membrane Injury by Pore-Forming Proteins. Curr. Opin. Cell Biol. 2009, 21, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Diz-Muñoz, A.; Fletcher, D.A.; Weiner, O.D. Use the Force: Membrane Tension as an Organizer of Cell Shape and Motility. Trends Cell Biol. 2013, 23, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, A.J.; Perez, F. Physico-Chemical and Biological Considerations for Membrane Wound Evolution and Repair in Animal Cells. Semin. Cell Dev. Biol. 2015, 45, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Zhelev, D.V.; Needham, D. Tension-Stabilized Pores in Giant Vesicles: Determination of Pore Size and Pore Line Tension. Biochim. Biophys. Acta Biomembr. 1993, 1147, 89–104. [Google Scholar] [CrossRef]
- Gilbert, R.J.C. Protein–lipid Interactions and Non-Lamellar Lipidic Structures in Membrane Pore Formation and Membrane Fusion. Biochim. Biophys. Acta (BBA)-Biomembr. 2016, 1858, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Bischofberger, M.; Iacovache, I.; Gisou Van Der Goot, F. Pathogenic Pore-Forming Proteins: Function and Host Response. Cell Host Microbe 2012, 12, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, A.J.; Perez, F. Plasma Membrane Repair: The Adaptable Cell Life-Insurance. Curr. Opin. Cell Biol. 2017, 47, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, M.; Miyake, K.; McNeil, P.L. Large Plasma Membrane Disruptions are Rapidly Resealed by Ca2+- Dependent Vesicle-Vesicle Fusion Events. J. Cell Biol. 1997, 139, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; Webster, P.; Ortego, J.; Andrews, N.W. Lysosomes Behave as Ca2+-Regulated Exocytic Vesicles in Fibroblasts and Epithelial Cells. J. Cell Biol. 1997, 137, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Togo, T.; Krasieva, T.B.; Steinhardt, R.A. A Decrease in Membrane Tension Precedes Successful Cell-Membrane Repair. Mol. Biol. Cell. 2000, 11, 4339–4346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babiychuk, E.B.; Draeger, A. Defying Death: Cellular Survival Strategies Following Plasmalemmal Injury by Bacterial Toxins. Semin. Cell Dev. Biol. 2015, 45, 39–47. [Google Scholar] [CrossRef] [PubMed]
- McNeil, A.K.; Rescher, U.; Gerke, V.; McNeil, P.L. Requirement for Annexin A1 in Plasma Membrane Repair. J. Biol. Chem. 2006, 281, 35202–35207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demonbreun, A.R.; McNally, E.M. Plasma Membrane Repair in Health and Disease. Curr. Top. Membr. 2016, 77, 67–96. [Google Scholar] [PubMed] [Green Version]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Intracellular Ca2+ Operates a Switch between Repair and Lysis of Streptolysin O-Perforated Cells. Cell Death Differ. 2009, 16, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An Actin-Dependent Annexin Complex Mediates Plasma Membrane Repair in Muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Bouter, A.; Gounou, C.; Bérat, R.; Tan, S.; Gallois, B.; Granier, T.; D’Estaintot, B.L.; Pöschl, E.; Brachvogel, B.; Brisson, A.R. Annexin-A5 Assembled into Two-Dimensional Arrays Promotes Cell Membrane Repair. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potez, S.; Luginbühl, M.; Monastyrskaya, K.; Hostettler, A.; Draeger, A.; Babiychuk, E.B. Tailored Protection against Plasmalemmal Injury by Annexins with Different Ca2+ Sensitivities. J. Biol. Chem. 2011, 286, 17982–17991. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskaya, K.; Babiychuk, E.B.; Draeger, A. The Annexins: Spatial and Temporal Coordination of Signaling Events during Cellular Stress. Cell Mol. Life Sci. 2009, 66, 2623–2642. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskaya, K.; Babiychuk, E.B.; Hostettler, A.; Rescher, U.; Draeger, A. Annexins as Intracellular Calcium Sensors. Cell Calcium. 