1. Introduction
Lysenin is a toxic protein produced by
Eisenia fetida that plays important roles in this earthworm’s innate immunity and defense strategies against parasites [
1,
2]. Lysenin belongs to the family of aerolysin-like pore-forming proteins (aβ-PFPs), of which the founding member (aerolysin) is a well-studied toxin produced by the pathogenic bacterium
Aeromonas hydrophila. The aβ-PFP family includes numerous toxins present throughout all kingdoms of life: bacteria, fungi, plants, and animals [
3,
4,
5,
6,
7,
8,
9]. While many aβ-PFPs family members produced by pathogenic bacteria (e.g.,
Aeromonas,
Clostridium, and
Bacillus) act as virulence factors in food poisoning and infections, aβ-PFP family members produced by eukaryotes serve in defense against pathogens or parasites [
6,
10,
11]. The amino acid sequences of all aerolysin-like proteins exhibit low homology but share a similar monomeric architecture, particularly a conserved N-terminal domain involved in β-barrel pore formation.
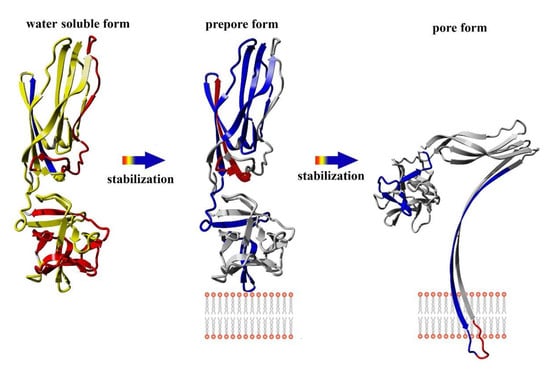
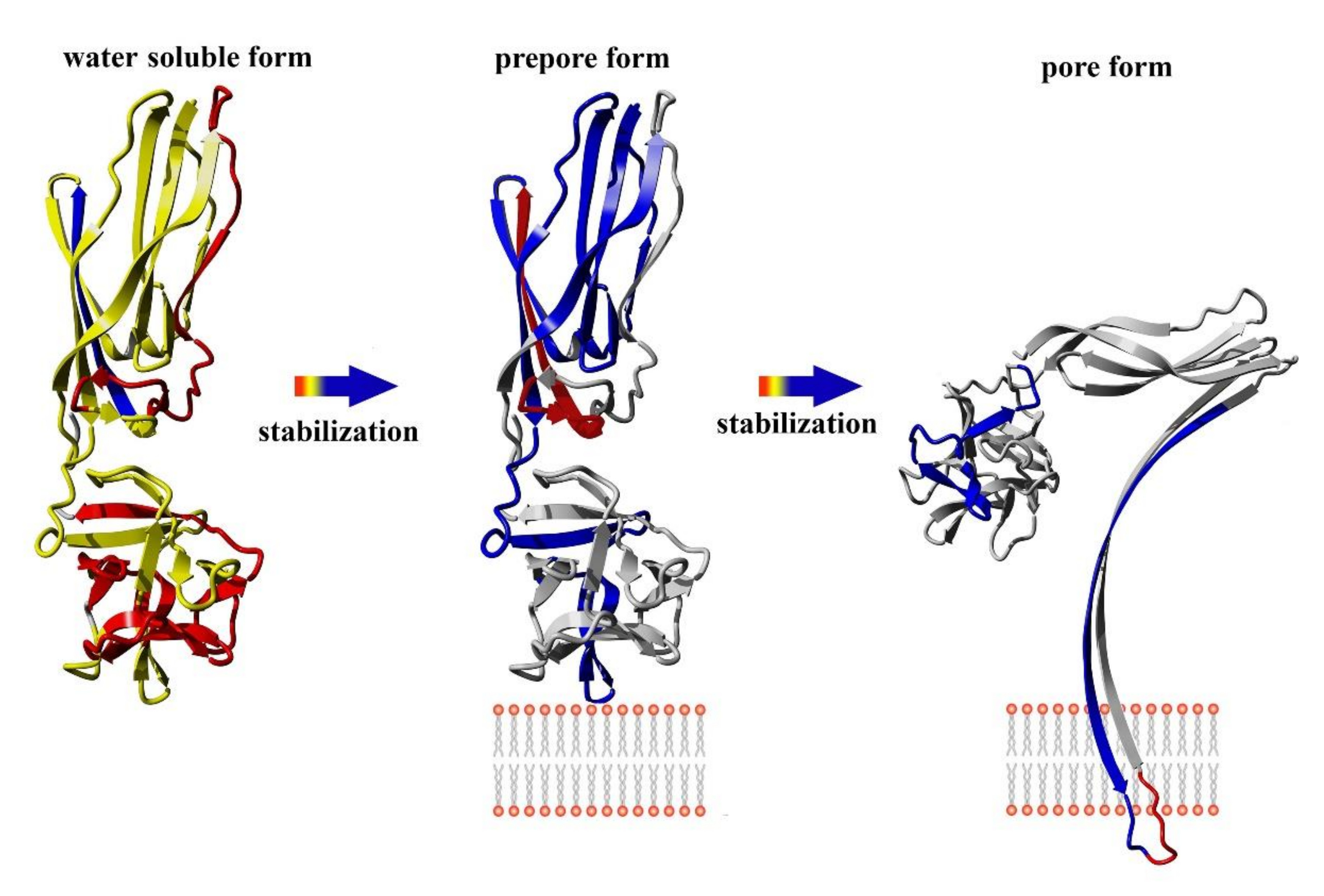
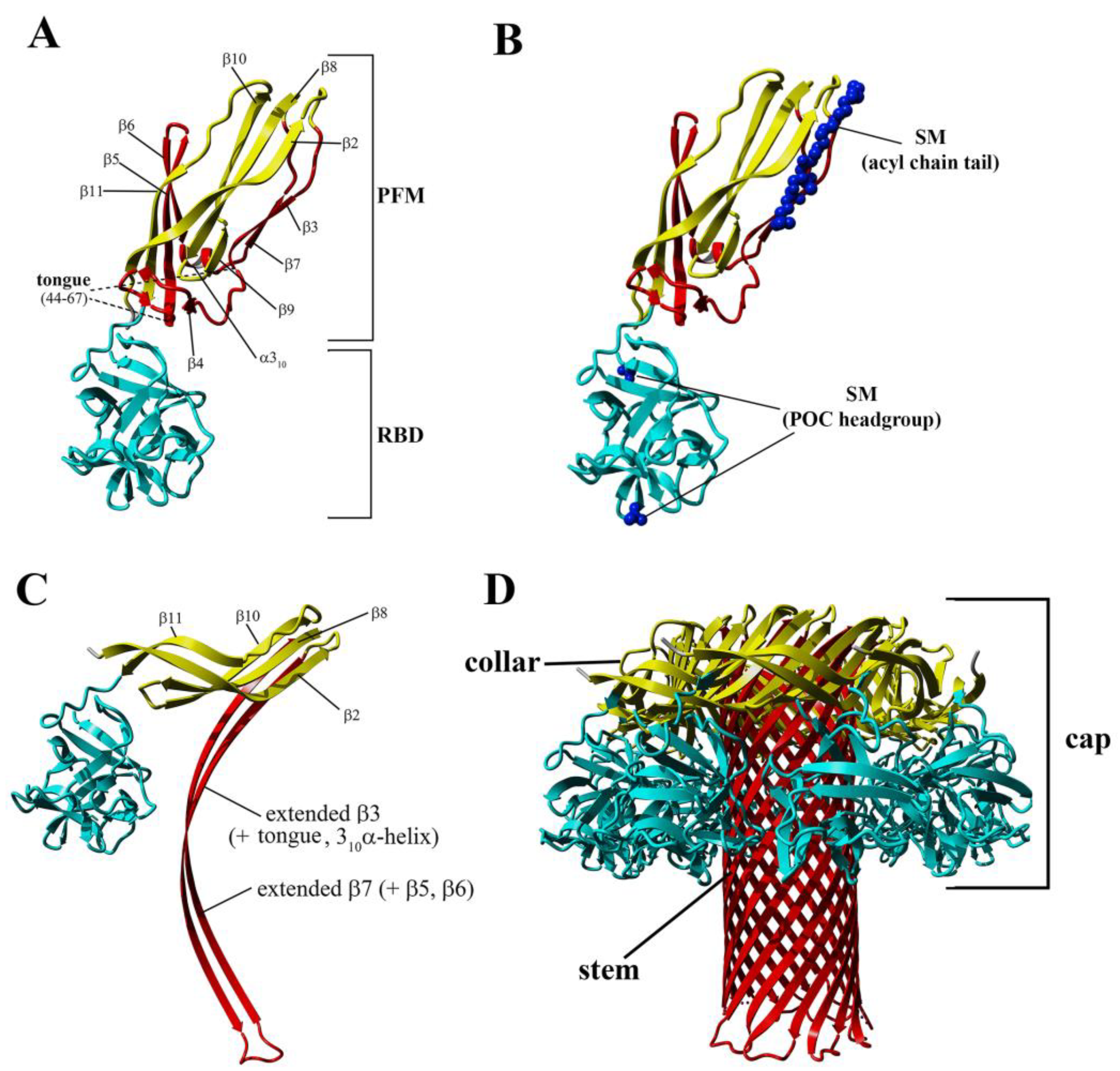
Like most pore-forming toxins, lysenin is produced and secreted as a soluble monomer. Crystallographic analyses reveal that lysenin comprises two main structural domains: an N-terminus pore-forming module (PFM) at amino acid position 1–160, which contains a SM-binding site and the membrane insertion loop (position 34–107), and the C-terminal β-trefoil receptor-binding domain (RBD) at amino acid position 161–297, which is responsible for initial binding with the lipid membrane [
9] (
Figure 1A). The crystal structure of the monomeric form of lysenin bound to SM reveals that toxin binding requires both the hydrophilic (phosphocholine head group; POC) and hydrophobic (acyl chain tail) lipid portions [
9] (
Figure 1B). Previous crystallographic studies and analyses of the binding of deletion mutants have revealed that lysenin interacts with membranes in a two-stage process [
9,
12]. First, a patch of positively charged amino acids in the C-terminal domain attracts toxin to the membrane surface and allows the initial binding of the C-terminal domain to the POC of SM via both a hydrogen bond network (including S227, Y233, and Y282) and a salt bridge interaction (K185). Second, two critical tyrosine residues (Y24 and Y26) located in the PFM of the N-terminal domain bind the acyl chain of SM via ring stacking–like interactions [
9]. Detailed analysis of the crystal structure of the lysenin/SM complex has revealed that other PFM residues, including K21, Q117, and E128, also interact with the SM head group, creating an additional hydrophilic binding site. The multivalent SM binding site at the C-terminal domain facilitates the interaction of lysenin with clustered SM organized into the SM-rich domains of the plasma membrane, thus increasing the probability of a monomer-monomer encounter [
13,
14,
15,
16,
17]. In turn, this enables lysenin monomers to create oligomeric structures (prepores) on the lipid membrane surface, which undergo a large conformational rearrangement, leading to insertion into the lipid membrane and pore formation.
