Intracellular Trafficking and Translocation of Pertussis Toxin


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Order: The PT Holotoxin
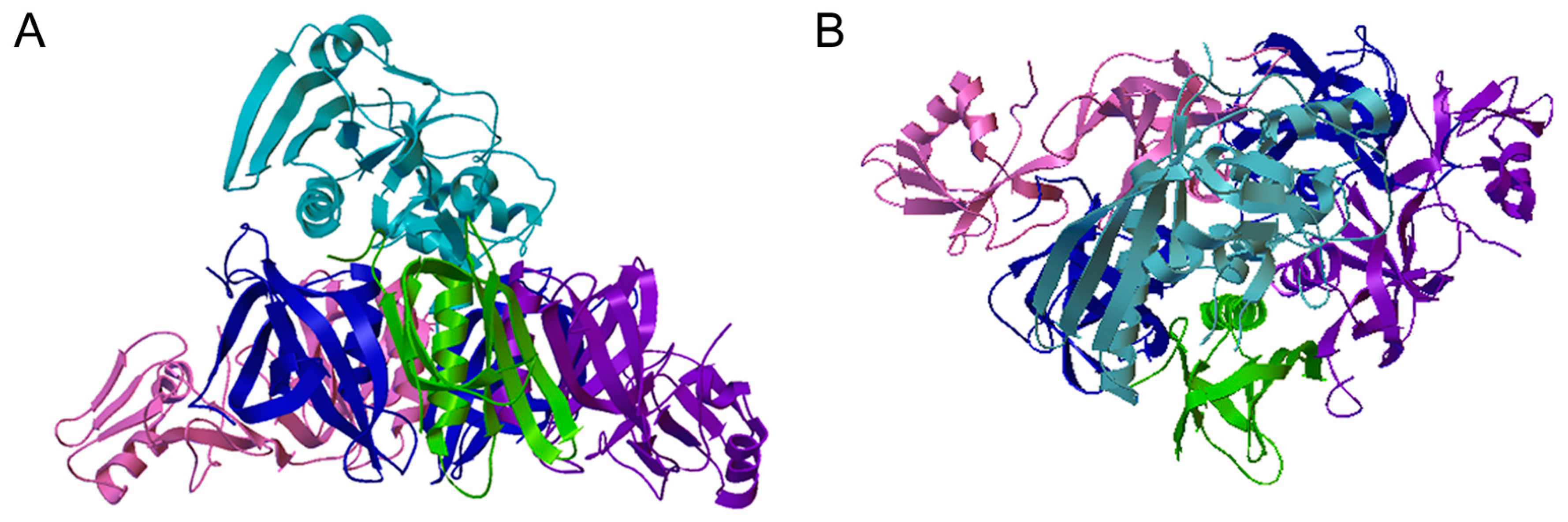
2.1. Assembly and Stability of the PT Holotoxin
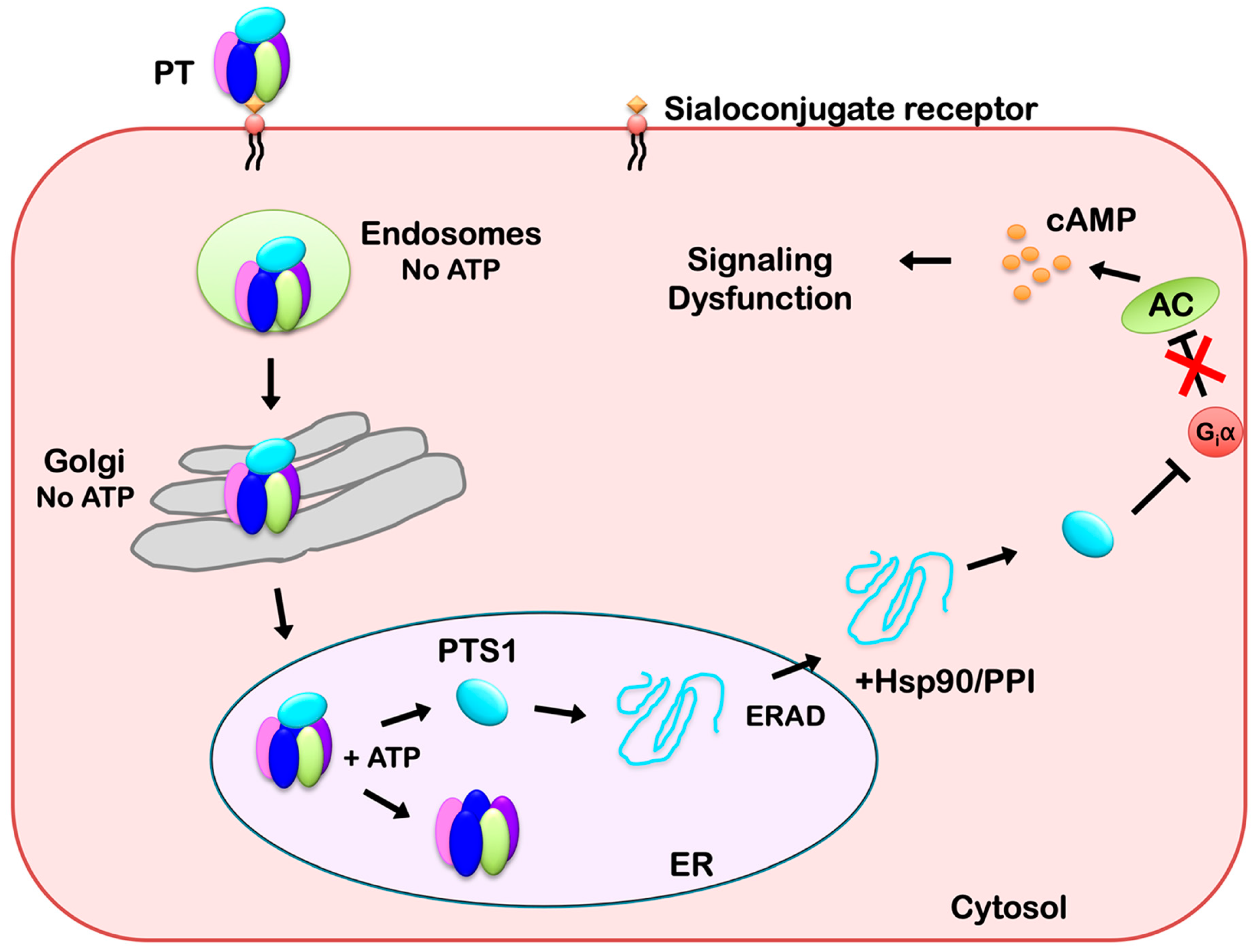
2.2. PT Recognition of Target Cells and Receptor-Mediated Endocytosis
2.3. PT Transport from the Golgi Apparatus to the ER
3. Disorder: The Free PTS1 Subunit
3.1. PT Disassembly in the ER
3.2. ERAD Processing of PTS1
3.3. PTS1 Extraction from the ER
4. Order: The Cytosolic PTS1 Subunit
4.1. The ADP-Ribosyltransferase Activity of PTS1
4.2. Refolding and Activation of Cytosolic PTS1
4.3. PTS1 Clearance from the Cytosol
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Melvin, J.A.; Scheller, E.V.; Miller, J.F.; Cotter, P.A. Bordetella pertussis pathogenesis: Current and future challenges. Nat. Rev. Microbiol. 2014, 2, 274–288. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Immunization, Vaccines and Biologicals; Pertussis. Available online: http://www.who.int/immunization/monitoring_surveillance/burden/vpd/surveillance_type/passive/pertussis/e/ (accessed on 23 May 2019).
- Centers for Disease Control and Prevention. Epidemiology and Prevention of Vaccine-Preventable Diseases; Hamborsky, J., Kroger, A., Wolfe, S., Eds.; Public Health Foundation: Washington, DC, USA, 2015; pp. 261–277.
