The Impact of Uremic Toxins on Cerebrovascular and Cognitive Disorders

 and
and 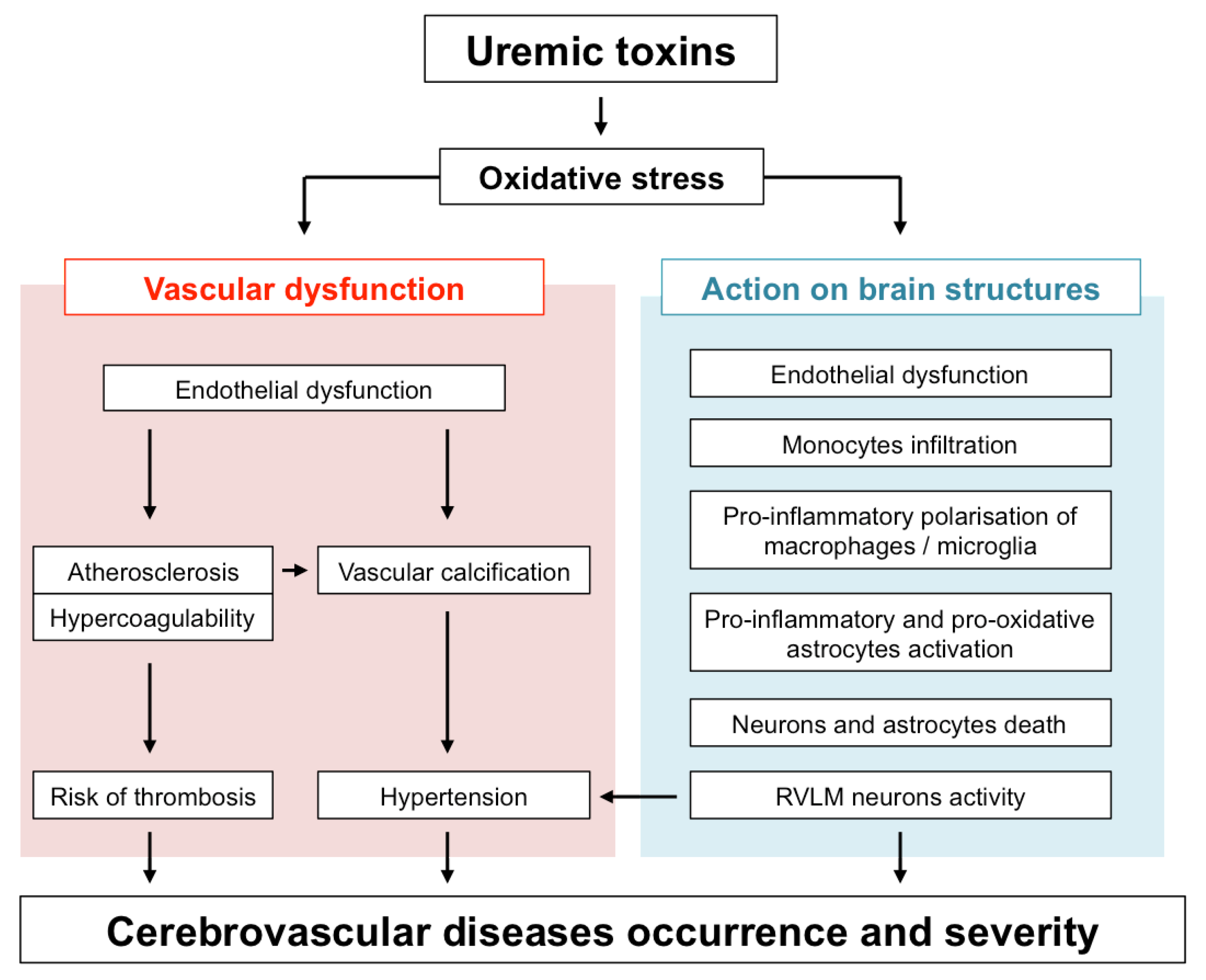{kind=link}
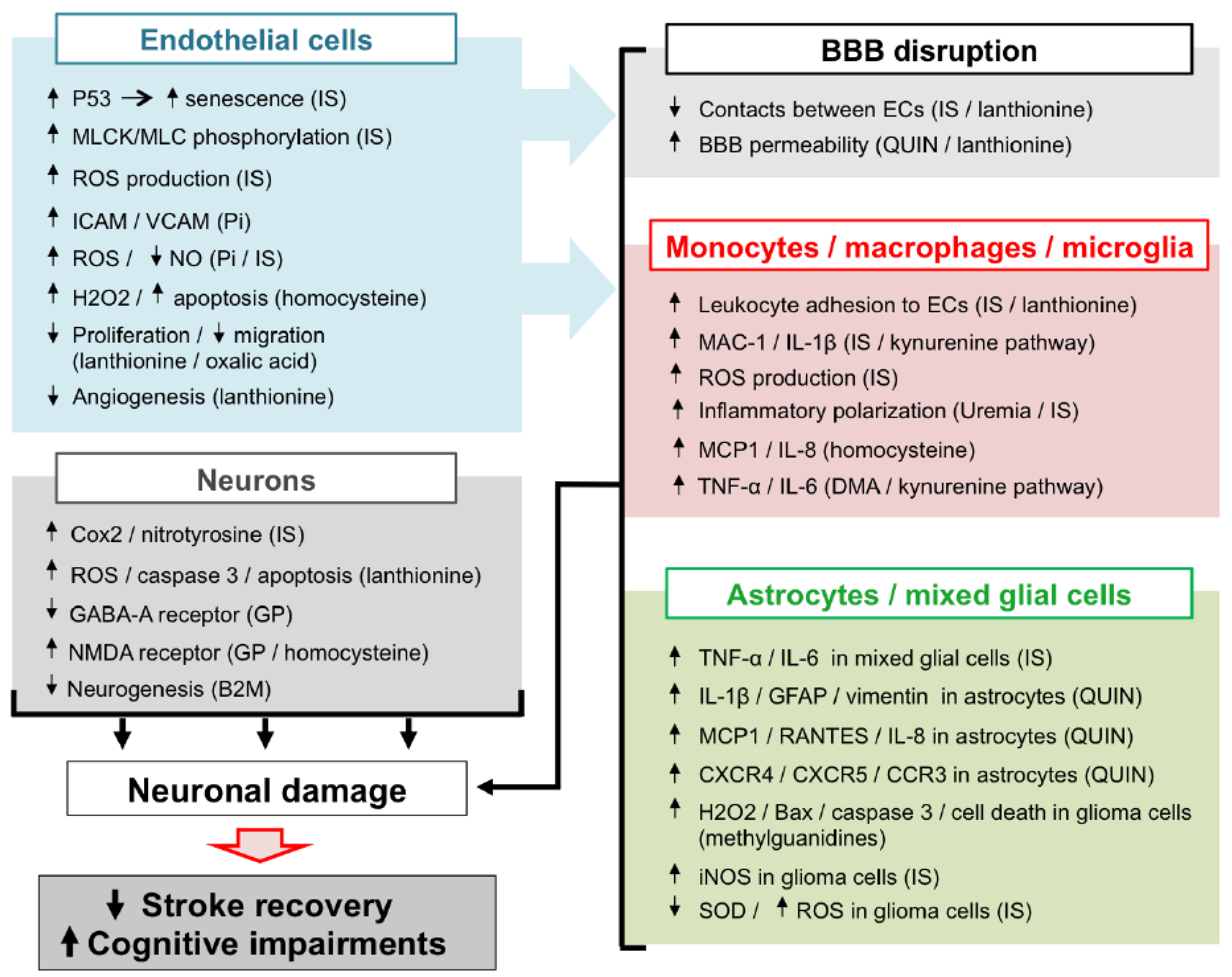{kind=link}
Abstract
:1. Introduction
2. Impact of Uremic Toxins on Large Vessels Functionality
2.1. Regulation of Blood Pressure
2.1.1. IS, Uric Acid, and Methylguanidine
2.1.2. Lanthionine
2.2. Vascular Dysfunction
2.3. Hemostasis
2.3.1. Indoxyl Sulfate
2.3.2. Homocysteine
2.3.3. Other UTs
2.4. Atrial Fibrillation
Indoxyl Sulfate
3. Impact of Uremic Toxins on Brain Microcirculation
3.1. Endothelial Cells
3.1.1. Uremic Toxins and Vasoreactivity
Phosphate and Indoxyl Sulfate
Homocysteine
3.1.2. Uremic Toxins and Endothelial Cell Integrity
Phosphate, Indoxyl Sulfate, Oxalic Acid, and Homocysteine
Lanthionine
3.1.3. Uremic Toxins and Angiogenesis
Lanthionine
3.2. Monocytes/Macrophages
3.2.1. Indoxyl Sulfate
3.2.2. Dimethylarginines
3.2.3. Guanidino Compounds
3.2.4. Quinolinic Acid
3.2.5. Homocysteine
3.2.6. Lanthionine
3.2.7. Other UTs
4. Impact of Uremic Toxins on Brain Resident Cells
4.1. Microglia
Kynurenine Pathway
4.2. Astrocytes
4.2.1. Methylguanidines
4.2.2. Indoxyl Sulfate
4.2.3. Quinolinic Acid
4.3. Neurons
4.3.1. Guanidino Compounds
4.3.2. Homocysteine
4.3.3. β-2-Microglobulin
4.3.4. Lanthionine
5. Conclusions
Funding
Conflicts of Interest
References
- Moodalbail, D.G.; Reiser, K.A.; Detre, J.A.; Schultz, R.T.; Herrington, J.D.; Davatzikos, C.; Doshi, J.J.; Erus, G.; Liu, H.S.; Radcliffe, J.; et al. Systematic review of structural and functional neuroimaging findings in children and adults with CKD. Clin. J. Am. Soc. Nephrol. 2013, 8, 1429–1448. [Google Scholar] [CrossRef] [PubMed]
- Seliger, S.L.; Siscovick, D.S.; Stehman-Breen, C.O.; Gillen, D.L.; Fitzpatrick, A.; Bleyer, A.; Kuller, L.H. Moderate renal impairment and risk of dementia among older adults: The cardiovascular health cognition study. J. Am. Soc. Nephrol. 2004, 15, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.M. Cognitive impairment in the aging dialysis and chronic kidney disease populations: An occult burden. Adv. Chronic Kidney Dis. 2008, 15, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Weiner, D.E.; Gaussoin, S.A.; Nord, J.; Auchus, A.P.; Chelune, G.J.; Chonchol, M.; Coker, L.; Haley, W.E.; Killeen, A.A.; Kimmel, P.L.; et al. Cognitive function and kidney disease: Baseline data from the systolic blood pressure intervention trial (sprint). Am. J. Kidney Dis. 2017, 70, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Lass, P.; Buscombe, J.R.; Harber, M.; Davenport, A.; Hilson, A.J. Cognitive impairment in patients with renal failure is associated with multiple-infarct dementia. Clin. Nucl. Med. 1999, 24, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Bugnicourt, J.M.; Godefroy, O.; Chillon, J.M.; Choukroun, G.; Massy, Z.A. Cognitive disorders and dementia in ckd: The neglected kidney-brain axis. J. Am. Soc. Nephrol. 2013, 24, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Chillon, J.M.; Massy, Z.A.; Stengel, B. Neurological complications in chronic kidney disease patients. Nephrol. Dial. Transplant. 2015, 31, 1606–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.C.; Lee, C.T.; Ho, S.C.; Kuo, H.W.; Wu, T.N.; Yang, C.Y. Haemodialysis and the risk of stroke: A population-based cohort study in taiwan, a country of high incidence of end-stage renal disease. Nephrology 2012, 17, 243–248. [Google Scholar] [CrossRef] [PubMed]
- El Husseini, N.; Kaskar, O.; Goldstein, L.B. Chronic kidney disease and stroke. Adv. Chronic Kidney Dis. 2014, 21, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, K.; Ninomiya, T. Stroke and cerebrovascular diseases in patients with chronic kidney disease. Lancet Neurol. 2014, 13, 823–833. [Google Scholar] [CrossRef]
- Hojs Fabjan, T.; Hojs, R. Stroke and renal dysfunction. Eur. J. Intern. Med. 2014, 25, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Koren-Morag, N.; Goldbourt, U.; Tanne, D. Renal dysfunction and risk of ischemic stroke or tia in patients with cardiovascular disease. Neurology 2006, 67, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Kumai, Y.; Kamouchi, M.; Hata, J.; Ago, T.; Kitayama, J.; Nakane, H.; Sugimori, H.; Kitazono, T. Proteinuria and clinical outcomes after ischemic stroke. Neurology 2012, 78, 1909–1915. [Google Scholar] [CrossRef] [PubMed]
- Sozio, S.M.; Armstrong, P.A.; Coresh, J.; Jaar, B.G.; Fink, N.E.; Plantinga, L.C.; Powe, N.R.; Parekh, R.S. Cerebrovascular disease incidence, characteristics, and outcomes in patients initiating dialysis: The choices for healthy outcomes in caring for esrd (choice) study. Am. J. Kidney Dis. 2009, 54, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Hojs Fabjan, T.; Hojs, R.; Tetickovic, E.; Pecovnik Balon, B. Ischaemic stroke--impact of renal dysfunction on in-hospital mortality. Eur. J. Neurol. 2007, 14, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Tsagalis, G.; Akrivos, T.; Alevizaki, M.; Manios, E.; Stamatellopoulos, K.; Laggouranis, A.; Vemmos, K.N. Renal dysfunction in acute stroke: An independent predictor of long-term all combined vascular events and overall mortality. Nephrol. Dial. Transplant. 2009, 24, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.L.; Bayraktutan, U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int. J. Stroke 2009, 4, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Liang, Y.; Zheng, S.; Zhang, H. Inhibition of myeloperoxidase by n-acetyl lysyltyrosylcysteine amide reduces oxidative stress-mediated inflammation, neuronal damage, and neural stem cell injury in a murine model of stroke. J. Pharmacol. Exp. Ther. 2018, 364, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Stetler, R.A.; Leak, R.K.; Shi, Y.; Li, Y.; Yu, W.; Bennett, M.V.L.; Chen, J. Oxidative stress and DNA damage after cerebral ischemia: Potential therapeutic targets to repair the genome and improve stroke recovery. Neuropharmacology 2018, 134, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.L.; Yang, C.M. Role of redox signaling in neuroinflammation and neurodegenerative diseases. BioMed Res. Int. 2013, 2013, 18. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.T.; Yu, J.; Grass, D.; de Beer, F.C.; Kindy, M.S. Inflammation-dependent cerebral deposition of serum amyloid a protein in a mouse model of amyloidosis. J. Neurosci. 2002, 22, 5900–5909. [Google Scholar] [CrossRef] [PubMed]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.A.; Richey, P.L.; Taneda, S.; Kutty, R.K.; Sayre, L.M.; Monnier, V.M.; Perry, G. Advanced maillard reaction end products, free radicals, and protein oxidation in alzheimer’s disease. Ann. N.Y. Acad. Sci. 1994, 738, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Miyamoto, Y.; Otagiri, M.; Maruyama, T. Update on the pharmacokinetics and redox properties of protein-bound uremic toxins. J. Pharm. Sci. 2011, 100, 3682–3695. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Saigusa, D.; Mishima, E.; Uchida, T.; Miura, D.; Morikawa-Ichinose, T.; Kisu, K.; Sekimoto, A.; Saito, R.; Oe, Y.; et al. Impact of the oral adsorbent ast-120 on organ-specific accumulation of uremic toxins: LC-MS/MS and MS imaging techniques. Toxins 2017, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- De Deyn, P.P.; Vanholder, R.; Eloot, S.; Glorieux, G. Guanidino compounds as uremic (neuro)toxins. Semin. Dial. 2009, 22, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.B. Hypertension mechanisms causing stroke. Clin. Exp. Pharmacol. Physiol. 1999, 26, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Madden, C.J.; Sved, A.F. Rostral ventrolateral medulla C1 neurons and cardiovascular regulation. Cell Mol. Neurobiol. 2003, 23, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Oshima, N.; Onimaru, H.; Matsubara, H.; Uchida, T.; Watanabe, A.; Takechi, H.; Nishida, Y.; Kumagai, H. Uric acid, indoxyl sulfate, and methylguanidine activate bulbospinal neurons in the rvlm via their specific transporters and by producing oxidative stress. Neuroscience 2015, 304, 133–145. [Google Scholar] [CrossRef] [PubMed]
- In't Veld, B.A.; Ruitenberg, A.; Hofman, A.; Stricker, B.H.; Breteler, M.M. Antihypertensive drugs and incidence of dementia: The rotterdam study. Neurobiol. Aging 2001, 22, 407–412. [Google Scholar] [CrossRef]
- Duron, E.; Hanon, O. Antihypertensive treatments, cognitive decline, and dementia. J. Alzheimers Dis. 2010, 20, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Stefani, A.; Sancesario, G.; Pierantozzi, M.; Leone, G.; Galati, S.; Hainsworth, A.H.; Diomedi, M. Csf biomarkers, impairment of cerebral hemodynamics and degree of cognitive decline in alzheimer’s and mixed dementia. J. Neurol. Sci. 2009, 283, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Vicenzini, E.; Ricciardi, M.C.; Altieri, M.; Puccinelli, F.; Bonaffini, N.; Di Piero, V.; Lenzi, G.L. Cerebrovascular reactivity in degenerative and vascular dementia: A transcranial doppler study. Eur. Neurol. 2007, 58, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Di Nunzio, A.; Amoresano, A.; Pane, F.; Fontanarosa, C.; Pucci, P.; Vigorito, C.; Cirillo, G.; Zacchia, M.; Trepiccione, F.; et al. Divergent behavior of hydrogen sulfide pools and of the sulfur metabolite lanthionine, a novel uremic toxin, in dialysis patients. Biochimie 2016, 126, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, A.F.; Zacchia, M.; Trepiccione, F.; Ingrosso, D. The sulfur metabolite lanthionine: Evidence for a role as a novel uremic toxin. Toxins 2017, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Chiku, T.; Padovani, D.; Zhu, W.; Singh, S.; Vitvitsky, V.; Banerjee, R. H2s biogenesis by human cystathionine gamma-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 2009, 284, 11601–11612. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Padovani, D.; Leslie, R.A.; Chiku, T.; Banerjee, R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to h2s biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 2009, 284, 22457–22466. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.X.; Wang, Y.B.; Peng, L.; Ge, X.Z.; Zhang, J.; Liu, S.S.; Zhang, X.N.; Xu, Z.H.; Chen, Z.; Luo, J.H. Lanthionine synthetase c-like protein 1 interacts with and inhibits cystathionine beta-synthase: A target for neuronal antioxidant defense. J. Biol. Chem. 2012, 287, 34189–34201. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Two’s company, three's a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M. Hydrogen sulfide as a vasodilator. IUBMB Life 2005, 57, 603–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S. Molecular mechanisms underlying uremic toxin-related systemic disorders in chronic kidney disease: Focused on β2-microglobulin-related amyloidosis and indoxyl sulfate-induced atherosclerosis-Oshima Award Address 2016. Clin. Exp. Nephrol. 2018, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Zuo, Y.; Ma, J.; Yancey, P.G.; Hunley, T.E.; Motojima, M.; Fogo, A.B.; Linton, M.F.; Fazio, S.; Ichikawa, I.; et al. Oral activated charcoal adsorbent (AST-120) ameliorates extent and instability of atherosclerosis accelerated by kidney disease in apolipoprotein e-deficient mice. Nephrol. Dial. Transplant. 2011, 26, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Li, L.C.; Chen, J.B.; Chang, H.W. Indoxyl sulfate-induced oxidative stress, mitochondrial dysfunction, and impaired biogenesis are partly protected by vitamin C and N-acetylcysteine. Sci. World J. 2015, 2015, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Carmona, A.; Guerrero, F.; Buendia, P.; Obrero, T.; Aljama, P.; Carracedo, J. Microvesicles derived from indoxyl sulfate treated endothelial cells induce endothelial progenitor cells dysfunction. Front. Physiol. 2017, 8, 666. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Wu, C.J.; Wu, P.C.; Pan, C.F.; Wang, T.J.; Sun, F.J.; Liu, H.L.; Chen, H.H.; Yeh, H.I. Indoxyl sulfate impairs endothelial progenitor cells and might contribute to vascular dysfunction in patients with chronic kidney disease. Kidney Blood Press. Res. 2016, 41, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; Van Kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Domaniewski, T.; Mysliwiec, M.; Pawlak, D. Kynurenines and oxidative status are independently associated with thrombomodulin and von willebrand factor levels in patients with end-stage renal disease. Thromb. Res. 2009, 124, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.R.; Beiser, A.; Seshadri, S.; Benjamin, E.J.; Polak, J.F.; Vasan, R.S.; Au, R.; DeCarli, C.; Wolf, P.A. Carotid artery atherosclerosis, MRI indices of brain ischemia, aging, and cognitive impairment: The framingham study. Stroke 2009, 40, 1590–1596. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.S.; Sigurdsson, S.; Jonsdottir, M.K.; Eiriksdottir, G.; Thorgeirsson, G.; Kjartansson, O.; Garcia, M.E.; van Buchem, M.A.; Harris, T.B.; Gudnason, V.; et al. Coronary artery calcium, brain function and structure: The ages-reykjavik study. Stroke 2010, 41, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Henaut, L.; Chillon, J.M.; Kamel, S.; Massy, Z.A. Updates on the mechanisms and the care of cardiovascular calcification in chronic kidney disease. Semin. Nephrol. 2018, 38, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Henaut, L.; Mary, A.; Chillon, J.M.; Kamel, S.; Massy, Z.A. The impact of uremic toxins on vascular smooth muscle cell function. Toxins 2018, 10, 218. [Google Scholar] [CrossRef] [PubMed]
- Bugnicourt, J.M.; Leclercq, C.; Chillon, J.M.; Diouf, M.; Deramond, H.; Canaple, S.; Lamy, C.; Massy, Z.A.; Godefroy, O. Presence of intracranial artery calcification is associated with mortality and vascular events in patients with ischemic stroke after hospital discharge: A cohort study. Stroke 2011, 42, 3447–3453. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.J. Brain regulation of thrombosis and hemostasis: From theory to practice. Stroke 2013, 44, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Mysliwiec, M.; Pawlak, D. Hypercoagulability is independently associated with kynurenine pathway activation in dialysed uraemic patients. Thromb. Haemost. 2009, 102, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Vallet, B.; Wiel, E. Endothelial cell dysfunction and coagulation. Crit. Care Med. 2001, 29, S36–S41. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, T.W.; Pawlak, K.; Karbowska, M.; Mysliwiec, M.; Pawlak, D. Indoxyl sulfate—The uremic toxin linking hemostatic system disturbances with the prevalence of cardiovascular disease in patients with chronic kidney disease. BMC Nephrol. 2017, 18, 35. [Google Scholar] [CrossRef] [PubMed]
- Massy, Z.A. Importance of homocysteine, lipoprotein (a) and non-classical cardiovascular risk factors (fibrinogen and advanced glycation end-products) for atherogenesis in uraemic patients. Nephrol. Dial. Transplant. 2000, 15 (Suppl. 5), 81–91. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Mielke, O.; Bertsch, T.; Nafe, B.; Froschen, S.; Hennerici, M. Homocysteine in cerebral macroangiography and microangiopathy. Lancet 1999, 353, 1586–1587. [Google Scholar] [CrossRef]
- Den Heijer, M.; Koster, T.; Blom, H.J.; Bos, G.M.; Briet, E.; Reitsma, P.H.; Vandenbroucke, J.P.; Rosendaal, F.R. Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N. Engl. J. Med. 1996, 334, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Hennekens, C.H.; Selhub, J.; Miletich, J.P.; Malinow, M.R.; Stampfer, M.J. Interrelation of hyperhomocyst(e)inemia, factor v leiden, and risk of future venous thromboembolism. Circulation 1997, 95, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Sauls, D.L.; Boyd, L.C.; Allen, J.C.; Hoffman, M. Differences in the metabolic response to exogenous homocysteine in juvenile and adult rabbits. J. Nutr. Biochem. 2004, 15, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Sauls, D.L.; Wolberg, A.S.; Hoffman, M. Elevated plasma homocysteine leads to alterations in fibrin clot structure and stability: Implications for the mechanism of thrombosis in hyperhomocysteinemia. J. Thromb. Haemost. 2003, 1, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Sauls, D.L.; Arnold, E.K.; Bell, C.W.; Allen, J.C.; Hoffman, M. Pro-thrombotic and pro-oxidant effects of diet-induced hyperhomocysteinemia. Thromb. Res. 2007, 120, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Harker, L.A.; Slichter, S.J.; Scott, C.R.; Ross, R. Homocystinemia. Vascular injury and arterial thrombosis. N. Engl. J. Med. 1974, 291, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Wall, R.T.; Harlan, J.M.; Harker, L.A.; Striker, G.E. Homocysteine-induced endothelial cell injury in vitro: A model for the study of vascular injury. Thromb. Res. 1980, 18, 113–121. [Google Scholar] [CrossRef]
- Lentz, S.R.; Sadler, J.E. Inhibition of thrombomodulin surface expression and protein c activation by the thrombogenic agent homocysteine. J. Clin. Investig. 1991, 88, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Nishinaga, M.; Ozawa, T.; Shimada, K. Homocysteine, a thrombogenic agent, suppresses anticoagulant heparan sulfate expression in cultured porcine aortic endothelial cells. J. Clin. Investig. 1993, 92, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, K.A. Homocysteine-induced modulation of tissue plasminogen activator binding to its endothelial cell membrane receptor. J. Clin. Investig. 1993, 91, 2873–2879. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, K.A.; Mauri, L.; Jacovina, A.T.; Zhong, F.; Mirza, U.A.; Padovan, J.C.; Chait, B.T. Tissue plasminogen activator binding to the annexin ii tail domain. Direct modulation by homocysteine. J. Biol. Chem. 1998, 273, 9987–9993. [Google Scholar] [CrossRef] [PubMed]
- Fryer, R.H.; Wilson, B.D.; Gubler, D.B.; Fitzgerald, L.A.; Rodgers, G.M. Homocysteine, a risk factor for premature vascular disease and thrombosis, induces tissue factor activity in endothelial cells. Arterioscler.Thromb. Vasc. Biol. 1993, 13, 1327–1333. [Google Scholar] [CrossRef]
- Khajuria, A.; Houston, D.S. Induction of monocyte tissue factor expression by homocysteine: A possible mechanism for thrombosis. Blood 2000, 96, 966–972. [Google Scholar] [PubMed]
- Mayer, O.; Filipovsky, J.; Hromadka, M.; Svobodova, V.; Racek, J.; Mayer, O., Jr.; Stehlik, P.; Trefil, L.; Zarybnicka, M. Treatment of hyperhomocysteinemia with folic acid: Effects on homocysteine levels, coagulation status, and oxidative stress markers. J. Cardiovasc. Pharm. 2002, 39, 851–857. [Google Scholar] [CrossRef]