Blue native electrophoresis has revealed that stable oligomer formation in SM-containing membranes correlates with lysenin self-assembly into trimers that act as the functional unit of the protein bound to SM [
13,
14]. Lysenin oligomerization, but not binding, is facilitated by cholesterol presence, which increases the fluidity of SM-containing membranes and promotes separation of the SM-rich liquid-ordered phase [
14,
17,
18,
19]. Previous studies based on electron microscopy, atomic force microscopy (AFM), and two-dimensional electron crystallography have revealed that lysenin oligomers assemble into a hexagonal close-packed (hcp) structure with an external diameter of ~10–11 nm and an inner pore of ~6 nm [
9,
16,
19]. AFM measurements of hcp assemblies of lysenin on a SM-containing bilayer have elucidated the presence of two oligomer populations with different heights—2.6 and 5 nm. Investigations of the cholesterol-dependent cytolysin perfringolysin O (PFO) have also revealed the presence of two distinct oligomer populations, while the lower-height oligomers (2.6 nm) correspond to membrane-inserted oligomers (pores), and the taller oligomers (5 nm) represent the prepore state [
20,
21]. The height of the putative lysenin’s prepore corresponds to the height of the water-soluble lysenin monomer, suggesting that lysenin protomers in the prepore state do not undergo radical structural rearrangements that change the protein’s orientation to the membrane surface.
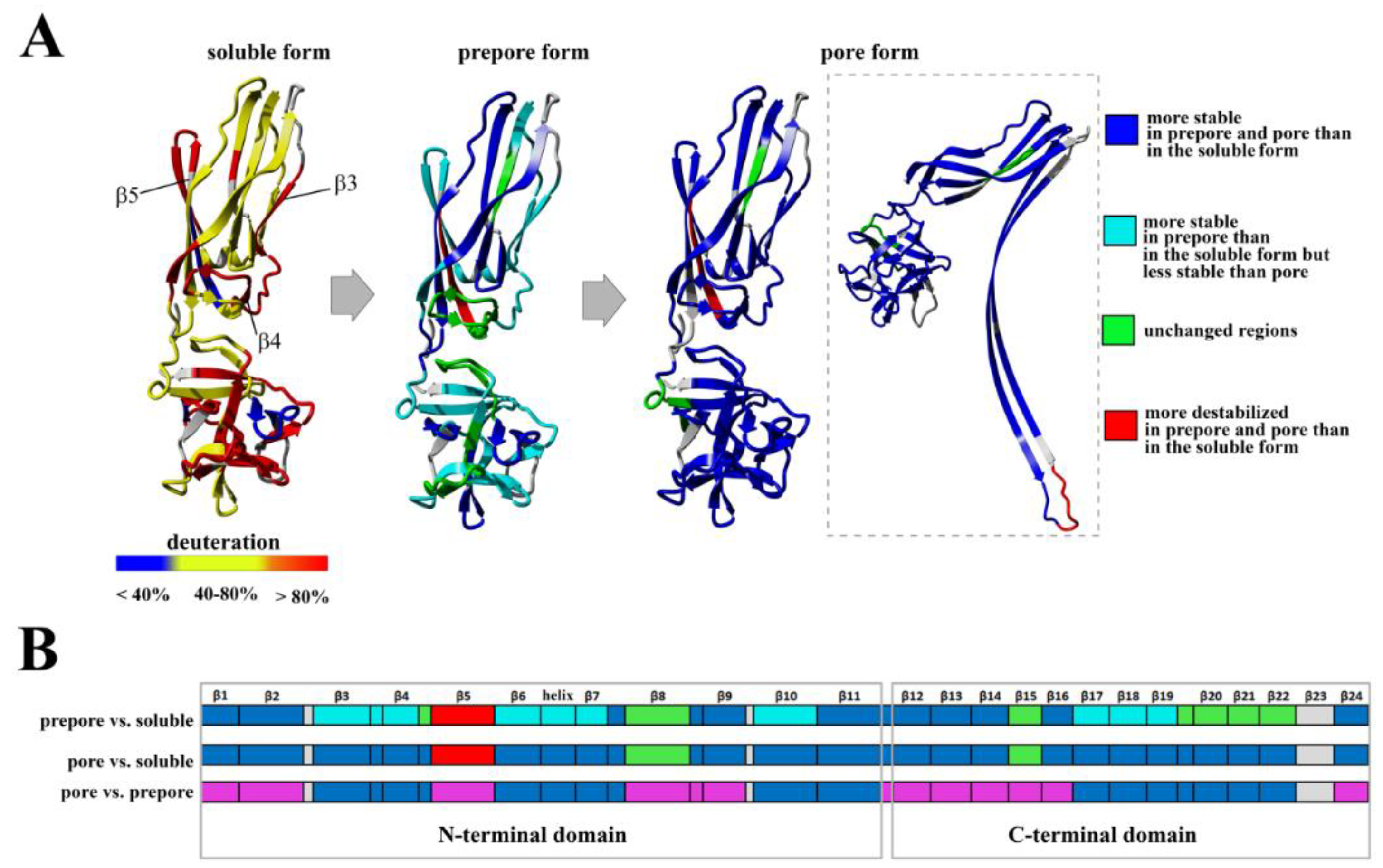
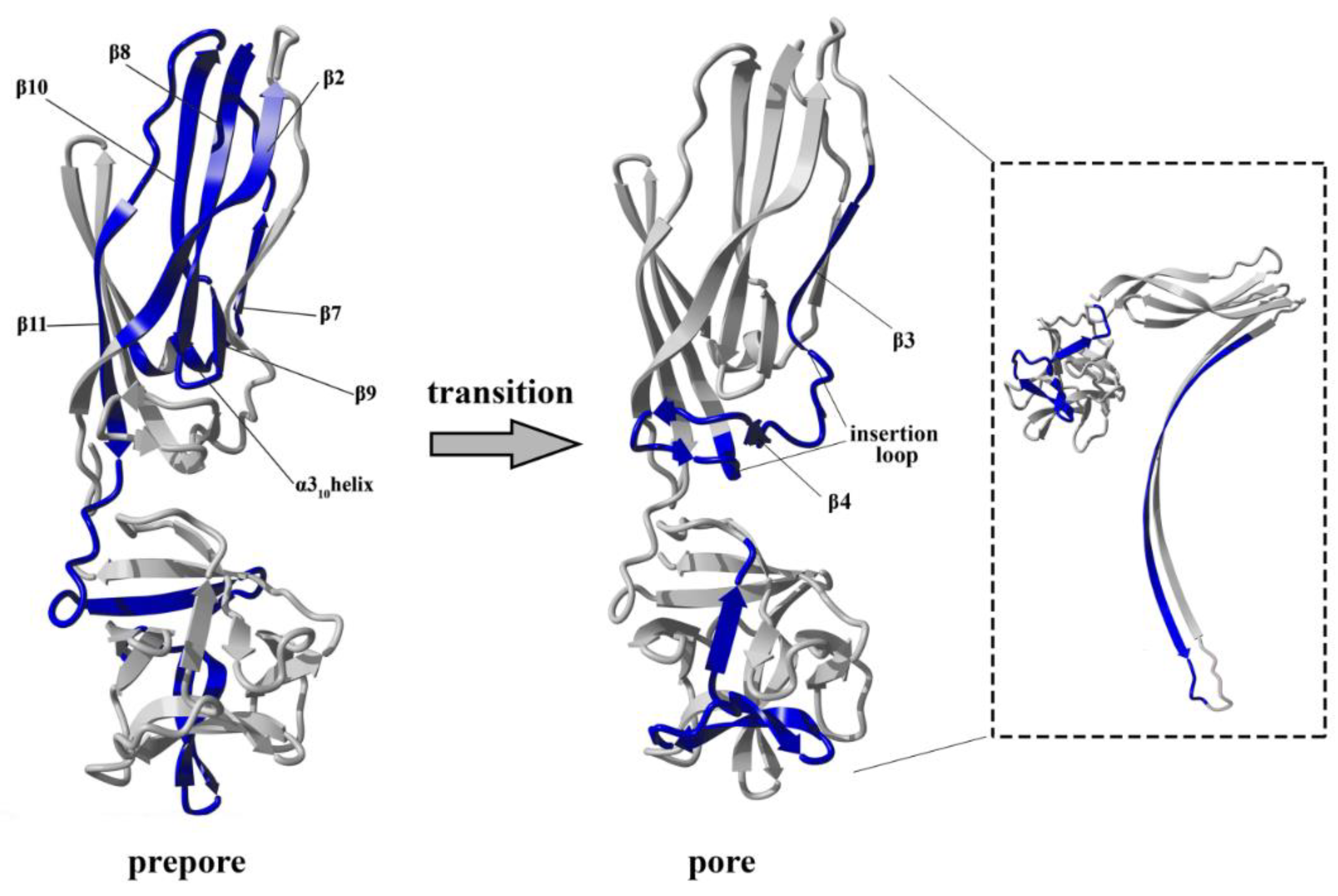
The conversion from the prepore to the pore state entails critical conformational reorganizations of lysenin structure, which only involve the N-terminal domain of lysenin (
Figure 1C). It has been suggested that the contacts between neighboring monomers in the prepore state, and the geometrically and energetically conductive intramolecular interactions between β-strands, are triggers for displacement of the insertion loop covering the region from amino acid residues 44–67 (the tongue). Insertion loop movement leads to breakage of the hydrogen bonds between the β-strand pairs β3 (pos. 38–44) and β9 (pos. 125–128), and β5 (pos. 68–81) and β11 (pos. 147–156), as well as displacement of the strands β5 and β6 (pos. 84–90). This leads to insertion loop unfolding, bringing together the parallel-oriented strands β2 (pos. 12–27) and β10 (pos.129–138) of adjacent monomers, and tilting the twisted β-sheet containing the strands β2, β8 (pos. 111–121), β10, and β11 by a 45-degree angle relative to the receptor-binding domain. The reorganization of the N-terminal domain within the strands β4 (pos. 54–57), β5, and β6 (pos. 84–90), as well as a single α3
10 helix (pos. 91–96) of each protomer, forces the extending strands β3 and β7 (pos. 99–103) to reassemble into tilted and twisted β-hairpins (amino acid residues 34–107). Finally, nine β-hairpins curve nearly 270° during incorporation into the lipid membrane, constructing a final β-barrel structure. Overall, the lysenin pore exhibits a mushroom-like architecture with the β-barrel creating a central stem passing through the lipid membrane, and the mushroom cap formed by the C-terminal receptor-binding domains (bottom part of the cap) and β-strands (β2, β8, β10, and β11) originating from the N-terminal domains (upper part of the cap, collar) (
Figure 1D).