- Güriş, D.; Strebel, P.M.; Bardenheier, B.; Brennan, M.; Tachdjian, R.; Finch, E.; Wharton, M.; Livengood, J.R. Changing epidemiology of pertussis in the United States: Increasing reported incidence among adolescents and adults, 1990–1996. Clin. Infect. Dis. 1999, 28, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Mattoo, S.; Cherry, J.D. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin. Microbiol. Rev. 2005, 18, 326–382. [Google Scholar] [CrossRef] [PubMed]
- Carbonetti, N.H. Contribution of pertussis toxin to the pathogenesis of pertussis disease. Pathog. Dis. 2015, 73, ftv073. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, K.; Skerry, C.; Carbonetti, N. Association of pertussis toxin with severe pertussis disease. Toxins 2019, 11, 373. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Nogimori, K.; Murai, S.; Yajima, M.; Ito, K.; Katada, T.; Ui, M.; Ishii, S. Subunit structure of islet-activating protein, pertussis toxin, in conformity with the A-B model. Biochemistry 1982, 21, 5516–5522. [Google Scholar] [CrossRef]
- Tamura, M.; Nogimori, K.; Yajima, M.; Ase, K.; Ui, M. A role of the B-oligomer moiety of islet-activating protein, pertussis toxin, in development of the biological effects on intact cells. J. Biol. Chem. 1983, 258, 6756–6761. [Google Scholar]
- Stein, P.E.; Boodhoo, A.; Armstrong, G.D.; Cockle, S.A.; Klein, M.H.; Read, R.J. The crystal structure of pertussis toxin. Structure 1994, 2, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Locht, C.; Keith, J.M. Pertussis toxin gene: Nucleotide sequence and genetic organization. Science 1986, 232, 1258–1264. [Google Scholar] [CrossRef]
- Nicosia, A.; Perugini, M.; Franzini, C.; Casagli, M.C.; Borri, M.G.; Antoni, G.; Almoni, M.; Neri, P.; Ratti, G.; Rappuoli, R. Cloning and sequencing of the pertussis toxin genes: Operon structure and gene duplication. Proc. Natl. Acad. Sci. USA 1986, 83, 4631–4635. [Google Scholar] [CrossRef]
- Locht, C.; Coutte, L.; Mielcarek, N. The ins and outs of pertussis toxin. FEBS J. 2011, 278, 4668–4682. [Google Scholar] [CrossRef] [PubMed]
- Sekura, R.D.; Fish, F.; Manclark, C.R.; Meade, B.; Zhang, Y.L. Pertussis toxin. Affinity purification of a new ADP-ribosyltransferase. J. Biol. Chem. 1983, 58, 14647–14651. [Google Scholar]
- Krell, T.; Greco, F.; Nicolai, M.C.; Dubayle, J.; Renauld-Mongenie, G.; Poisson, N.; Bernard, I. The use of microcalorimetry to characterize tetanus neurotoxin, pertussis toxin and filamentous haemagglutinin. Biotechnol. Appl. Biochem. 2003, 38, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mou, J.; Shao, Z. Structure and stability of pertussis toxin studied by in situ atomic force microscopy. FEBS Lett. 1994, 338, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Stein, P.E.; Boodhoo, A.; Armstrong, G.D.; Heerze, L.D.; Cockle, S.A.; Klein, M.H.; Read, R.J. Structure of a pertussis toxin-sugar complex as a model for receptor binding. Nat. Struct. Biol. 1994, 1, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.D.; Howard, L.A.; Peppler, M.S. Use of glycosyltransferases to restore pertussis toxin receptor activity to asialoagalactofetuin. J. Biol. Chem. 1988, 263, 8677–8684. [Google Scholar] [PubMed]
- el Baya, A.; Bruckener, K.; Schmidt, M.A. Nonrestricted differential intoxication of cells by pertussis toxin. Infect. Immun. 1999, 67, 433–435. [Google Scholar] [PubMed]
- Carbonetti, N.H. Pertussis toxin and adenylate cyclase toxin: Key virulence factors of Bordetella pertussis and cell biology tools. Future Microbiol. 2010, 5, 455–469. [Google Scholar] [CrossRef]
- Kaslow, H.R.; Burns, D.L. Pertussis toxin and target eukaryotic cells: Binding, entry, and activation. FASEB J. 1992, 6, 2684–2690. [Google Scholar] [CrossRef]