- Huo, Y.; Li, J.; Qin, X.; Huang, Y.; Wang, X.; Gottesman, R.F.; Tang, G.; Wang, B.; Chen, D.; He, M.; et al. Efficacy of folic acid therapy in primary prevention of stroke among adults with hypertension in China: The CSPPT randomized clinical trial. JAMA 2015, 313, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Ingrosso, D.; Satta, E.; Lombardi, C.; Galletti, P.; D’Aniello, A.; De Santo, N.G. Plasma protein aspartyl damage is increased in hemodialysis patients: Studies on causes and consequences. J. Am. Soc. Nephrol. 2004, 15, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Watanabe, T.; Sasaki, S.; Nagai, K.; Roden, D.M.; Aizawa, Y. Close bidirectional relationship between chronic kidney disease and atrial fibrillation: The niigata preventive medicine study. Am. Heart J. 2009, 158, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Beyerbach, D.M.; Zipes, D.P. Mortality as an endpoint in atrial fibrillation. Heart Rhythm 2004, 1, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.T.; Chen, Y.C.; Hsieh, M.H.; Huang, S.Y.; Kao, Y.H.; Chen, Y.A.; Lin, Y.K.; Chen, S.A.; Chen, Y.J. The uremic toxin indoxyl sulfate increases pulmonary vein and atrial arrhythmogenesis. J. Cardiovasc. Electrophysiol. 2015, 26, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Lekawanvijit, S.; Adrahtas, A.; Kelly, D.J.; Kompa, A.R.; Wang, B.H.; Krum, H. Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur. Heart J. 2010, 31, 1771–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, A.; Hunt, B.J.; O’Sullivan, M.; Bell, R.; D’Souza, R.; Jeffery, S.; Bamford, J.M.; Markus, H.S. Homocysteine is a risk factor for cerebral small vessel disease, acting via endothelial dysfunction. Brain 2004, 127, 212–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, M.; Leite, C.D.C.; Lucato, L.T. Neuroimaging in cerebral small vessel disease: Update and new concepts. Dement. Neuropsychol. 2017, 11, 336–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.; Harakalova, M.; den Ruijter, H.; Pasterkamp, G.; Duncker, D.J.; Verhaar, M.C.; Asselbergs, F.W.; Cheng, C. Cardiorenal disease connection during post-menopause: The protective role of estrogen in uremic toxins induced microvascular dysfunction. Int. J. Cardiol. 2017, 238, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Bugnicourt, J.M.; Da Silveira, C.; Bengrine, A.; Godefroy, O.; Baumbach, G.; Sevestre, H.; Bode-Boeger, S.M.; Kielstein, J.T.; Massy, Z.A.; Chillon, J.M. Chronic renal failure alters endothelial function in cerebral circulation in mice. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1143–H1152. [Google Scholar] [CrossRef] [PubMed]
- Stinghen, A.E.; Chillon, J.M.; Massy, Z.A.; Boullier, A. Differential effects of indoxyl sulfate and inorganic phosphate in a murine cerebral endothelial cell line (bend.3). Toxins 2014, 6, 1742–1760. [Google Scholar] [CrossRef] [PubMed]
- Kielstein, J.T.; Donnerstag, F.; Gasper, S.; Menne, J.; Kielstein, A.; Martens-Lobenhoffer, J.; Scalera, F.; Cooke, J.P.; Fliser, D.; Bode-Boger, S.M. Adma increases arterial stiffness and decreases cerebral blood flow in humans. Stroke 2006, 37, 2024–2029. [Google Scholar] [CrossRef] [PubMed]
- Fujisaki, K.; Tsuruya, K.; Yamato, M.; Toyonaga, J.; Noguchi, H.; Nakano, T.; Taniguchi, M.; Tokumoto, M.; Hirakata, H.; Kitazono, T. Cerebral oxidative stress induces spatial working memory dysfunction in uremic mice: Neuroprotective effect of tempol. Nephrol. Dial. Transplant. 2014, 29, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Wolf, P.A.; Beiser, A.S.; Selhub, J.; Au, R.; Jacques, P.F.; Yoshita, M.; Rosenberg, I.H.; D’Agostino, R.B.; DeCarli, C. Association of plasma total homocysteine levels with subclinical brain injury: Cerebral volumes, white matter hyperintensity, and silent brain infarcts at volumetric magnetic resonance imaging in the framingham offspring study. Arch. Neurol. 2008, 65, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S. Elevated plasma homocysteine levels: Risk factor or risk marker for the development of dementia and alzheimer’s disease? J. Alzheimers Dis. 2006, 9, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.B.; Paik, M.C.; Brown, T.R.; Stabler, S.P.; Allen, R.H.; Sacco, R.L.; DeCarli, C. Total homocysteine is associated with white matter hyperintensity volume: The northern manhattan study. Stroke 2005, 36, 1207–1211. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Osborne, J.A.; Jaraki, O.; Rabbani, L.E.; Mullins, M.; Singel, D.; Loscalzo, J. Adverse vascular effects of homocysteine are modulated by endothelium-derived relaxing factor and related oxides of nitrogen. J. Clin. Investig. 1993, 91, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Tawakol, A.; Omland, T.; Gerhard, M.; Wu, J.T.; Creager, M.A. Hyperhomocyst(e)inemia is associated with impaired endothelium-dependent vasodilation in humans. Circulation 1997, 95, 1119–1121. [Google Scholar] [CrossRef] [PubMed]
- Woo, K.S.; Chook, P.; Lolin, Y.I.; Cheung, A.S.; Chan, L.T.; Sun, Y.Y.; Sanderson, J.E.; Metreweli, C.; Celermajer, D.S. Hyperhomocyst(e)inemia is a risk factor for arterial endothelial dysfunction in humans. Circulation 1997, 96, 2542–2544. [Google Scholar] [CrossRef] [PubMed]
- Six, I.; Maizel, J.; Barreto, F.C.; Rangrez, A.Y.; Dupont, S.; Slama, M.; Tribouilloy, C.; Choukroun, G.; Maziere, J.C.; Bode-Boeger, S.; et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc. Res. 2012, 96, 130–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pletinck, A.; Glorieux, G.; Schepers, E.; Cohen, G.; Gondouin, B.; Van Landschoot, M.; Eloot, S.; Rops, A.; Van de Voorde, J.; De Vriese, A.; et al. Protein-bound uremic toxins stimulate crosstalk between leukocytes and vessel wall. J. Am. Soc. Nephrol. 2013, 24, 1981–1994. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of e-selectin. J. Biol. Chem. 2010, 285, 38869–38875. [Google Scholar] [CrossRef] [PubMed]