2007, 41, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Campbell, K.P. Dysferlin and Muscle Membrane Repair. Curr. Opin. Cell Biol. 2007, 19, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Atanassoff, A.P.; Wolfmeier, H.; Schoenauer, R.; Hostettler, A.; Ring, A.; Draeger, A.; Babiychuk, E.B. Microvesicle Shedding and Lysosomal Repair Fulfill Divergent Cellular Needs during the Repair of Streptolysin O-Induced Plasmalemmal Damage. PLoS ONE 2014, 9, e89743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyel, P.A.; Loultcheva, L.; Roth, R.; Salter, R.D.; Watkins, S.C.; Yokoyama, W.M.; Heuser, J.E. Streptolysin O Clearance through Sequestration into Blebs that Bud Passively from the Plasma Membrane. J. Cell Sci. 2011, 124, 2414–2423. [Google Scholar] [CrossRef] [PubMed]
- Walev, I.; Palmer, M.; Martin, E.; Jonas, D.; Weller, U.; Höhn-Bentz, H.; Husmann, M.; Bhakdi, S. Recovery of Human Fibroblasts from Attack by the Pore-Forming a-Toxin of Staphylococcus aureus. Microb. Pathog. 1994, 17, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Husmann, M.; Beckmann, E.; Boller, K.; Kloft, N.; Tenzer, S.; Bobkiewicz, W.; Neukirch, C.; Bayley, H.; Bhakdi, S. Elimination of a Bacterial Pore-Forming Toxin by Sequential Endocytosis and Exocytosis. FEBS Lett. 2009, 583, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT Machinery Is Required for Plasma Membrane Repair. Science 2014, 343, 1247136. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, L.L.; Sreetama, S.C.; Sharma, N.; Medikayala, S.; Brown, K.J.; Defour, A.; Jaiswal, J.K. Mechanism of Ca2-Triggered ESCRT Assembly and Regulation of Cell Membrane Repair. Nat. Commun. 2014, 5, 5646. [Google Scholar] [CrossRef] [PubMed]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of Injured Plasma Membrane by Rapid Ca2+-Dependent Endocytosis. J. Cell Biol. 2008, 180, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.; Keefe, D.; Saffarian, S.; Martinvalet, D.; Walch, M.; Boucrot, E.; Kirchhausen, T.; Lieberman, J. Perforin Activates Clathrin- and Dynamin-Dependent Endocytosis, which is Required for Plasma Membrane Repair and Delivery of Granzyme B for Granzyme-Mediated Apoptosis. Blood 2010, 115, 1582–1593. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.; Idone, V.; Devlin, C.; Fernandes, M.C.; Flannery, A.; He, X.; Schuchman, E.; Tabas, I.; Andrews, N.W. Exocytosis of Acid Sphingomyelinase by Wounded Cells Promotes Endocytosis and Plasma Membrane Repair. J. Cell Biol. 2010, 189, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Saka, H.A.; Chinen, I.; Zoppino, F.C.M.; Yoshimori, T.; Bocco, J.L.; Colombo, M.I. Protective Role of Autophagy against Vibrio cholerae Cytolysin, a Pore-Forming Toxin from V. cholerae. Proc. Natl. Acad. Sci. USA 2007, 104, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.; Flannery, A.R.; Andrews, N. Live Imaging Assay for Assessing the Roles of Ca2+ and Sphingomyelinase in the Repair of Pore-Forming Toxin Wounds. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed]
- Corrotte, M.; Almeida, P.E.; Tam, C.; Castro-Gomes, T.; Fernandes, M.C.; Millis, B.A.; Cortez, M.; Miller, H.; Song, W.; Maugel, T.K.; et al. Caveolae Internalization Repairs Wounded Cells and Muscle Fibers. Elife 2013. [Google Scholar] [CrossRef] [PubMed]
- Corrotte, M.; Fernandes, M.C.; Tam, C.; Andrews, N.W. Toxin Pores Endocytosed during Plasma Membrane Repair Traffic into the Lumen of MVBs for Degradation. Traffic 2012, 13, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, J.K.; Andrews, N.W.; Simon, S.M. Membrane Proximal Lysosomes are the Major Vesicles Responsible for Calcium-Dependent Exocytosis in Nonsecretory Cells. J. Cell Biol. 2002, 159, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma Membrane Repair is Mediated by Ca2+-Regulated Exocytosis of Lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef]