- Brennan, M.J.; David, J.L.; Kenimer, J.G.; Manclark, C.R. Lectin-like binding of pertussis toxin to a 165-kilodalton Chinese hamster ovary cell glycoprotein. J. Biol. Chem. 1988, 263, 4895–4899. [Google Scholar]
- Witvliet, M.H.; Burns, D.L.; Brennan, M.J.; Poolman, J.T.; Manclark, C.R. Binding of pertussis toxin to eucaryotic cells and glycoproteins. Infect. Immun. 1989, 57, 3324–3330. [Google Scholar] [PubMed]
- el Baya, A.; Linnemann, R.; von Olleschik-Elbheim, L.; Robenek, H.; Schmidt, M.A. Endocytosis and retrograde transport of pertussis toxin to the Golgi complex as a prerequisite for cellular intoxication. Eur. J. Cell Biol. 1997, 73, 40–48. [Google Scholar] [PubMed]
- Kugler, S.; Bocker, K.; Heusipp, G.; Greune, L.; Kim, K.S.; Schmidt, M.A. Pertussis toxin transiently affects barrier integrity, organelle organization and transmigration of monocytes in a human brain microvascular endothelial cell barrier model. Cell Microbiol. 2007, 9, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R. Multiple routes of protein transport from endosomes to the trans Golgi network. FEBS Lett. 2009, 583, 3811–3816. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Barbieri, J.T. Pertussis toxin-catalyzed ADP-ribosylation of Gi-2 and Gi-3 in CHO cells is modulated by inhibitors of intracellular trafficking. Infect. Immun. 1996, 64, 593–599. [Google Scholar] [PubMed]
- Xu, Y.; Barbieri, J.T. Pertussis toxin-mediated ADP-ribosylation of target proteins in Chinese hamster ovary cells involves a vesicle trafficking mechanism. Infect. Immun. 1995, 63, 825–832. [Google Scholar] [PubMed]
- Schmid, S.L.; Smythe, E. Stage-specific assays for coated pit formation and coated vesicle budding in vitro. J. Cell Biol. 1991, 114, 869–880. [Google Scholar] [CrossRef]
- Punnonen, E.L.; Ryhänen, K.; Marjomäki, V.S. At reduced temperature, endocytic membrane traffic is blocked in multivesicular carrier endosomes in rat cardiac myocytes. Eur. J. Cell Biol. 1998, 75, 344–352. [Google Scholar] [CrossRef]
- Granger, E.; McNee, G.; Allan, V.; Woodman, P. The role of the cytoskeleton and molecular motors in endosomal dynamics. Semin. Cell Dev. Biol. 2014, 31, 20–29. [Google Scholar] [CrossRef]
- Egea, G.; Serra-Peinado, C.; Gavilan, M.P.; Rios, R.M. Cytoskeleton and Golgi-apparatus interactions: A two-way road of function and structure. Cell Health Cytoskelet. 2015, 7, 37–54. [Google Scholar] [CrossRef]
- Plaut, R.D.; Carbonetti, N.H. Retrograde transport of pertussis toxin in the mammalian cell. Cell Microbiol. 2008, 10, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- Nabi, I.R.; Le, P.U. Caveolae/raft-dependent endocytosis. J. Cell Biol. 2003, 161, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Stüven, E.; Porat, A.; Shimron, F.; Fass, E.; Kaloyanova, D.; Brügger, B.; Wieland, F.T.; Elazar, Z.; Helms, J.B. Intra-Golgi protein transport depends on a cholesterol balance in the lipid membrane. J. Biol. Chem. 2003, 278, 53112–53122. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.A.; Smith, D.C.; Easton, A.J.; Roberts, L.M.; Lord, J.M. Retrograde transport pathways utilised by viruses and protein toxins. Virol. J. 2006, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.M.; Spooner, R.A. Ricin trafficking in plant and mammalian cells. Toxins 2011, 3, 787–801. [Google Scholar] [CrossRef]
- Sowa-Rogozińska, N.; Sominka, H.; Nowakowska-Golacka, J.; Sandvig, K.; Słomińska-Wojewódzka, M. Intracellular transport and cytotoxicity of the protein toxin ricin. Toxins 2019, 11, 350. [Google Scholar] [CrossRef]