- Adelibieke, Y.; Shimizu, H.; Muteliefu, G.; Bolati, D.; Niwa, T. Indoxyl sulfate induces endothelial cell senescence by increasing reactive oxygen species production and p53 activity. J. Ren. Nutr. 2012, 22, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.S.; Lin, Y.T.; Chen, Y.; Hung, K.Y.; Wang, S.M. Effects of indoxyl sulfate on adherens junctions of endothelial cells and the underlying signaling mechanism. J. Cell. Biochem. 2012, 113, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Levin, R.I.; Kantoff, P.W.; Jaffe, E.A. Uremic levels of oxalic acid suppress replication and migration of human endothelial cells. Arteriosclerosis 1990, 10, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Starkebaum, G.; Harlan, J.M. Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. J. Clin. Investig. 1986, 77, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Blundell, G.; Jones, B.G.; Rose, F.A.; Tudball, N. Homocysteine mediated endothelial cell toxicity and its amelioration. Atherosclerosis 1996, 122, 163–172. [Google Scholar] [CrossRef]
- Tyagi, N.; Ovechkin, A.V.; Lominadze, D.; Moshal, K.S.; Tyagi, S.C. Mitochondrial mechanism of microvascular endothelial cells apoptosis in hyperhomocysteinemia. J. Cell. Biochem. 2006, 98, 1150–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yoshizumi, M.; Lai, K.; Tsai, J.C.; Perrella, M.A.; Haber, E.; Lee, M.E. Inhibition of growth and p21ras methylation in vascular endothelial cells by homocysteine but not cysteine. J. Biol. Chem. 1997, 272, 25380–25385. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, J.; Ao, G.; Hu, L.; Liu, H.; Xiao, Y.; Du, H.; Alkayed, N.J.; Liu, C.F.; Cheng, J. Hydrogen sulfide protects blood-brain barrier integrity following cerebral ischemia. J. Neurochem. 2014, 129, 827–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, H.; Plate, K.H. Angiogenesis after cerebral ischemia. Acta Neuropathol. 2009, 117, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Krupinski, J.; Kaluza, J.; Kumar, P.; Kumar, S.; Wang, J.M. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 1994, 25, 1794–1798. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, S.; Xiong, X.; Ling, L.; He, R.; Wang, M.; Deng, W.; Liu, Z.; Li, Y. Lipoprostaglandin e1 modifies cognitive impairment in rats with vascular cognitive impairment by promoting angiogenesis via the VEGF/VEGFR pathway. Mol. Med. Rep. 2017, 16, 3117–3124. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.P.; Liu, H.J.; Liu, X.F. Vegf promotes angiogenesis and functional recovery in stroke rats. J. Surg. Res. 2010, 23, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.J.; Wang, M.J.; Moore, P.K.; Jin, H.M.; Yao, T.; Zhu, Y.C. The novel proangiogenic effect of hydrogen sulfide is dependent on akt phosphorylation. Cardiovasc. Res. 2007, 76, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Papapetropoulos, A. Hydrogen sulphide and angiogenesis: Mechanisms and applications. Br. J. Pharmacol. 2011, 164, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. Association between albuminuria, kidney function, and inflammatory biomarker profile in ckd in cric. Clin. J. Am. Soc. Nephrol. 2012, 7, 1938–1946. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.J.; Stenvinkel, P. Inflammation in end-stage renal disease--what have we learned in 10 years? Semin. Dial. 2010, 23, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Gouroju, S.; Rao, P.; Bitla, A.R.; Vinapamula, K.S.; Manohar, S.M.; Vishnubhotla, S. Role of gut-derived uremic toxins on oxidative stress and inflammation in patients with chronic kidney disease. Indian J. Nephrol. 2017, 27, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Bologa, R.M.; Levine, D.M.; Parker, T.S.; Cheigh, J.S.; Serur, D.; Stenzel, K.H.; Rubin, A.L. Interleukin-6 predicts hypoalbuminemia, hypocholesterolemia, and mortality in hemodialysis patients. Am. J. Kidney Dis. 1998, 32, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Barreto, D.V.; Barreto, F.C.; Liabeuf, S.; Temmar, M.; Lemke, H.D.; Tribouilloy, C.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Plasma interleukin-6 is independently associated with mortality in both hemodialysis and pre-dialysis patients with chronic kidney disease. Kidney Int. 2010, 77, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Krane, V.; Winkler, K.; Drechsler, C.; Lilienthal, J.; Marz, W.; Wanner, C. Effect of atorvastatin on inflammation and outcome in patients with type 2 diabetes mellitus on hemodialysis. Kidney Int. 2008, 74, 1461–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo-Rodriguez, E.; Pizarro-Sanchez, S.; Sanz, A.B.; Ramos, A.M.; Sanchez-Nino, M.D.; Martin-Cleary, C.; Fernandez-Fernandez, B.; Ortiz, A. Inflammatory cytokines as uremic toxins: “Ni son todos los que estan, ni estan todos los que son”. Toxins 2017, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Osaka, M.; Edamatsu, T.; Itoh, Y.; Yoshida, M. Crucial role of the aryl hydrocarbon receptor (AhR) in indoxyl sulfate-induced vascular inflammation. J. Atheroscler. Thromb. 2016, 23, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Higuchi, Y.; Yagi, Y.; Nishijima, F.; Yamato, H.; Ishii, H.; Osaka, M.; Yoshida, M. Reduction of indoxyl sulfate by ast-120 attenuates monocyte inflammation related to chronic kidney disease. J. Leukoc. Biol. 2013, 93, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: A family of cell surface receptors. Cell 1987, 48, 549–554. [Google Scholar] [CrossRef]
- Sheppard, D. In vivo functions of integrins: Lessons from null mutations in mice. Matrix Biol. 2000, 19, 203–209. [Google Scholar] [CrossRef]
- Li, C.; Ding, X.Y.; Xiang, D.M.; Xu, J.; Huang, X.L.; Hou, F.F.; Zhou, Q.G. Enhanced M1 and impaired M2 macrophage polarization and reduced mitochondrial biogenesis via inhibition of AMP kinase in chronic kidney disease. Cell. Physiol. Biochem. 2015, 36, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, T.; Yamamoto, S.; Ito, T.; Sato, Y.; Matsuo, K.; Takahashi, Y.; Kaneko, Y.; Goto, S.; Kazama, J.J.; Gejyo, F.; et al. Indoxyl sulfate promotes macrophage IL-1β production by activating aryl hydrocarbon receptor/nf-kappa/mapk cascades, but the nlrp3 inflammasome was not activated. Toxins 2018, 10, 124. [Google Scholar] [CrossRef] [PubMed]