The transition of a toxin from a water-soluble form to a membrane-anchored form requires dramatic structural reorganization. Although recent studies have elucidated several aspects of pore formation by lysenin [
8,
22], the structural characterization of intermediate steps of the pore-formation pathway remains incomplete. To gain insight into the structural dynamics of lysenin in the pre-pore state, and to characterize structural changes accompanying its insertion into the lipid membrane, we applied hydrogen–deuterium exchange mass spectrometry (HDX-MS).
HDX is a complex process whose kinetics depends on both protein dynamics and intrinsic amide hydrogen exchange rates. Two factors may affect the hydrogen–deuterium exchange: solvent accessibility and H-bonding. However, water molecules penetrate protein structures quite efficiently. Therefore, it is believed the main factor protecting from exchange is the involvement of amide in H-bonding, i.e., the stability of local secondary structure elements, while solvent accessibility has minor influence [
23]. Generally, due to the fact that protection against hydrogen–deuterium exchange may be best correlated with the “order” in a given region of protein, we interpret the results in terms of structural stability in a given region. Therefore, this method provides useful information regarding the dynamics of structural elements of pore-forming proteins, both in solution and upon interaction with a lipid membrane and enables investigation of the conformational changes accompanying transitions between protein states with high reproducibility and precision [
24,
25,
26].
Previous studies have indicated that introducing the double-cysteine mutation V88C/Y131C in lysenin enables creation of a disulfide bond that locks together the β6 and β10 strands and prevents β6 displacement from the rest of the toxin molecule. Consequently, this mutation abolishes lytic activity while preserving oligomer formation [
22], such that lysenin
V88C/Y131C, is an ideal candidate for characterizing the structural dynamics of lysenin oligomers arrested in the pre-pore state. To obtain structural information regarding the mechanism of lysenin oligomerization, we compared the patterns of the exchange of amide protons in lysenin
WT with those in lysenin
V88C/Y131C, both in aqueous solution and upon binding to SM-containing membranes. This approach enabled mapping of the regions in the lysenin molecule where the kinetics of hydrogen–deuterium exchange are substantially altered after interaction with the model lipid membranes.
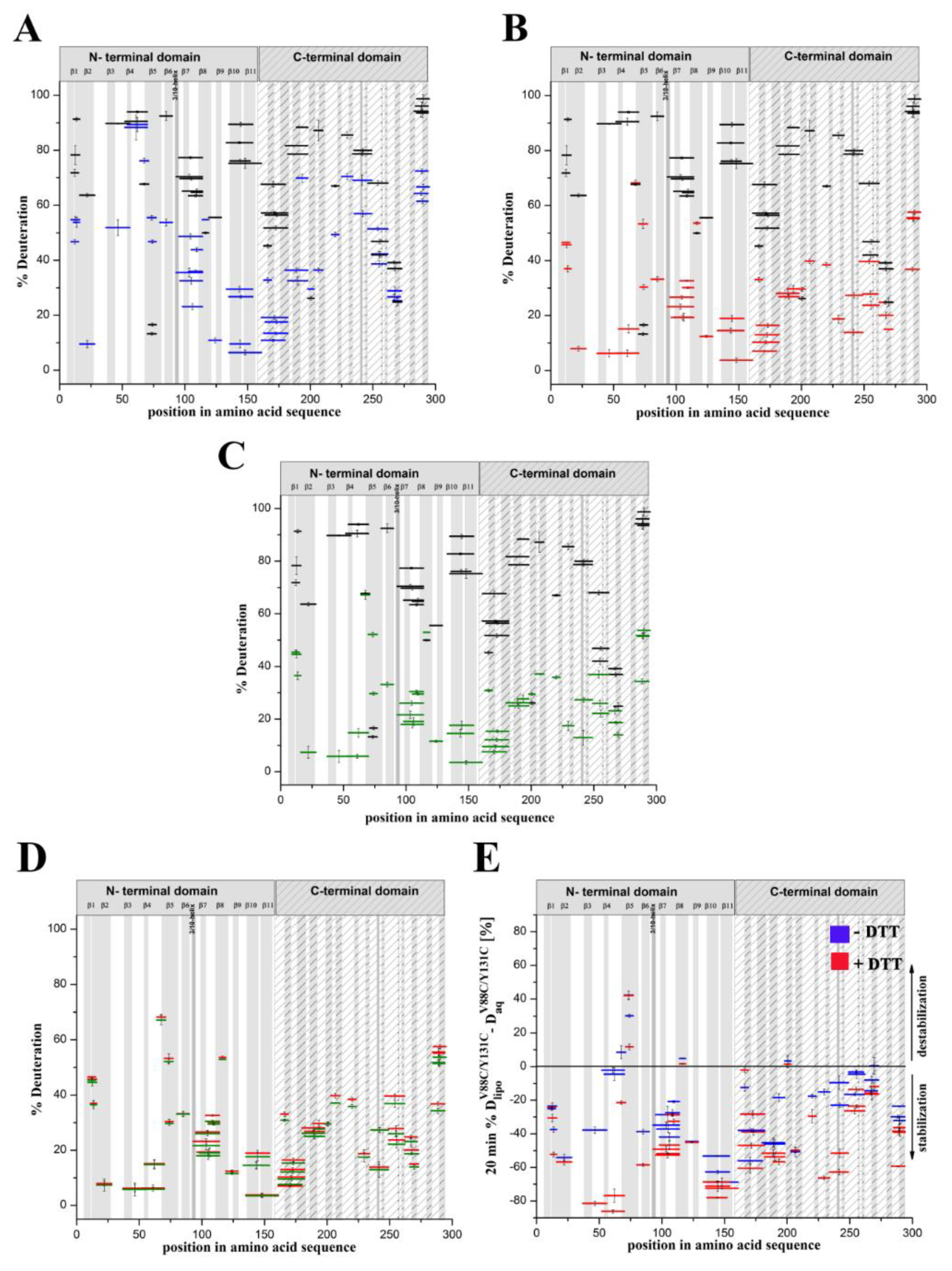
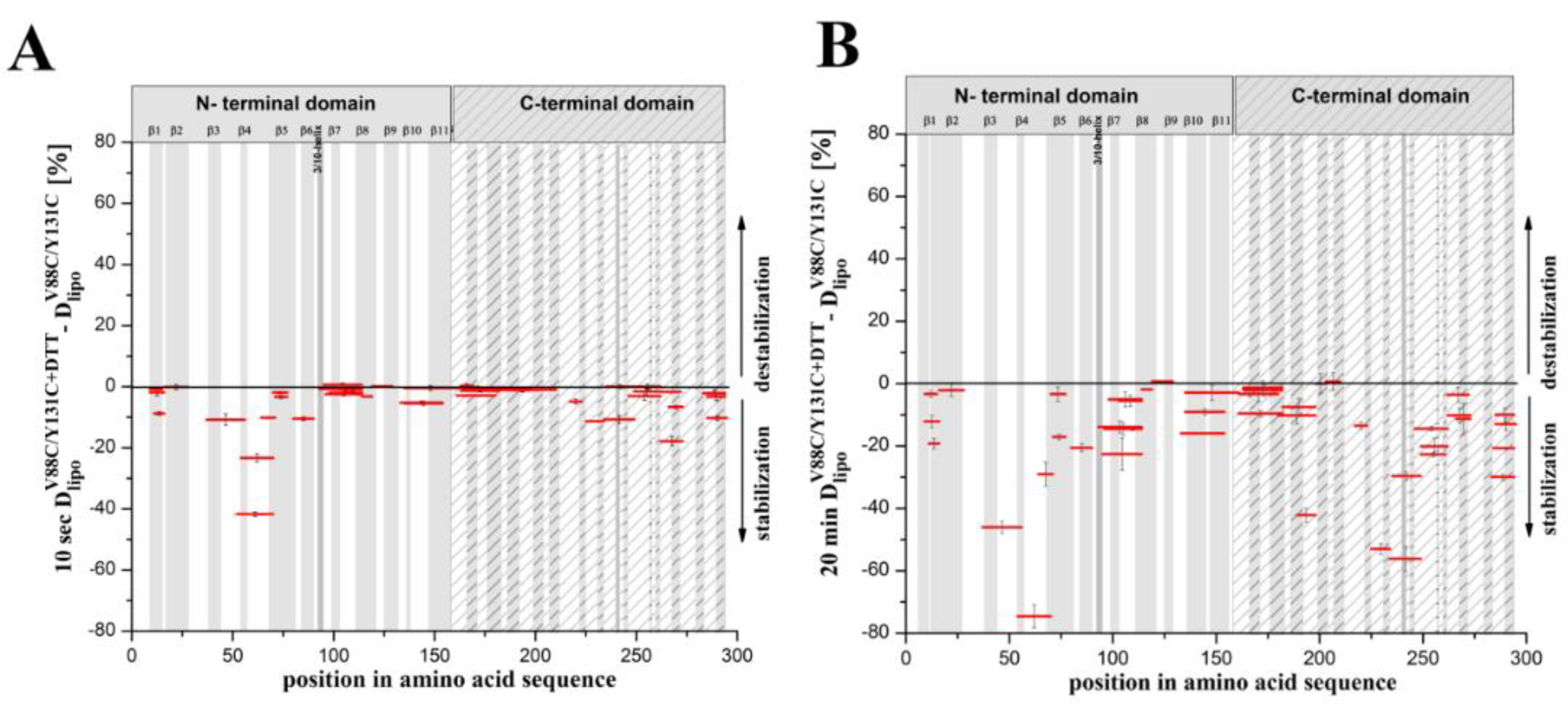
Our present results indicate the structural dynamics of the lysenin regions involved in the two stages of pore formation: transition of the water-soluble lysenin monomer into the ring-shaped oligomer and its conversion into the β-barrel membrane-inserted pore. We demonstrated that the double-cysteine mutation promoted stabilization of the majority of lysenin regions upon binding the lipid membrane. Moreover, the structural stabilization of lyseninV88C/Y131C was more pronounced in the regions that form the N-terminal cap domain in the lysenin pore state, which have not previously been considered to be crucial in the oligomerization and pore formation processes. These findings suggest that increased stability in these regions supports the maintenance of lysenin in the correct orientation relative to the membrane and adjacent monomers in the pre-pore complex and forces further structural rearrangements that drive pore formation.