- Acquaye-Seedah, E.; Huang, Y.; Sutherland, J.N.; DiVenere, A.M.; Maynard, J.A. Humanised monoclonal antibodies neutralise pertussis toxin by receptor blockade and reduced retrograde trafficking. Cell Microbiol. 2018, 20, e12948. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Oda, K.; Yokota, S.; Takatsuki, A.; Ikehara, Y. Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J. Biol. Chem. 1988, 263, 18545–18552. [Google Scholar]
- Banerjee, T.; Cilenti, L.; Taylor, M.; Showman, A.; Tatulian, S.A.; Teter, K. Thermal unfolding of the pertussis toxin S1 subunit facilitates toxin translocation to the cytosol by the mechanism of endoplasmic reticulum-associated degradation. Infect. Immun. 2016, 84, 3388–3398. [Google Scholar] [CrossRef]
- Plaut, R.D.; Scanlon, K.M.; Taylor, M.; Teter, K.; Carbonetti, N.H. Intracellular disassembly and activity of pertussis toxin require interaction with ATP. Pathog. Dis. 2016, 74, ftw065. [Google Scholar] [CrossRef] [PubMed]
- Bagola, K.; Mehnert, M.; Jarosch, E.; Sommer, T. Protein dislocation from the ER. Biochim. Biophys. Acta 2011, 1808, 925–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazes, B.; Boodhoo, A.; Cockle, S.A.; Read, R.J. Crystal structure of the pertussis toxin-ATP complex: A molecular sensor. J. Mol. Biol. 1996, 258, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.L.; Manclark, C.R. Adenine nucleotides promote dissociation of pertussis toxin subunits. J. Biol. Chem. 1986, 261, 4324–4327. [Google Scholar] [PubMed]
- Kaslow, H.R.; Lim, L.K.; Moss, J.; Lesikar, D.D. Structure-activity analysis of the activation of pertussis toxin. Biochemistry 1987, 26, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Kaslow, H.R.; Lesikar, D.D. Sulfhydryl-alkylating reagents inactivate the NAD glycohydrolase activity of pertussis toxin. Biochemistry 1987, 26, 4397–4402. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Stanley, S.J.; Watkins, P.A.; Burns, D.L.; Manclark, C.R.; Kaslow, H.R.; Hewlett, E.L. Stimulation of the thiol-dependent ADP-ribosyltransferase and NAD glycohydrolase activities of Bordetella pertussis toxin by adenine nucleotides, phospholipids, and detergents. Biochemistry 1986, 25, 2720–2725. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Helenius, J.; Helenius, A. Role of ATP and disulphide bonds during protein folding in the endoplasmic reticulum. Nature 1992, 356, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Clairmont, C.A.; De Maio, A.; Hirschberg, C.B. Translocation of ATP into the lumen of rough endoplasmic reticulum-derived vesicles and its binding to luminal proteins including BiP (GRP 78) and GRP 94. J. Biol. Chem. 1992, 267, 3983–3990. [Google Scholar]
- Pande, A.H.; Moe, D.; Jamnadas, M.; Tatulian, S.A.; Teter, K. The pertussis toxin S1 subunit is a thermally unstable protein susceptible to degradation by the 20S proteasome. Biochemistry 2006, 45, 13734–13740. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.L.; Manclark, C.R. Role of cysteine 41 of the A subunit of pertussis toxin. J. Biol. Chem. 1989, 264, 564–568. [Google Scholar] [PubMed]
- Taylor, M.; Curtis, D.; Teter, K. A conformational shift in the dissociated cholera toxin A1 subunit prevents reassembly of the cholera holotoxin. Toxins 2015, 7, 2674–2684. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.G.; McNamara, U.; Carbonetti, N.H. Expression, activity and cytotoxicity of pertussis toxin S1 subunit in transfected mammalian cells. Cell Microbiol. 2001, 3, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Veithen, A.; Raze, D.; Locht, C. Intracellular trafficking and membrane translocation of pertussis toxin into host cells. Int J. Med. Microbiol. 2000, 290, 409–413. [Google Scholar] [CrossRef]