- Barisione, C.; Garibaldi, S.; Furfaro, A.L.; Nitti, M.; Palmieri, D.; Passalacqua, M.; Garuti, A.; Verzola, D.; Parodi, A.; Ameri, P.; et al. Moderate increase of indoxyl sulfate promotes monocyte transition into profibrotic macrophages. PloS ONE 2016, 11, e0149276. [Google Scholar] [CrossRef] [PubMed]
- Worthmann, H.; Chen, S.; Martens-Lobenhoffer, J.; Li, N.; Deb, M.; Tryc, A.B.; Goldbecker, A.; Dong, Q.; Kielstein, J.T.; Bode-Boger, S.M.; et al. High plasma dimethylarginine levels are associated with adverse clinical outcome after stroke. J. Atheroscler. Thromb. 2011, 18, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Brouns, R.; Marescau, B.; Possemiers, I.; Sheorajpanday, R.; De Deyn, P.P. Dimethylarginine levels in cerebrospinal fluid of hyperacute ischemic stroke patients are associated with stroke severity. Neurochem. Res. 2009, 34, 1642–1649. [Google Scholar] [CrossRef] [PubMed]
- Luneburg, N.; von Holten, R.A.; Topper, R.F.; Schwedhelm, E.; Maas, R.; Boger, R.H. Symmetric dimethylarginine is a marker of detrimental outcome in the acute phase after ischaemic stroke: Role of renal function. Clin. Sci. 2012, 122, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Schepers, E.; Barreto, D.V.; Liabeuf, S.; Glorieux, G.; Eloot, S.; Barreto, F.C.; Massy, Z.; Vanholder, R. Symmetric dimethylarginine as a proinflammatory agent in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 2374–2383. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Martens-Lobenhoffer, J.; Weissenborn, K.; Kielstein, J.T.; Lichtinghagen, R.; Deb, M.; Li, N.; Tryc, A.B.; Goldbecker, A.; Dong, Q.; et al. Association of dimethylarginines and mediators of inflammation after acute ischemic stroke. J. Neuroinflamm. 2012, 9, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glorieux, G.L.; Dhondt, A.W.; Jacobs, P.; Van Langeraert, J.; Lameire, N.H.; De Deyn, P.P.; Vanholder, R.C. In vitro study of the potential role of guanidines in leukocyte functions related to atherogenesis and infection. Kidney Int. 2004, 65, 2184–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, K.K.; Brew, B.J.; Guillemin, G.J. Effect of quinolinic acid on human astrocytes morphology and functions: Implications in alzheimer's disease. J. Neuroinflamm. 2009, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- St'astny, F.; Skultetyova, I.; Pliss, L.; Jezova, D. Quinolinic acid enhances permeability of rat brain microvessels to plasma albumin. Brain Res. Bull. 2000, 53, 415–420. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Brew, B.J. Implications of the kynurenine pathway and quinolinic acid in alzheimer’s disease. Redox Rep. 2002, 7, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Brew, B.J.; Noonan, C.E.; Takikawa, O.; Cullen, K.M. Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in alzheimer's disease hippocampus. Neuropathol. Appl. Neurobiol. 2005, 31, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, P.; Luthi-Carter, R.E.; Augood, S.J.; Schwarcz, R. Neostriatal and cortical quinolinate levels are increased in early grade huntington’s disease. Neurobiol. Dis. 2004, 17, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Dantzer, R.; Kelley, K.W.; Lawson, M.A.; Woolwine, B.J.; Vogt, G.; Spivey, J.R.; Saito, K.; Miller, A.H. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-α: Relationship to CNS immune responses and depression. Mol. Psychiatry 2010, 15, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Rassoulpour, A.; Wu, H.Q.; Medoff, D.; Tamminga, C.A.; Roberts, R.C. Increased cortical kynurenate content in schizophrenia. Biol. Psychiatry 2001, 50, 521–530. [Google Scholar] [CrossRef]
- Ogawa, T.; Matson, W.R.; Beal, M.F.; Myers, R.H.; Bird, E.D.; Milbury, P.; Saso, S. Kynurenine pathway abnormalities in parkinson’s disease. Neurology 1992, 42, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Dai, J.; Remick, D.G.; Wang, X. Homocysteine mediated expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human monocytes. Circ. Res. 2003, 93, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Antonelli, E.; Distrutti, E.; Rizzo, G.; Mencarelli, A.; Orlandi, S.; Zanardo, R.; Renga, B.; Di Sante, M.; Morelli, A.; et al. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology 2005, 129, 1210–1224. [Google Scholar] [CrossRef] [PubMed]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Mariggio, M.A.; Minunno, V.; Riccardi, S.; Santacroce, R.; De Rinaldis, P.; Fumarulo, R. Sulfide enhancement of pmn apoptosis. Immunopharmacol. Immunotoxicol. 1998, 20, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Kanellis, J.; Watanabe, S.; Li, J.H.; Kang, D.H.; Li, P.; Nakagawa, T.; Wamsley, A.; Sheikh-Hamad, D.; Lan, H.Y.; Feng, L.; et al. Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension 2003, 41, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.; Raupachova, J.; Horl, W.H. The uraemic toxin phenylacetic acid contributes to inflammation by priming polymorphonuclear leucocytes. Nephrol. Dial. Transplant. 2013, 28, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Domaniewski, T.; Mysliwiec, M.; Pawlak, D. The kynurenines are associated with oxidative stress, inflammation and the prevalence of cardiovascular disease in patients with end-stage renal disease. Atherosclerosis 2009, 204, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Rist, P.M.; Jimenez, M.C.; Rexrode, K.M. Prospective association between β2-microglobulin levels and ischemic stroke risk among women. Neurology 2017, 88, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Kuragano, T.; Kida, A.; Furuta, M.; Nanami, M.; Otaki, Y.; Hasuike, Y.; Nonoguchi, H.; Nakanishi, T. The impact of β2-microglobulin clearance on the risk factors of cardiovascular disease in hemodialysis patients. ASAIO J. 2010, 56, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, K.; Tachikawa, M. Roles of organic anion/cation transporters at the blood-brain and blood-cerebrospinal fluid barriers involving uremic toxins. Clin. Exp. Nephrol. 2011, 15, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Sekine, T.; Watanabe, N.; Hosoyamada, M.; Kanai, Y.; Endou, H. Expression cloning and characterization of a novel multispecific organic anion transporter. J. Biol. Chem. 1997, 272, 18526–18529. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Takanaga, H.; Ohtsuki, S.; Deguchi, T.; Kang, Y.S.; Hosoya, K.; Terasaki, T. Rat organic anion transporter 3 (roat3) is responsible for brain-to-blood efflux of homovanillic acid at the abluminal membrane of brain capillary endothelial cells. J. Cereb. Blood Flow Metab. 2003, 23, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Sweet, D.H.; Miller, D.S.; Pritchard, J.B.; Fujiwara, Y.; Beier, D.R.; Nigam, S.K. Impaired organic anion transport in kidney and choroid plexus of organic anion transporter 3 (oat3 (slc22a8)) knockout mice. J. Biol. Chem. 2002, 277, 26934–26943. [Google Scholar] [CrossRef] [PubMed]