3. Discussion
The interaction of lysenin with SM-containing membranes forces structural changes within the toxin molecule, leading to transmembrane pore formation. Lytic pore formation is a complex process that involves binding of lysenin’s monomeric form to the SM-containing membrane, oligomerization on the plasma membrane surface, and insertion of lysenin into the lipid membrane. In a previously proposed model, lytic pore formation is initiated by the binding of lysenin to a SM-enriched lipid membrane via the C-terminal domain of the toxin molecule. The lysenin molecule concentration at the membrane surface promotes interactions between neighboring monomers, leading to the formation of arc-shape oligomers that eventually grow into complete nonameric ring-shaped prepores [
22]. It has not previously been shown that prepore formation is accompanied by structural changes in the lysenin molecule, although conformational changes during the monomer-to-prepore transition have been observed for other members of the aerolysin family, including aerolysin and epsilon toxin [
29,
30,
31]. Finally, significant structural rearrangement within lysenin’s N-terminal domain enables insertion of the protein into the lipid membrane, and formation of a β-barrel transmembrane pore. Prior analyses of the crystal and cryo-EM structures of the lysenin pore clearly reveal the structural rearrangement that occurs during the transition of the soluble monomer structure to the membrane-inserted oligomeric state [
8,
22]. Conformational changes accompanying lytic pore formation lead to reorganization of a region of the N-terminal domain including three β strands (β4, β5, and β6), which consequently forces extension of the β3 and β7 strands and a single 3
10 α-helix, promoting their assembly into the twisted β-hairpin. This rearrangement results in downward rotation of the N-terminal domain and a bend in the middle of the structurally intact region of the N-terminal domain (collar), allowing insertion of the twisted β-hairpins of adjacent monomers into the membrane and formation of the β-barrel. Although analysis of the lysenin pore structure has provided new insights into the final membrane-inserted state, structural characterization of the intermediate steps along the pore-formation pathway is required to derive the complete molecular mechanism.