- Ruggiano, A.; Foresti, O.; Carvalho, P. Quality control: ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Hazes, B.; Read, R.J. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 1997, 36, 11051–11054. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.M.; Roberts, L.M. Toxin entry: Retrograde transport through the secretory pathway. J. Cell Biol. 1998, 140, 733–736. [Google Scholar] [CrossRef]
- Perlmutter, D.H. Chemical chaperones: A pharmacological strategy for disorders of protein folding and trafficking. Pediatr. Res. 2002, 52, 832–836. [Google Scholar] [CrossRef]
- Cortez, L.; Sim, V. The therapeutic potential of chemical chaperones in protein folding diseases. Prion 2014, 8, 28938. [Google Scholar] [CrossRef]
- Teter, K.; Holmes, R.K. Inhibition of endoplasmic reticulum-associated degradation in CHO cells resistant to cholera toxin, Pseudomonas aeruginosa exotoxin A, and ricin. Infect. Immun. 2002, 70, 6172–6179. [Google Scholar] [CrossRef]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system. Biochim. Biophys. Acta 2014, 1843, 182–196. [Google Scholar] [CrossRef] [PubMed]
- London, E.; Luongo, C.L. Domain-specific bias in arginine/lysine usage by protein toxins. Biochem. Biophys. Res. Commun. 1989, 160, 333–339. [Google Scholar] [CrossRef]
- Worthington, Z.E.; Carbonetti, N.H. Evading the proteasome: Absence of lysine residues contributes to pertussis toxin activity by evasion of proteasome degradation. Infect. Immun. 2007, 75, 2946–2953. [Google Scholar] [CrossRef] [PubMed]
- Ernst, K.; Schnell, L.; Barth, H. Host cell chaperones Hsp70/Hsp90 and peptidyl-prolyl cis/trans isomerases are required for the membrane translocation of bacterial ADP-ribosylating toxins. Curr. Top. Microbiol. Immunol. 2017, 406, 163–198. [Google Scholar] [PubMed]
- Taylor, M.; Navarro-Garcia, F.; Huerta, J.; Burress, H.; Massey, S.; Ireton, K.; Teter, K. Hsp90 is required for transfer of the cholera toxin A1 subunit from the endoplasmic reticulum to the cytosol. J. Biol. Chem. 2010, 285, 31261–31267. [Google Scholar] [CrossRef] [PubMed]
- Burress, H.; Taylor, M.; Banerjee, T.; Tatulian, S.A.; Teter, K. Co- and post-translocation roles for Hsp90 in cholera intoxication. J. Biol. Chem. 2014, 289, 33644–33654. [Google Scholar] [CrossRef] [PubMed]
- Peskin, C.S.; Odell, G.M.; Oster, G.F. Cellular motions and thermal fluctuations: The Brownian ratchet. Biophys. J. 1993, 65, 316–324. [Google Scholar] [CrossRef]
- Kellner, A.; Taylor, M.; Banerjee, T.; Britt, C.B.T.; Teter, K. A binding motif for Hsp90 in the A chains of ADP-ribosylating toxins that move from the endoplasmic reticulum to the cytosol. Cell Microbiol. 2019, e13074. [Google Scholar] [CrossRef]
- Barth, H. Exploring the role of host cell chaperones/PPIases during cellular up-take of bacterial ADP-ribosylating toxins as basis for novel pharmacological strategies to protect mammalian cells against these virulence factors. Naunyn Schmiedebergs Arch. Pharmacol. 2011, 383, 237–245. [Google Scholar] [CrossRef]
- Ernst, K.; Eberhardt, N.; Mittler, A.K.; Sonnabend, M.; Anastasia, A.; Freisinger, S.; Schiene-Fischer, C.; Malešević, M.; Barth, H. Pharmacological cyclophilin inhibitors prevent intoxication of mammalian cells with Bordetella pertussis toxin. Toxins 2018, 10, 181. [Google Scholar] [CrossRef]
- Burress, H.; Kellner, A.; Guyette, J.; Tatulian, S.A.; Teter, K. HSC70 and HSP90 chaperones perform complementary roles in translocation of the cholera toxin A1 subunit from the endoplasmic reticulum to the cytosol. J. Biol. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.A.; Hart, P.J.; Cook, J.P.; Pietroni, P.; Rogon, C.; Hohfeld, J.; Roberts, L.M.; Lord, J.M. Cytosolic chaperones influence the fate of a toxin dislocated from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2008, 105, 17408–17413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.; Britt, C.B.T.; Fundora, J.; Teter, K. Modulation of cholera toxin structure/function by Hsp90. In Physical Biology of Proteins and Peptides; Olivares-Quiroz, L., Guzman-Lopez, O., Jardón-Valadez, E., Eds.; Springer: New York, NY, USA, 2015; pp. 67–80. [Google Scholar]