- Castro-Gomes, T.; Corrotte, M.; Tam, C.; Andrews, N.W. Plasma Membrane Repair is Regulated Extracellularly by Proteases Released from Lysosomes. PLoS ONE 2016, 11, e0152583. [Google Scholar] [CrossRef] [PubMed]
- Vieira, O.V. Rab3a and Rab10 are Regulators of Lysosome Exocytosis and Plasma Membrane Repair. Small GTPases. 2016, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.R.; Bischofberger, M.; Frêche, B.; Ho, S.; Parton, R.G.; Van der Goot, F.G. Pore-Forming Toxins Induce Multiple Cellular Responses Promoting Survival. Cell. Microbiol. 2011, 13, 1026–1043. [Google Scholar] [CrossRef] [PubMed]
- Von Hoven, G.; Rivas, A.J.; Neukirch, C.; Meyenburg, M.; Qin, Q.; Parekh, S.; Hellmann, N.; Husmann, M. Repair of a Bacterial Small ß-Barrel Toxin Pore Depends on Channel Width. mBio 2017, 8, e02083-16. [Google Scholar] [CrossRef] [PubMed]
- Hotze, E.M.; Tweten, R.K. Membrane Assembly of the Cholesterol-Dependent Cytolysin Pore Complex. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Pathak-Sharma, S.; Zhang, X.; Lam, J.G.T.; Weisleder, N.; Seveau, S.M. High-Throughput Microplate-Based Assay to Monitor Plasma Membrane Wounding and Repair. Front. Cell. Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfmeier, H.; Schoenauer, R.; Atanassoff, A.P.; Neill, D.R.; Kadioglu, A.; Draeger, A.; Babiychuk, E.B. Ca2-Dependent Repair of Pneumolysin Pores: A New Paradigm for Host Cellular Defense Against Bacterial Pore-Forming Toxins. Biochim. Biophys. Acta 2015, 1853, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Wolfmeier, H.; Radecke, J.; Schoenauer, R.; Koeffel, R.; Babiychuk, V.S.; Drücker, P.; Hathaway, L.J.; Mitchell, T.J.; Zuber, B.; Draeger, A.; et al. Active Release of Pneumolysin Prepores and Pores by Mammalian Cells Undergoing a Streptococcus pneumoniae Attack. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 2498–2509. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.; Keyel, M.; Shi, G.; Bhattacharjee, P.; Roth, R.; Heuser, J.E.; Keyel, P.A. Intrinsic Repair Protects Cells from Pore-Forming Toxins by Microvesicle Shedding. Cell Death Differ. 2017, 24, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Iacovache, I.; De Carlo, S.; Cirauqui, N.; Dal Peraro, M.; Van Der Goot, F.G.; Zuber, B. Cryo-EM Structure of Aerolysin Variants Reveals a Novel Protein Fold and the Pore-Formation Process. Nat. Commun. 2016, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valeva, A.; Walev, I.; Gerber, A.; Klein, J.; Palmer, M.; Bhakdi, S. Staphylococcal a-Toxin: Repair of a Calcium-Impermeable Pore in the Target Cell Membrane. Mol. Microbiol. 2000, 36, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Zitzer, A.; Wassenaar, T.M.; Walev, I.; Bhakdi, S. Potent Membrane-Permeabilizing and Cytocidal Action of Vibrio cholerae Cytolysin on Human Intestinal Cells. Infect. Immun. 1997, 65, 1293–1298. [Google Scholar] [PubMed]
- Rivas, A.J.; Hoven, G.; Neukirch, C.; Meyenburg, M.; Qin, Q.; Füser, S.; Boller, K.; Lemos, M.L.; Osorio, C.R.; Husmanna, M. Phobalysin, a Small ß-Pore-Forming Toxin of Photobacterium damselae Subsp. Damselae Infect. Immun. 2015, 83, 4335–4348. [Google Scholar] [CrossRef] [PubMed]