In our present study, we endeavored to gain information about the dynamics of conformational changes occurring during prepore and pore formation. To this end, we characterized the interaction of the V88C/Y131C lysenin mutant with lipid membranes and determined its structural dynamics in both the water-soluble and membrane-bound forms. This double mutation enables the creation of a disulfide bridge between cysteine residues at positions 88 and 131, which prevents insertion loop displacement and consequently abolishes pore formation. On the other hand, this disulfide bond does not affect lysenin’s ability to form nonameric oligomers. Reduction of the disulfide bond leads to disengagement and unfolding of the insertion loop, which completely restores pore-forming activity to the level observed for lysenin
WT. As previously shown for another pore-forming protein, perfringolysin O, the introduction of single point mutations induces local changes in the protein structure and may also cause global changes in the protein structure without influencing the secondary structure [
25,
32]. It has also been shown that the introduction of a point mutation outside of the area involved in membrane binding can alter the interaction between the receptor-binding motif (domain) and the lipid bilayer, indicating both structural and functional coupling of distant and spatially separated domains of PFO [
33]. These prior findings indicated a need to verify the binding properties of lysenin
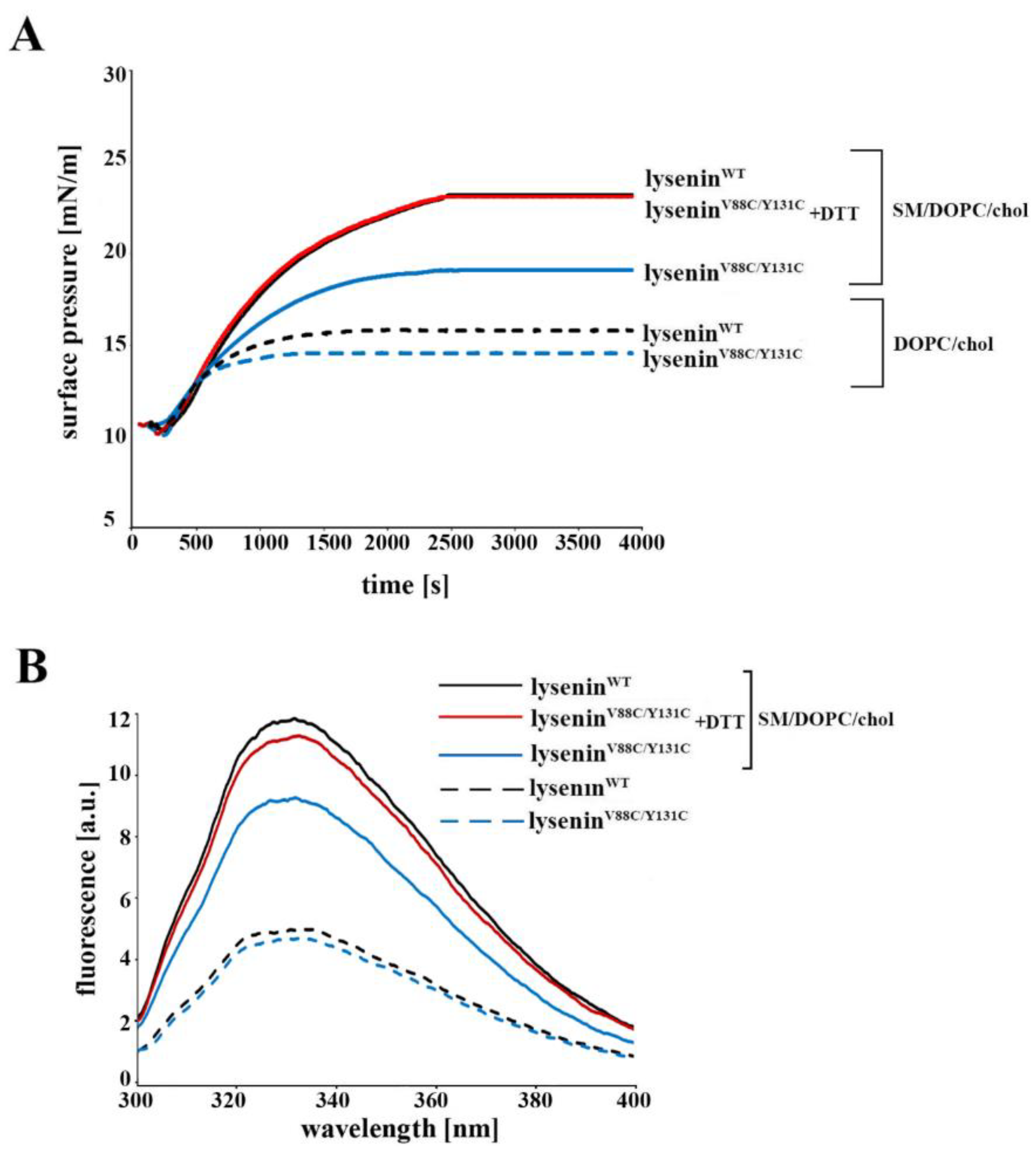
V88C/Y131C. Extensive analysis of lysenin’s interaction with the lipid bilayer revealed that the double-cysteine mutation and the creation of a cysteine bridge between strands β6 and β10 did not affect lysenin’s recognition and initial binding to SM or oligomer formation. However, subtle differences in the stability of this interaction were detectable depending on the redox state of the V88C and Y131C residues. The decreased surface pressure of the lipid monolayer in the presence of the oxidized form of lysenin
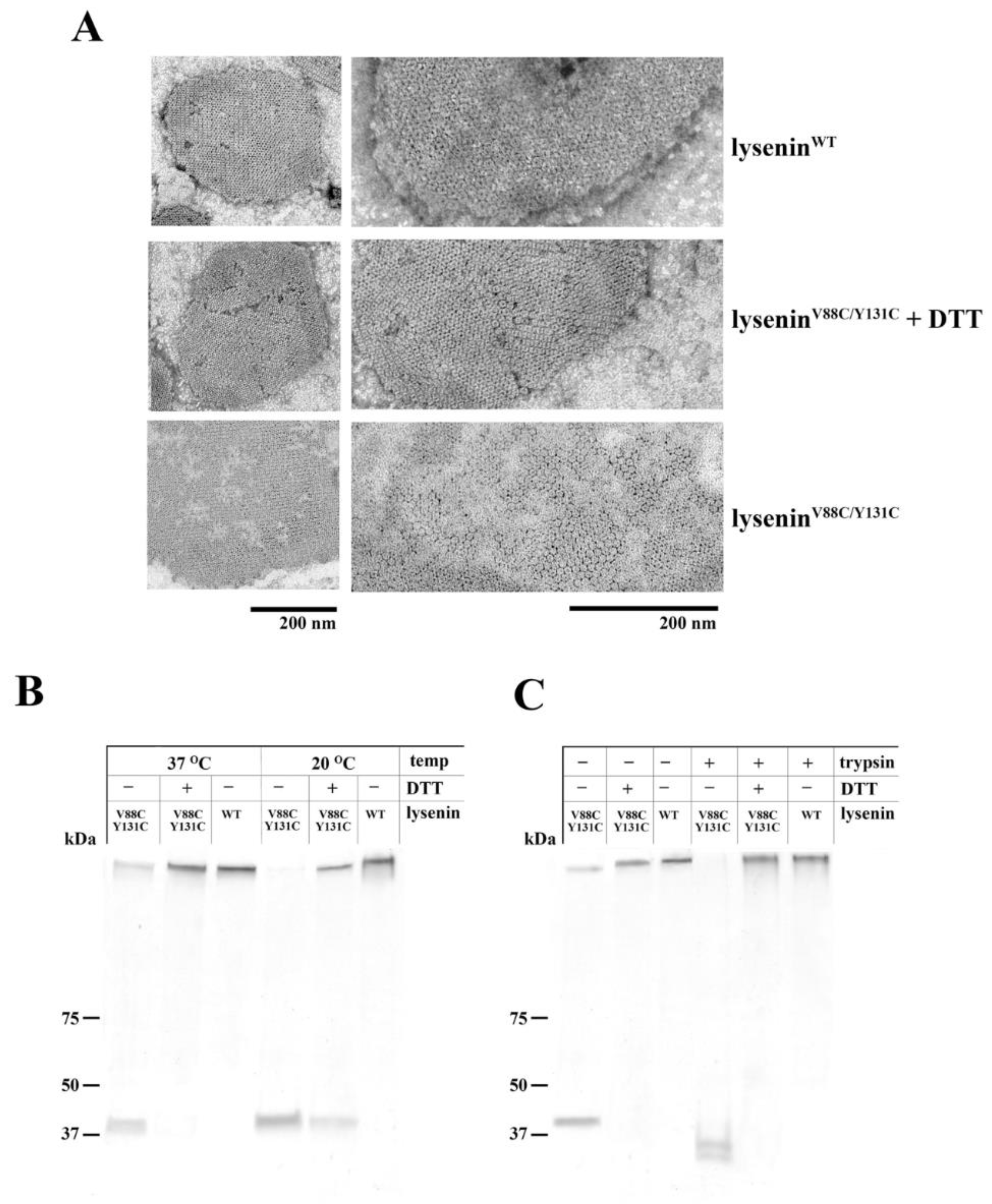
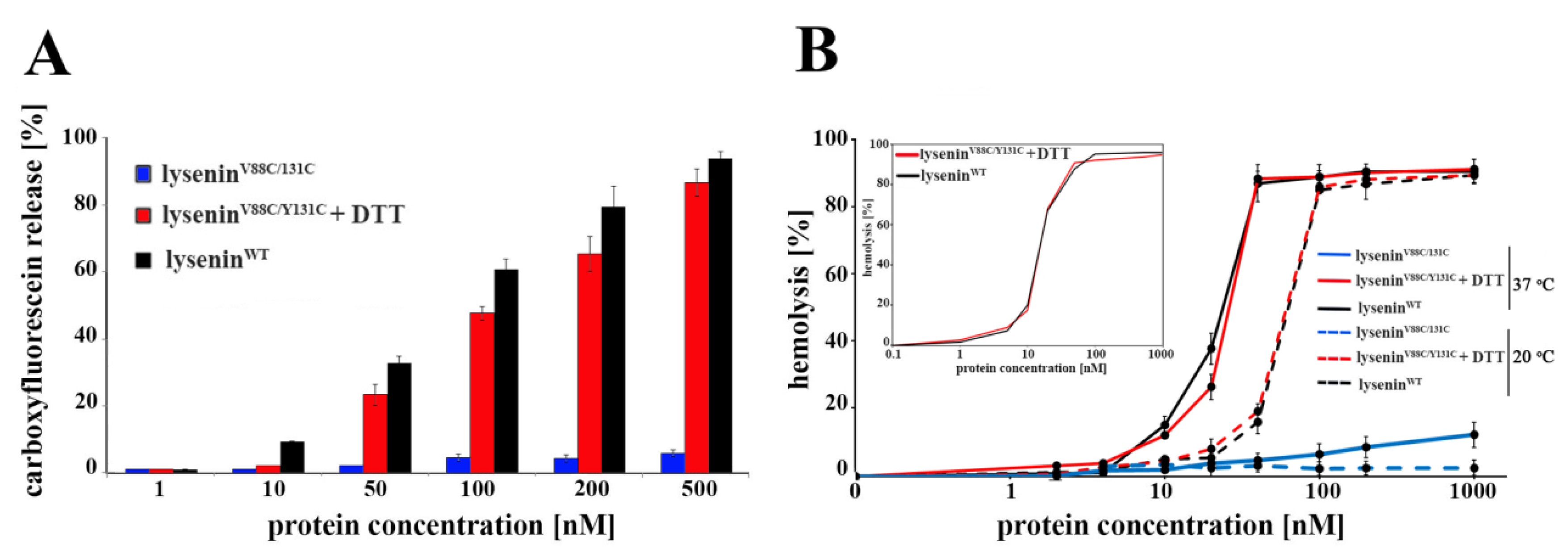
V88C/Y131C, the increased dissociation of this protein upon binding to SM-containing liposomes, less contrasting oligomers in ultrastructural analysis, and higher susceptibility to proteolysis indicated weaker anchoring of lysenin oligomers in the lipid membrane, thus confirming impairment of lysenin’s insertion into the lipid membrane in the prepore state. Based on these properties of lysenin
V88C/Y131C, we decided to use the soluble form of this protein as a model for studying the starting structure during pore formation, and to use the liposome-bound lysenin
V88C/Y131C under different redox conditions as model to study the prepore and pore forms.