- Hsia, J.A.; Tsai, S.C.; Adamik, R.; Yost, D.A.; Hewlett, E.L.; Moss, J. Amino acid-specific ADP-ribosylation. Sensitivity to hydroxylamine of [cysteine(ADP-ribose)]protein and [arginine(ADP-ribose)]protein linkages. J. Biol. Chem. 1985, 260, 16187–16191. [Google Scholar] [PubMed]
- Mangmool, S.; Kurose, H. Gi/o protein-dependent and -independent actions of pertussis toxin (Ptx). Toxins 2011, 3, 884–899. [Google Scholar] [CrossRef] [PubMed]
- Koulen, P.; Liu, J.; Nixon, E.; Madry, C. Interaction between mGluR8 and calcium channels in photoreceptors is sensitive to pertussis toxin and occurs via G protein betagamma subunit signaling. Investig. Ophthalmol. Vis. Sci. 2005, 46, 287–291. [Google Scholar] [CrossRef]
- Wu, E.H.; Wong, Y.H. Pertussis toxin-sensitive Gi/o proteins are involved in nerve growth factor-induced pro-survival Akt signaling cascade in PC12 cells. Cell Signal. 2005, 17, 881–890. [Google Scholar] [CrossRef]
- Bokoch, G.M.; Gilman, A.G. Inhibition of receptor-mediated release of arachidonic acid by pertussis toxin. Cell 1984, 39, 301–308. [Google Scholar] [CrossRef]
- Moss, J.; Stanley, S.J.; Burns, D.L.; Hsia, J.A.; Yost, D.A.; Myers, G.A.; Hewlett, E.L. Activation by thiol of the latent NAD glycohydrolase and ADP-ribosyltransferase activities of Bordetella pertussis toxin (islet-activating protein). J. Biol. Chem. 1983, 258, 11879–11882. [Google Scholar]
- Kaslow, H.R.; Schlotterbeck, J.D.; Mar, V.L.; Burnette, W.N. Alkylation of cysteine 41, but not cysteine 200, decreases the ADP-ribosyltransferase activity of the S1 subunit of pertussis toxin. J. Biol. Chem. 1989, 264, 6386–6390. [Google Scholar]
- Murayama, T.; Hewlett, E.L.; Maloney, N.J.; Justice, J.M.; Moss, J. Effect of temperature and host factors on the activities of pertussis toxin and Bordetella adenylate cyclase. Biochemistry 1994, 33, 15293–15297. [Google Scholar] [CrossRef]
- Murayama, T.; Tsai, S.C.; Adamik, R.; Moss, J.; Vaughan, M. Effects of temperature on ADP-ribosylation factor stimulation of cholera toxin activity. Biochemistry 1993, 32, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Taylor, M.; Banerjee, T.; Tatulian, S.A.; Teter, K. Lipid rafts alter the stability and activity of the cholera toxin A1 subunit. J. Biol. Chem. 2012, 287, 30395–30405. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Taylor, M.; Jobling, M.G.; Burress, H.; Yang, Z.; Serrano, A.; Holmes, R.K.; Tatulian, S.A.; Teter, K. ADP-ribosylation factor 6 acts as an allosteric activator for the folded but not disordered cholera toxin A1 polypeptide. Mol. Microbiol. 2014, 94, 898–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [PubMed]
- Ampapathi, R.S.; Creath, A.L.; Lou, D.I.; Craft, J.W., Jr.; Blanke, S.R.; Legge, G.B. Order-disorder-order transitions mediate the activation of cholera toxin. J. Mol. Biol. 2008, 377, 748–760. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teter, K. Intracellular Trafficking and Translocation of Pertussis Toxin. Toxins 2019, 11, 437. https://doi.org/10.3390/toxins11080437
Teter K. Intracellular Trafficking and Translocation of Pertussis Toxin. Toxins. 2019; 11(8):437. https://doi.org/10.3390/toxins11080437
Chicago/Turabian StyleTeter, Ken. 2019. "Intracellular Trafficking and Translocation of Pertussis Toxin" Toxins 11, no. 8: 437. https://doi.org/10.3390/toxins11080437
APA StyleTeter, K. (2019). Intracellular Trafficking and Translocation of Pertussis Toxin. Toxins, 11(8), 437. https://doi.org/10.3390/toxins11080437