- Moschioni, M.; Tombola, F.; De Bernard, M.; Coelho, A.; Zitzer, A.; Zoratti, M.; Montecucco, C. The Vibrio cholerae Haemolysin Anion Channel Is Required for Cell Vacuolation and Death. Cell. Microbiol. 2002, 4, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Husmann, M.; Dersch, K.; Bobkiewicz, W.; Beckmann, E.; Veerachato, G.; Bhakdi, S. Differential Role of p38 Mitogen Activated Protein Kinase for Cellular Recovery from Attack by Pore-Forming S. aureus α-Toxin or Streptolysin O. Biochem. Biophys. Res. Commun. 2006, 344, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Kloft, N.; Busch, T.; Neukirch, C.; Weis, S.; Boukhallouk, F.; Bobkiewicz, W.; Cibis, I.; Bhakdi, S.; Husmann, M. Pore-Forming Toxins Activate MAPK p38 by Causing Loss of Cellular Potassium. Biochem. Biophys. Res. Commun. 2009, 385, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Gurcel, L.; Abrami, L.; Girardin, S.; Tschopp, J.; van der Goot, F.G. Caspase-1 Activation of Lipid Metabolic Pathways in Response to Bacterial Pore-Forming Toxins Promotes Cell Survival. Cell 2006, 126, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Carbonetti, N.H. Pertussis Toxin and Adenylate Cyclase Toxin: Key Virulence Factors of Bordetella pertussis and Cell Biology Tools. Future Microbiol. 2010, 5, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Welch, R.A. RTX Toxin Structure and Function: A Story of Numerous Anomalies and Few Analogies in Toxin Biology. Curr. Top. Microbiol. Immunol. 2000, 257, 85–111. [Google Scholar]
- Hackett, M.; Guo, L.; Shabanowitz, J.; Hunt, D.F.; Hewlett, E.L. Internal Lysine Palmitoylation in Adenylate Cyclase Toxin from Bordetella pertussis. Science 1994, 266, 433–435. [Google Scholar] [CrossRef] [PubMed]
- El-Azami-El-Idrissi, M.; Bauche, C.; Loucka, J.; Osicka, R.; Sebo, P.; Ladant, D.; Leclerc, C. Interaction of Bordetella pertussis Adenylate Cyclase with CD11b/CD18. Role of Toxin Acylation and Identification of the Main Integrin Interaction Domain. J. Biol. Chem. 2003, 278, 38514–38521. [Google Scholar] [CrossRef] [PubMed]
- Masin, J.; Osickova, A.; Sukova, A.; Fiser, R.; Halada, P.; Bumba, L.; Linhartova, I.; Osicka, R.; Sebo, P. Negatively Charged Residues of the Segment Linking the Enzyme and Cytolysin Moieties Restrict the Membrane-Permeabilizing Capacity of Adenylate Cyclase Toxin. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Subrini, O.; Sotomayor-Pérez, A.-C.; Hessel, A.; Spiaczka-Karst, J.; Selwa, E.; Sapay, N.; Veneziano, R.; Pansieri, J.; Chopineau, J.; Ladant, D.; et al. Characterization of a Membrane-Active Peptide from the Bordetella pertussis CyaA Toxin. J. Biol. Chem. 2013, 288, 32585–32598. [Google Scholar] [CrossRef] [PubMed]
- Cannella, S.E.; Ntsogo Enguéné, V.Y.; Davi, M.; Malosse, C.; Sotomayor Pérez, A.C.; Chamot-Rooke, J.; Vachette, P.; Durand, D.; Ladant, D.; Chenal, A. Stability, Structural and Functional Properties of a Monomeric, Calcium-Loaded Adenylate Cyclase Toxin, CyaA, from Bordetella pertussis. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.; Cook, G.H.; Goldhammer, A.R.; Londos, C.; Hewlett, E.L. Bordetella pertussis: Multiple Attacks on Host Cell Cyclic AMP Regulation. Adv. Cyclic Nucleotide Protein Phosphorylation Res. 1984, 17, 161–172. [Google Scholar] [PubMed]
- Vojtova, J.; Kamanova, J.; Sebo, P. Bordetella Adenylate Cyclase Toxin: A Swift Saboteur of Host Defense. Curr. Opin. Microbiol. 2006, 9, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Masin, J.; Osicka, R.; Sebo, P. Pore-Forming and Enzymatic Activities of Bordetella pertussis Adenylate Cyclase Toxin Synergize in Promoting Lysis of Monocytes. Infect. Immun. 2006, 74, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- González-Bullón, D.; Uribe, K.B.; Martín, C.; Ostolaza, H. Phospholipase A Activity of Adenylate Cyclase Toxin Mediates Translocation of Its Adenylate Cyclase Domain. Proc. Natl. Acad. Sci. USA 2017, 114, E6784–E6793. [Google Scholar] [CrossRef] [PubMed]