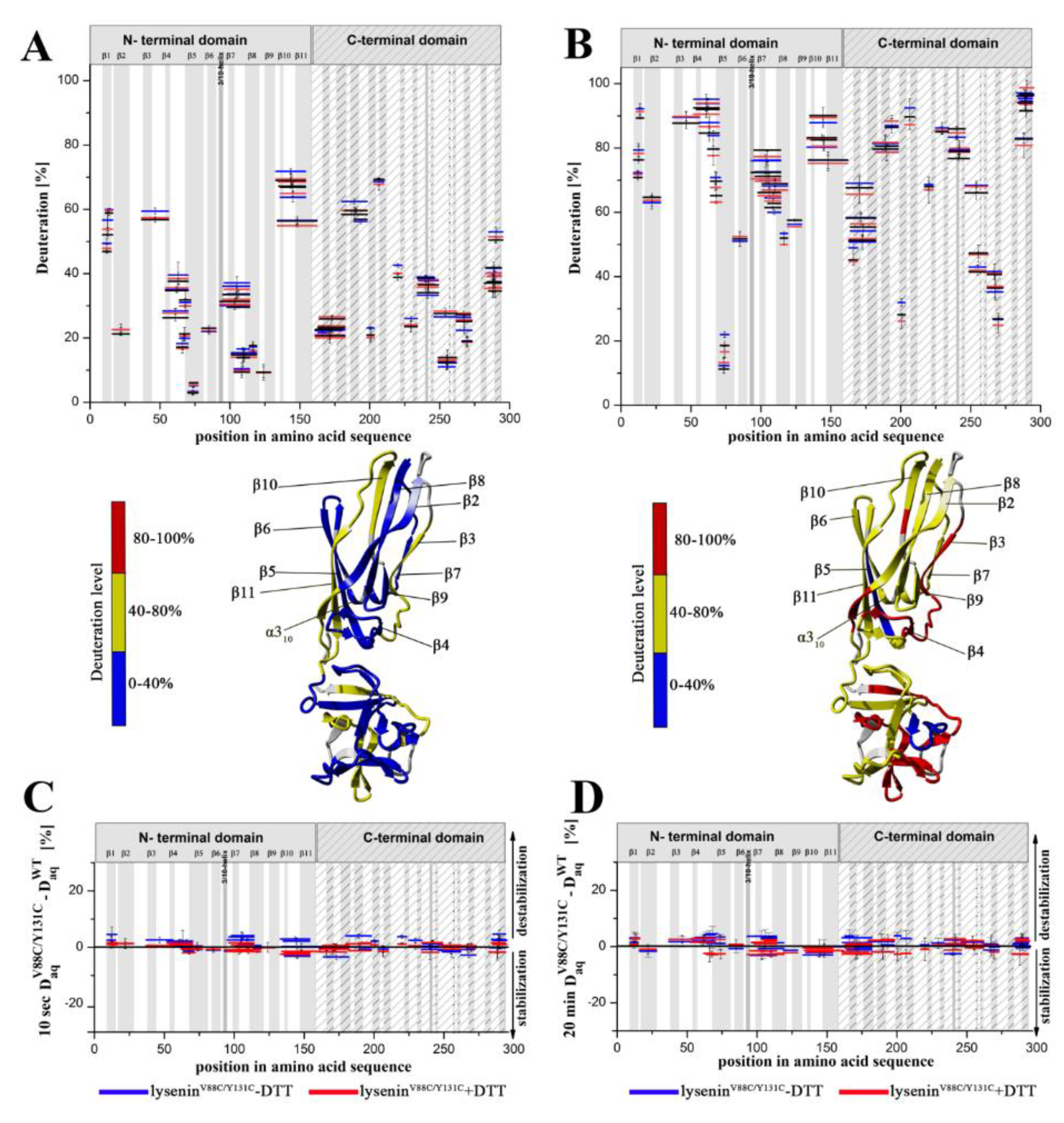
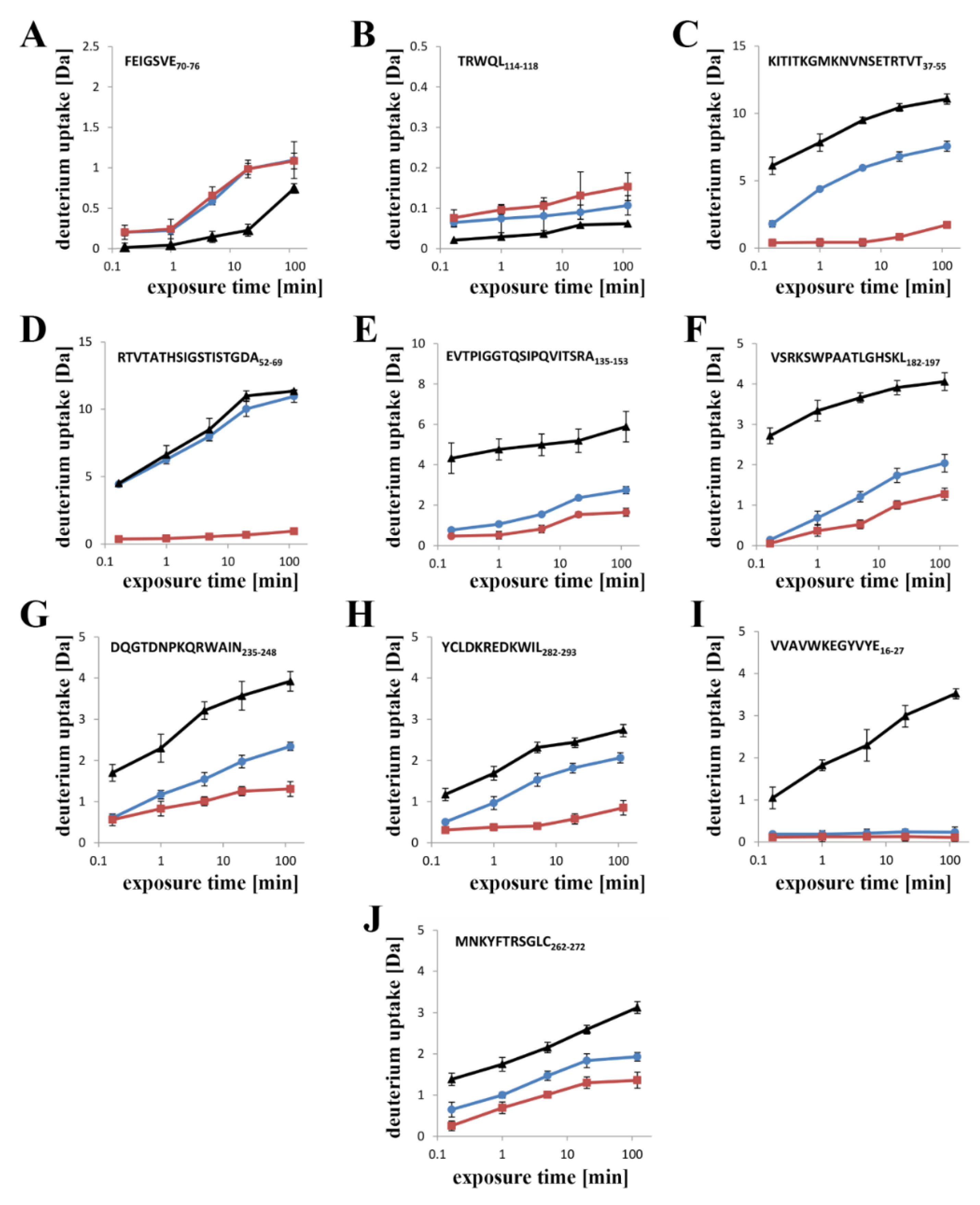
Previous data have revealed that the flexibility of two regions: the random coil region (V157–E167) connecting the C-terminal domain to the structurally intact region of the N-terminal domain (collar) and the tongue region (M44–G67) play crucial roles in the transition from the soluble monomer to the membrane-inserted state [
22]. The high dynamics of the linker between the N-terminal and C-terminal domains allows lysenin to collapse toward the lipid membrane and the flexibility of the tongue enables formation of the β-hairpin that incorporates into the lipid bilayer. Our present HDX results confirmed that both regions were highly dynamic in the soluble form of lysenin. However, both were strongly stabilized upon incorporation into the lipid bilayer and pore formation (
Figure 10). Interestingly, the linker between the N-terminal and C-terminal domains was strongly stabilized already in the prepore state, and the stabilization was similar as in the pore. In contrast, the tongue region showed a similar (region between strands β4 and β5) or slightly reduced (region between strands β3 and β4) flexibility in the prepore state compared to the soluble monomer of lysenin. We also demonstrated that prepore formation was accompanied by stabilization of β strands embedded in the collar region, and exposure of the hydrophobic β5 strand to the solvent, which ultimately created the transmembrane β-hairpin and inserted it into the lipid bilayer. These findings indicated that the gradual decrease in flexibility of the tongue fragment, and the stability of the β-strand network in the collar region, play crucial roles in the prepore-to-pore transition.
In the previously described prepore model, the N-terminal twisted β-sheet remains structurally intact relative to lysenin in its soluble state. Therefore, the β2 strand is faced toward the insertion loop of the neighboring lysenin molecule and interacts and arranges itself parallel to the β10 strand of the adjacent monomer only upon unwinding of the tongue and tilting by 45° from the collar [
22]. The network of interactions between amino acid residues maintains the correct dynamics and conformation of lysenin that are required for the structural rearrangement involved in lytic pore formation.
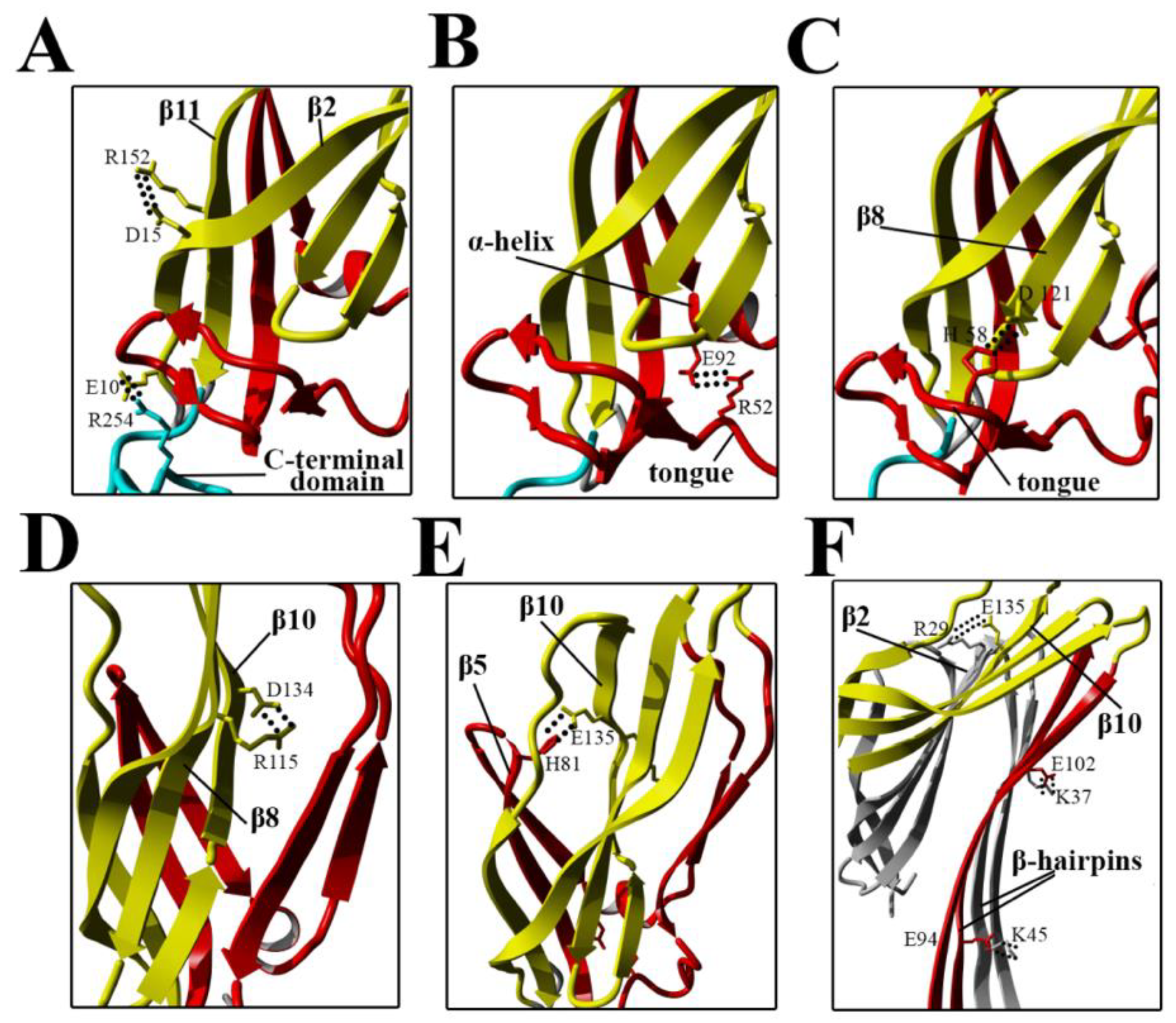
Our present findings support the possibility that subtle intramolecular rearrangements in lysenin occur at prepore formation step, which ultimately facilitate membrane insertion and pore formation. Despite the salt bridges are not dominant factors that governs lysenin’s stability in the soluble monomeric form, they are one type of interaction crucial for structural rearrangement of monomer during pore formation (
Figure 11A–E) [
8].