- Hayer-Zillgen, M.; Bruss, M.; Bonisch, H. Expression and pharmacological profile of the human organic cation transporters hoct1, hoct2 and hoct3. Br. J. Pharmacol. 2002, 136, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Organic cation transporters in intestine, kidney, liver, and brain. Annu. Rev. Physiol. 1998, 60, 243–266. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Asaba, H.; Takanaga, H.; Deguchi, T.; Hosoya, K.; Otagiri, M.; Terasaki, T. Role of blood-brain barrier organic anion transporter 3 (OAT3) in the efflux of indoxyl sulfate, a uremic toxin: Its involvement in neurotransmitter metabolite clearance from the brain. J. Neurochem. 2002, 83, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, M.; Kasai, Y.; Takahashi, M.; Fujinawa, J.; Kitaichi, K.; Terasaki, T.; Hosoya, K. The blood-cerebrospinal fluid barrier is a major pathway of cerebral creatinine clearance: Involvement of transporter-mediated process. J. Neurochem. 2008, 107, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011, 12, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.A.; Van Enoo, A.A.; Ikezu, T. Alzheimer’s disease: The role of microglia in brain homeostasis and proteopathy. Front. Neurosci. 2017, 11, 680. [Google Scholar] [CrossRef] [PubMed]
- Garrison, A.M.; Parrott, J.M.; Tunon, A.; Delgado, J.; Redus, L.; O’Connor, J.C. Kynurenine pathway metabolic balance influences microglia activity: Targeting kynurenine monooxygenase to dampen neuroinflammation. Psychoneuroendocrinology 2018, 94, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Parrott, J.M.; O’Connor, J.C. Kynurenine 3-monooxygenase: An influential mediator of neuropathology. Front. Psychiatry 2015, 6, 116. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcock, D.M.; Vitek, M.P.; Colton, C.A. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with alzheimer's disease. Neuroscience 2009, 159, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Brambilla, L.; Valori, C.F.; Roncoroni, C.; Crugnola, A.; Yokota, T.; Bredesen, D.E.; Volterra, A. Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ. 2008, 15, 1691–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzocco, S.; Popolo, A.; Bianco, G.; Pinto, A.; Autore, G. Pro-apoptotic effect of methylguanidine on hydrogen peroxide-treated rat glioma cell line. Neurochem. Int. 2010, 57, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wu, Z.B.; Zhuge, Q.; Zheng, W.; Shao, B.; Wang, B.; Sun, F.; Jin, K. Glial scar formation occurs in the human brain after ischemic stroke. Int. J. Med. Sci. 2014, 11, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Grande, B.; Swana, M.; Nguyen, L.; Englezou, P.; Maysami, S.; Allan, S.M.; Rothwell, N.J.; Garlanda, C.; Denes, A.; Pinteaux, E. The acute-phase protein ptx3 is an essential mediator of glial scar formation and resolution of brain edema after ischemic injury. J. Cereb. Blood Flow Metab. 2014, 34, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Adesso, S.; Magnus, T.; Cuzzocrea, S.; Campolo, M.; Rissiek, B.; Paciello, O.; Autore, G.; Pinto, A.; Marzocco, S. Indoxyl sulfate affects glial function increasing oxidative stress and neuroinflammation in chronic kidney disease: Interaction between astrocytes and microglia. Front. Pharmacol. 2017, 8, 370. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Croitoru-Lamoury, J.; Dormont, D.; Armati, P.J.; Brew, B.J. Quinolinic acid upregulates chemokine production and chemokine receptor expression in astrocytes. GLIA 2003, 41, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar] [CrossRef] [PubMed]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975. [Google Scholar] [CrossRef]
- Okada, A.; Nangaku, M.; Jao, T.M.; Maekawa, H.; Ishimono, Y.; Kawakami, T.; Inagi, R. D-serine, a novel uremic toxin, induces senescence in human renal tubular cells via gcn2 activation. Sci. Rep. 2017, 7, 11168. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, M.; Tatebe, J.; Watanabe, I.; Yamazaki, J.; Ikeda, T.; Morita, T. Aryl hydrocarbon receptor mediates indoxyl sulfate-induced cellular senescence in human umbilical vein endothelial cells. J. Atheroscler. Thromb. 2014, 21, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Small, D.M.; Ng, K.L.; Vesey, D.A.; Vitetta, L.; Francis, R.S.; Gobe, G.C.; Morais, C. Indoxyl sulfate induces apoptosis and hypertrophy in human kidney proximal tubular cells. Toxicol. Pathol. 2018, 46. [Google Scholar] [CrossRef] [PubMed]
- Hunkerler, Z.; Koken, T.; Koca, B.; Kahraman, A. Role of uremic toxins on apoptosis with varying periods of hemodialysis. Ther. Apher. Dial. 2017, 21, 38–42. [Google Scholar] [CrossRef] [PubMed]