- Osicková, A.; Osicka, R.; Maier, E.; Benz, R.; Šebo, P. An Amphipathic a-Helix Including Glutamates 509 and 516 is Crucial for Membrane Translocation of Adenylate Cyclase Toxin and Modulates Formation and Cation Selectivity of its Membrane Channels. J. Biol. Chem. 1999, 274, 37644–37650. [Google Scholar] [PubMed]
- Basler, M.; Knapp, O.; Masin, J.; Fiser, R.; Maier, E.; Benz, R.; Sebo, P.; Osicka, R. Segments Crucial for Membrane Translocation and Pore-Forming Activity of Bordetella Adenylate Cyclase Toxin. J. Biol. Chem. 2007, 282, 12419–12429. [Google Scholar] [CrossRef] [PubMed]
- Benz, R.; Maier, E.; Ladant, D.; Ullmann, A.; Sebo, P. Adenylate Cyclase Toxin (CyaA) of Bordetella pertussis. Evidence for the Formation of Small Ion-Permeable Channels and Comparison with HlyA of Escherichia coli. J. Biol. Chem. 1994, 269, 27231–27239. [Google Scholar] [PubMed]
- Szabo, G.; Gray, M.C.; Hewlett, E.L. Adenylate Cyclase Toxin from Bordetella pertussis Produces Ion Conductance across Artificial Lipid Bilayers in a Calcium- and Polarity-Dependent Manner. J. Biol. Chem. 1994, 269, 22496–22499. [Google Scholar] [PubMed]
- Ehrmann, I.E.; Gray, M.C.; Gordon, V.M.; Gray, L.S.; Hewlett, E.L. Hemolytic Activity of Adenylate Cyclase Toxin from Bordetella pertussis. FEBS Lett. 1991, 278, 79–83. [Google Scholar] [PubMed]
- Vojtova-Vodolanova, J.; Basler, M.; Osicka, R.; Knapp, O.; Maier, E.; Cerny, J.; Benada, O.; Benz, R.; Sebo, P. Oligomerization is Involved in Pore Formation by Bordetella Adenylate Cyclase Toxin. FASEB J. 2009, 23, 2831–2843. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Welch, R.A. Effects of Temperature, Time, and Toxin Concentration on Lesion Formation by the Escherichia coli Hemolysin. Infect. Immun. 1994, 62, 4124–4134. [Google Scholar] [PubMed]
- Martin, C.; Requero, M.A.; Masin, J.; Konopasek, I.; Goni, F.M.; Sebo, P.; Ostolaza, H. Membrane Restructuring by Bordetella pertussis Adenylate Cyclase Toxin, a Member of the RTX Toxin Family. J. Bacteriol. 2004, 186, 3760–3765. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.C.; Boesze-Battaglia, K.; Du, Y.; Stefano, F.P.; Kieba, I.R.; Epand, R.F.; Kakalis, L.; Yeagle, P.L.; Epand, R.M.; Lally, E.T. Aggregatibacter actinomycetemcomitans Leukotoxin Cytotoxicity Occurs through Bilayer Destabilization. Cell. Microbiol. 2012, 14, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Bakás, L.; Chanturiya, A.; Herlax, V.; Zimmerberg, J. Paradoxical Lipid Dependence of Pores Formed by the Escherichia coli a-Hemolysin in Planar Phospholipid Bilayer Membranes. Biophys. J. 2006, 91, 3748–3755. [Google Scholar] [CrossRef] [PubMed]
- Etxaniz, A.; González-Bullón, D.; Alonso, M.; Martín, C.; Ostolaza, H. Irreversible vs. Reparairable Membrane Poration: Differences in Permeabilization elicited by Bordetella Adenylate Cyclase Toxin and its Hemolysin Domain in Macrophages. Manuscript under review.




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Etxaniz, A.; González-Bullón, D.; Martín, C.; Ostolaza, H. Membrane Repair Mechanisms against Permeabilization by Pore-Forming Toxins. Toxins 2018, 10, 234. https://doi.org/10.3390/toxins10060234
Etxaniz A, González-Bullón D, Martín C, Ostolaza H. Membrane Repair Mechanisms against Permeabilization by Pore-Forming Toxins. Toxins. 2018; 10(6):234. https://doi.org/10.3390/toxins10060234
Chicago/Turabian StyleEtxaniz, Asier, David González-Bullón, César Martín, and Helena Ostolaza. 2018. "Membrane Repair Mechanisms against Permeabilization by Pore-Forming Toxins" Toxins 10, no. 6: 234. https://doi.org/10.3390/toxins10060234
APA StyleEtxaniz, A., González-Bullón, D., Martín, C., & Ostolaza, H. (2018). Membrane Repair Mechanisms against Permeabilization by Pore-Forming Toxins. Toxins, 10(6), 234. https://doi.org/10.3390/toxins10060234