The formation of intermolecular salt bridges between adjacent monomers during oligomerization is the mechanism controlling the transition of prepore to pore in perfringolysin O and may commonly occur among pore-forming proteins [
34].
Electrostatic interactions are also important for maintaining contact between adjacent monomers and the stability of the β-barrel pore (
Figure 11F) [
22]. Our results suggested that stiffening of the N-terminal twisted β-sheet formed by β2, β8, β9, β10, and β11 may force the breaking of salt bridges between the α-helix and tongue (R52–E92), β8 strand and tongue (H58–D121), and β5 and β10 strands (H81–E135), thus leading to slight stabilization of the insertion loop and consequently exposing the β5 strand to solvent and increasing its conformational dynamics (
Figure 8A and
Figure 11). This orientation is energetically unfavorable; therefore, to decrease the energy state, the insertion loop must be displaced, thus allowing unfolding of the β-hairpin and formation of the β-barrel pore structure.
Based on our present findings and previous work, we also conclude that the interaction of lysenin with an SM-containing membrane in the prepore state moderately affect the structural dynamics of the C-terminal domain, including the regions that interact with the phosphocholine head group of SM, including peptide 180–192 (K185), peptide 225–233 (S227, Q229, Y233), and peptide 282–293 (Y282) [
9]. Surprisingly, the prepore-to-pore transition was accompanied by strong stabilization of the C-terminal part of lysenin, even at regions that are not critical for SM binding. This may be explained by the structural rearrangement within the N-terminal domain of lysenin during pore formation, which forces the proper orientation and stability of the lysenin pore in the lipid membrane, including stabilization of the C-terminal domain of lysenin. This observation indicates that the receptor-binding domain’s role is not limited only to the recognition of targets on cell membranes but is also crucial for stabilizing the oligomeric complex of the lysenin in the prepore form and the formation of functional pore structures. It can be assumed that initial electrostatic interaction between C-terminal domain of lysenin and POC forces changes in the structural dynamics of lysenin, which lead to stabilization of structural elements in the N-terminal domain, including the region responsible for the interaction of lysenin with the SM acyl chain tail (β2 strand containing Y24 and Y26). Stabilization of this region was found already at the prepore state and is maintained also in the pore form. Similar effect is observed for β10 strand that interacts with β2 of adjacent monomer during pore formation upon tilting from the collar and unwinding of the tongue [
22]. Stabilization of these two elements at the prepore stage may indicate that initial interaction of lysenin with the lipid membrane induces dynamic changes first in the regions responsible for assembly of oligomeric complex, and only then in the regions that participate in the insertion of lysenin into the membrane. Thus, we suggest that the initial interaction of β2 and β10 of adjacent monomers takes place even before unwinding of the tongue. However, despite the fact that a lot of detailed information on the structural dynamics of lysenin at various stages of pore formation has been obtained, extensive research is still required to fully understand the mechanism of pore formation.
The above-presented results demonstrate that HDX-MS is an excellent technique that provides valuable data regarding the structural dynamics of proteins at different steps of their interactions with lipid membranes. Our results obtained using this method show that binding to SM-containing membranes caused changes in the structural dynamics of lysenin, as early as in the prepore state.
At this stage, interaction between adjacent monomers induced conformational stabilization of lysenin, which was initiated at the membrane-binding site in the C-terminal domain of lysenin and was transmitted via the N-terminal twisted β-sheet to the region forming the transmembrane domain of the pore (
Figure 12). Structural stabilization of these regions forced a structural rearrangement within the PFM, which was accompanied by stabilization of insertion loop and was required for transmembrane lytic pore formation.
Overall, our present results demonstrated that the HDX-MS technique perfectly complements structural studies based on crystallography and high-resolution microscopy, bringing us closer to a full understanding of the molecular mechanism of pore formation by lysenin.
4. Materials and Methods
4.1. Expression and Purification of Recombinant Proteins
The plasmids expressing lysenin
WT and lysenin
V88C/Y131C with a HIS tag at the N terminus were generated from the pGEX4T-lysenin WT template, constructed as previously described [
17]. The V88C/Y131C substitution in the lysenin sequence was generated by site-directed mutagenesis using two primer pairs: 5′-CATGAAGAATCCCAATGTAGCATGACGGAAAC-3′ and 5′-GTTTCCGTCATGCTACATTGGGATTCTTCATG-3′ to introduce the V88C mutation, and 5′-GATATTGAATACATGTGTTTGATTGATGAAGTC-3′ and 5′-GACTTCATCAATCAAACACATGTATTCAATATC-3′ for the Y131C substitution. The cDNA fragments encoding the two proteins were amplified by PCR, and subcloned into the pET28a(+) vector (Novagen, Madison, USA) using BamHI/HindIII restriction sites. The recombinant vectors pET28a-lyseninWT and pET28-lyseninV88C/Y131C were transformed into the
E. coli BL21(DE3) and SHuffle T7 Express strains, respectively. The produced proteins (
Figure S6) were purified on a Ni Sepharose 6 Fast Flow column (GE Healthcare, Munich, Germany) and dialyzed against 30 mM Tris buffer (pH 7.5) containing 150 mM NaCl. Finally, the collected samples were mixed with 10% (
v/
v) glycerol, frozen in liquid nitrogen, and stored at −80 °C.
4.2. Protein-Lipid Overlay Assay
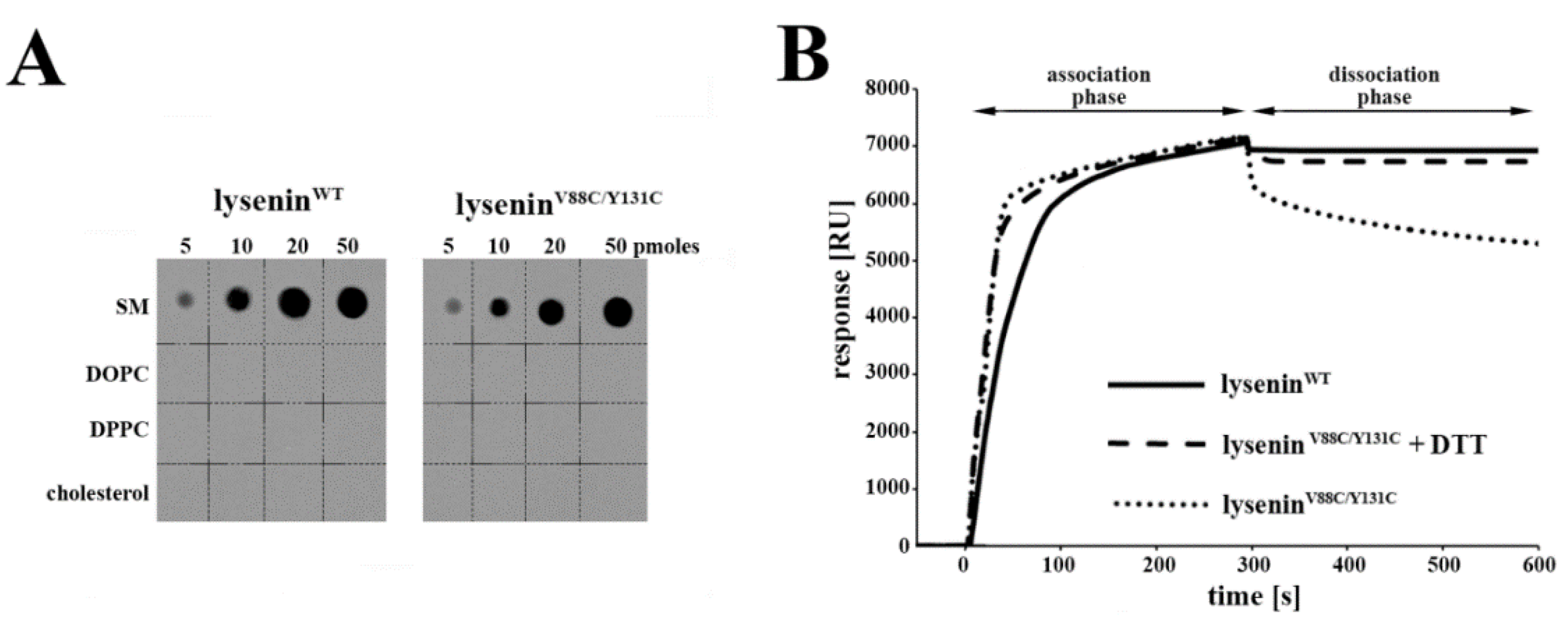
To test the specificity of lipid recognition by lysenin
V88C/Y131C, we spotted nitrocellulose membranes with 1 µL of lipid samples containing various amounts (5–50 pmol) of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC; Avanti), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC; Avanti), cholesterol (Sigma-Aldrich, Munich, Germany), or bovine brain SM (Sigma-Aldrich, Munich, Germany) in a chloroform:methanol:water mixture (1:1:0.3,
v/
v). The membranes were blocked for 1 h at 20 °C with 1% gelatin and 1% polyvinylpyrrolidone in Tris-buffered saline containing 0.03% Tween-20 (TBST) and then incubated for 2 h with 100 nM lysenin
WT or lysenin
V88C/Y131C and processed as previously described [
14].