- D’Hooge, R.; Van de Vijver, G.; Van Bogaert, P.P.; Marescau, B.; Vanholder, R.; De Deyn, P.P. Involvement of voltage- and ligand-gated Ca2+ channels in the neuroexcitatory and synergistic effects of putative uremic neurotoxins. Kidney Int. 2003, 63, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- De Deyn, P.P.; Vanholder, R.; D’Hooge, R. Nitric oxide in uremia: Effects of several potentially toxic guanidino compounds. Kidney Int. 2003, 63, S25–S28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, S.A.; Kim, W.K.; Choi, Y.B.; Kumar, S.; D'Emilia, D.M.; Rayudu, P.V.; Arnelle, D.R.; Stamler, J.S. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, R.P.; Swan, J.H.; Griffiths, T.; Meldrum, B.S. Blockade of n-methyl-d-aspartate receptors may protect against ischemic damage in the brain. Science 1984, 226, 850–852. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Rosenberg, P.A. Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; He, Y.; Park, J.S.; Bieri, G.; Snethlage, C.E.; Lin, K.; Gontier, G.; Wabl, R.; Plambeck, K.E.; Udeochu, J.; et al. β2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat. Med. 2015, 21, 932–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvidsson, A.; Collin, T.; Kirik, D.; Kokaia, Z.; Lindvall, O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 2002, 8, 963–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xu, D.; Qi, H.; Yuan, Y.; Liu, H.; Yao, S.; Yuan, S.; Zhang, J. Enriched environment promotes post-stroke neurogenesis through nf-kappab-mediated secretion of IL-17a from astrocytes. Brain Res. 2018, 1687, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, S.; Hu, Z.; Zhu, G.; Zheng, G.; Wang, G. Enriched housing promotes post-stroke neurogenesis through calpain 1-stat3/hif-1alpha/vegf signaling. Brain Res. Bull. 2018, 139, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.F.; Wang, Y.C.; Zong, L.; Chen, Z.Y.; Li, Y. Elevating integrin-linked kinase expression has rescued hippocampal neurogenesis and memory deficits in an ad animal model. Brain Res. 2018, 1695, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Armstrong, J.S.; Chu, S.H.; Jia-Ling, S.; Wong, B.S.; Cheung, N.S.; Halliwell, B.; Moore, P.K. The novel neuromodulator hydrogen sulfide: An endogenous peroxynitrite ‘scavenger’? J. Neurochem. 2004, 90, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem. Int. 2013, 63, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Tsugane, M.; Oka, J.; Kimura, H. Hydrogen sulfide induces calcium waves in astrocytes. FASEB J. 2004, 18, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yang, X.; Zhao, S.; Wei, C.; Yin, Y.; Liu, T.; Jiang, S.; Xie, J.; Wan, X.; Mao, M.; et al. Hydrogen sulfide prevents ogd/r-induced apoptosis via improving mitochondrial dysfunction and suppressing an ros-mediated caspase-3 pathway in cortical neurons. Neurochem. Int. 2013, 63, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Huang, Y.; Lin, W.; Gao, D.; Fei, Z. Protective effects of hydrogen sulfide in a rat model of traumatic brain injury via activation of mitochondrial adenosine triphosphate-sensitive potassium channels and reduction of oxidative stress. J. Surg. Res. 2013, 184, e27–e35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Shan, H.; Chang, P.; Wang, T.; Dong, W.; Chen, X.; Tao, L. Hydrogen sulfide offers neuroprotection on traumatic brain injury in parallel with reduced apoptosis and autophagy in mice. PloS ONE 2014, 9, e87241. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Shan, H.; Wang, T.; Liu, W.; Wang, Y.; Wang, L.; Zhang, L.; Chang, P.; Dong, W.; Chen, X.; et al. Dynamic change of hydrogen sulfide after traumatic brain injury and its effect in mice. Neurochem. Res. 2013, 38, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Tu, C.; Zhao, J.; Ou, D.; Chen, G.; Liu, Y.; Xiao, X. Exogenous hydrogen sulfide protects against global cerebral ischemia/reperfusion injury via its anti-oxidative, anti-inflammatory and anti-apoptotic effects in rats. Brain Res. 2013, 1491, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Bolati, D.; Adijiang, A.; Enomoto, A.; Nishijima, F.; Dateki, M.; Niwa, T. Senescence and dysfunction of proximal tubular cells are associated with activated p53 expression by indoxyl sulfate. Am. J. Physiol. Cell. Physiol. 2010, 299, C1110–C1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.C.; Lu, Y.C.; Chiu, C.A.; Yu, T.H.; Hung, W.C.; Wang, C.P.; Lu, L.F.; Chung, F.M.; Lee, Y.J.; Tsai, I.T. Levels of indoxyl sulfate are associated with severity of coronary atherosclerosis. Clin. Investig. Med. 2013, 36, E42–E49. [Google Scholar] [CrossRef]
- Ryu, J.H.; Yu, M.; Lee, S.; Ryu, D.R.; Kim, S.J.; Kang, D.H.; Choi, K.B. Ast-120 improves microvascular endothelial dysfunction in end-stage renal disease patients receiving hemodialysis. Yonsei Med. J. 2016, 57, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Inami, Y.; Hamada, C.; Seto, T.; Hotta, Y.; Aruga, S.; Inuma, J.; Azuma, K.; Io, H.; Kaneko, K.; Watada, H.; et al. Effect of ast-120 on endothelial dysfunction in adenine-induced uremic rats. Int. J. Nephrol. 2014, 2014, 164125. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Tanaka, A.; Oyama, J.; Yamasaki, A.; Shimomura, M.; Hiwatashi, A.; Ueda, Y.; Amaha, M.; Nomura, M.; Matsumura, D.; et al. Long-term effects of ast-120 on the progression and prognosis of pre-dialysis chronic kidney disease: A 5-year retrospective study. Heart Vessels 2016, 31, 1625–1632. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Song, Y.S.; Chan, P.H. Inhibition of nadph oxidase is neuroprotective after ischemia-reperfusion. J. Cereb. Blood Flow Metab. 2009, 29, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.M.; Pennypacker, K.R. Targeting antioxidant enzyme expression as a therapeutic strategy for ischemic stroke. Neurochem. Int. 2017, 107, 23–32. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Assem, M.; Lando, M.; Grissi, M.; Kamel, S.; Massy, Z.A.; Chillon, J.-M.; Hénaut, L. The Impact of Uremic Toxins on Cerebrovascular and Cognitive Disorders. Toxins 2018, 10, 303. https://doi.org/10.3390/toxins10070303
Assem M, Lando M, Grissi M, Kamel S, Massy ZA, Chillon J-M, Hénaut L. The Impact of Uremic Toxins on Cerebrovascular and Cognitive Disorders. Toxins. 2018; 10(7):303. https://doi.org/10.3390/toxins10070303
Chicago/Turabian StyleAssem, Maryam, Mathilde Lando, Maria Grissi, Saïd Kamel, Ziad A. Massy, Jean-Marc Chillon, and Lucie Hénaut. 2018. "The Impact of Uremic Toxins on Cerebrovascular and Cognitive Disorders" Toxins 10, no. 7: 303. https://doi.org/10.3390/toxins10070303
APA StyleAssem, M., Lando, M., Grissi, M., Kamel, S., Massy, Z. A., Chillon, J. -M., & Hénaut, L. (2018). The Impact of Uremic Toxins on Cerebrovascular and Cognitive Disorders. Toxins, 10(7), 303. https://doi.org/10.3390/toxins10070303