4.3. Surface Plasmon Resonance
We examined the interaction of lysenin
WT and lysenin
V88C/Y131C with large unilamellar vesicles (LUVs) using a BIAcore 3000 instrument (GE Healthcare, Munich, Germany) equipped with an L1 chip. LUVs composed of SM/DOPC/cholesterol (molar ratio 1:2:1) were prepared as previously described, yielding a final lipid concentration of 1 mM [
17]. The liposomes were deposited onto the L1 chip surface, at a flow rate of 1 μL/min for 10 min, using an amount that yielded 7000 resonance units (RU). Measurements were conducted using 1 μM protein at a flow rate of 5 μL/min for 300 s. Then, the dissociation of the deposited protein was followed for an additional 300 s.
4.4. Surface Pressure Measurement
These experiments were performed using a NIMA Technologyn tensiometer model PS3 (Coventry, UK) at 20 ± 1 °C in darkness under an argon atmosphere, as previously described [
13]. The water sub-phase was buffered with 30 mM Tris-HCl (pH 7.5). To measure the surface pressure of mixed lipid monomolecular layers, a lipid monolayer comprising SM/DOPC/cholesterol (molar ratio 1:2:1) or DOPC/cholesterol (molar ratio 2:1) was deposited at the argon–water interface from a chloroform solution. A 40 μM final concentration of lysenin
WT or lysenin
V88C/Y131C (with or without 10 mM DTT) was injected into the 12 mL of buffer sub-phase.
4.5. Oligomerization Assay
To prepare small unilamellar vesicles (SUVs) composed of SM/DOPC/cholesterol (1:2:1 molar ratio, 1 mM total phospholipid concentration), we resuspended the lipid film in buffer containing 30 mM Tris and 150 mM NaCl (pH 7.5). This liposome suspension was vortexed for 5 min and then subjected to six freeze-thaw cycles, and sonication (30 min, 0.3 cycle, amplitude 33%). After centrifugation at 10,000× g (30 min, 4 °C), the pelleted SUVs were resuspended in buffer containing 30 mM Tris and 150 mM NaCl (pH 7.5), and incubated with 1 μM lyseninWT or lyseninV88C/Y131C (with or without 10 mM DTT) for 30 min at 25 °C and 37 °C. The samples were then pelleted by centrifugation at 10,000× g (30 min), suspended in 20 μL of SDS loading buffer (40% glycerol (v/v), 25% SDS (w/v), and 0.1% bromophenol blue (w/v)), and analyzed by SDS-PAGE.
The oligomeric complexes of lysenin were then analyzed for their susceptibility to proteolysis. The samples of proteins bound to liposomes were suspended in buffer containing 30 mM Tris and 150 mM NaCl (pH 7.5) and supplemented with trypsin (trypsin:protein ratio 1:20, w/w; Promega). After incubation for 2 hours at 37 °C, the samples were diluted with SDS loading buffer and analyzed by SDS-PAGE.
4.6. Measurement of Intrinsic Protein Fluorescence
To determine the intrinsic protein fluorescence, we measured the tryptophan emission spectra of lyseninWT and lyseninV88C/Y131C, both in solution and bound to liposomes. SUV preparation and lysenin binding to lipid vesicles were performed as described above. Tryptophan emission was excited at 280 nm, and fluorescence emission was recorded from 300 to 400 nm at 1 nm/s. All measurements were made using a JASCO FP 6500 spectrofluorimeter at 25 °C. The spectral band widths were 3 nm for both excitation and emission.
4.7. Electron Microscopy
Ultrastructural studies of lysenin oligomeric complexes were performed as described by Kwiatkowska et al. [
14], with some modifications. To prepare multilamellar lipid vesicles (MLVs), the lipid film (composed of SM/DOPC/cholesterol in a 1:2:1 molar ratio) was suspended in buffer containing 30 mM Tris and 150 mM NaCl (pH 7.5), vortexed, and sonicated for 2 min at 4 °C (30 min, 0.3 cycle, amplitude 33%). After centrifugation at 15,000× g (15 min, 4 °C), the liposomes were resuspended in buffer containing 30 mM Tris and 150 mM NaCl (pH 7.5) and incubated for 30 min at 20 °C with 7 µM lysenin
WT or lysenin
V88C/Y131C, with or without 10 mM DTT. After incubation, the liposomes were fixed with 1% glutaraldehyde in PBS for 20 min at 20 °C and transferred onto poly-L-lysine-coated and formvar/carbon-treated grids for 20 min. Finally, the samples were washed twice with PBS and once with water, counterstained with 2% uranyl acetate, and examined under a JEM-1200EX (JEOL) microscope.
4.8. Pore-Forming Activity
SUVs composed of SM/DOPC/cholesterol (1:2:1 molar ratio) were prepared as described above, in the presence of 50 µM 6-carboxyfluorescein (Sigma, Munich, Germany). These SUVs were pelleted, resuspended in buffer containing 30 mM Tris and 150 mM NaCl (pH 7.5), and incubated with 1 μM lyseninWT or lyseninV88C/Y131C with or without 10 mM DTT. The suspension was incubated for 30 min at 20 °C, and then centrifuged. The level of released carboxyfluorescein in the supernatant was measured using a Jasco FP 6500 spectrofluorometer at an excitation/emission of 492/517 nm. The results were expressed as the percentage of maximum carboxyfluorescein release from SUVs, as triggered by addition of 0.1% Triton X-100.
4.9. Hemolytic Activity
The hemolytic activity of lysenin
WT and lysenin
V88C/Y131C was measured as previously described [
14]. Briefly, serial protein dilutions (up to 1 µM) were incubated for 45 min at 20 °C and 37 °C, with 7 × 10
7 sheep red blood cells (RBCs), with or without 10 mM DTT. Then the samples were centrifuged at 200×
g (5 min, 4 °C), and the level of hemoglobin released from RBCs was estimated by measurement of the supernatant’s absorbance at 405 nm using a NanoDrop 2000c Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The results were expressed as the percentage of maximal hemolysis (100%), obtained by osmotic lysis of RBCs with water.
4.10. Hydrogen–Deuterium Exchange Measurements
Samples for hydrogen–deuterium exchange were prepared following a previously described procedure, with some modifications [
24,
25]. Briefly, lipid-bound samples were prepared by incubation of 4 µM lysenin
WT and lysenin
V88C/Y131C (with or without 10 mM DTT) with 2 mM SUVs composed of SM/DOPC/cholesterol (molar ratio 1:2:1) at room temperature for 30 min. Then, the samples were centrifuged at 10,000×
g (10 min, 4 °C), and the pellets were resuspended in 30 mM Tris with 150 mM NaCl (pH 7.5) supplemented with 2% n-dodecyl-β-D-maltoside (DDM) and incubated at room temperature for 1 h with shaking. Next, the samples were again centrifuged at 10,000×
g (10 min, 4 °C), and the supernatant containing extracted oligomers was subjected to HDX-MS.
The HDX-MS measurements were compared between lysenin
V88C/Y131C and lysenin
WT, without liposome binding (aqueous solution) and upon binding to liposomes (extracted oligomers). The hydrogen–deuterium exchange reaction was initiated by adding 5 µL protein sample to 45 µL D
2O Reaction buffer (30 mM Tris and 150 mM NaCl, pH 7.5). The reaction was carried out for the required time period (10 s, 1 min, 5 min, 20 min, 120 min, or 24 h) and was quenched by adding the reaction mixture to 10 μL pre-chilled D
2O stopping buffer (2 M glycine and 150 mM NaCl, pH 2.4). Finally, the samples were injected onto an immobilized pepsin column (Poroszyme; ABI), and the obtained peptides were further separated using the nanoACQUITY ultra-performance liquid chromatography (UPLC) system, followed by mass measurements on the SYNAPT G2 HDMS mass spectrometer (Waters, Milford, MA, USA). Peptide identification was based on a list of peptic peptides obtained for a non-deuterated sample using ProteinLynx Global Server software (Waters, Milford, MA, USA), as previously described [
25]. HDX data analysis was performed using the DynamX 2.0 program (Waters, Milford, MA, USA). All measurements were repeated in triplicate. All controls were performed, including a back-exchange control and a carry-over effect control.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

 ), lyseninV88C/Y131C (●●●), and lyseninV88C/Y131C with 10 mM 1,4-Dithiothreitol (DTT) (▬ ▬) to SM-containing liposomes (composed of SM/1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)/cholesterol) that were immobilized on the surface of an L1 sensor chip. Sensorgrams were performed in triplicate, and one representative experiment is shown.
), lyseninV88C/Y131C (●●●), and lyseninV88C/Y131C with 10 mM 1,4-Dithiothreitol (DTT) (▬ ▬) to SM-containing liposomes (composed of SM/1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)/cholesterol) that were immobilized on the surface of an L1 sensor chip. Sensorgrams were performed in triplicate, and one representative experiment is shown.










