Chronic Low Dose Oral Exposure to Microcystin-LR Exacerbates Hepatic Injury in a Murine Model of Non-Alcoholic Fatty Liver Disease


 , , , ,
, , , , 
 ,
, 
 and add
Show full author list
and add
Show full author list
Abstract
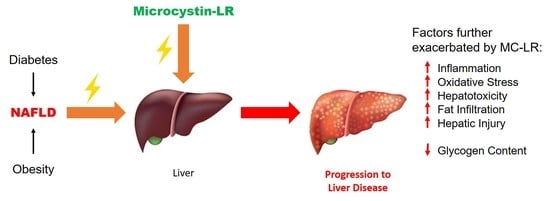
:
1. Introduction
2. Results
2.1. Survival, Appearance and Weight
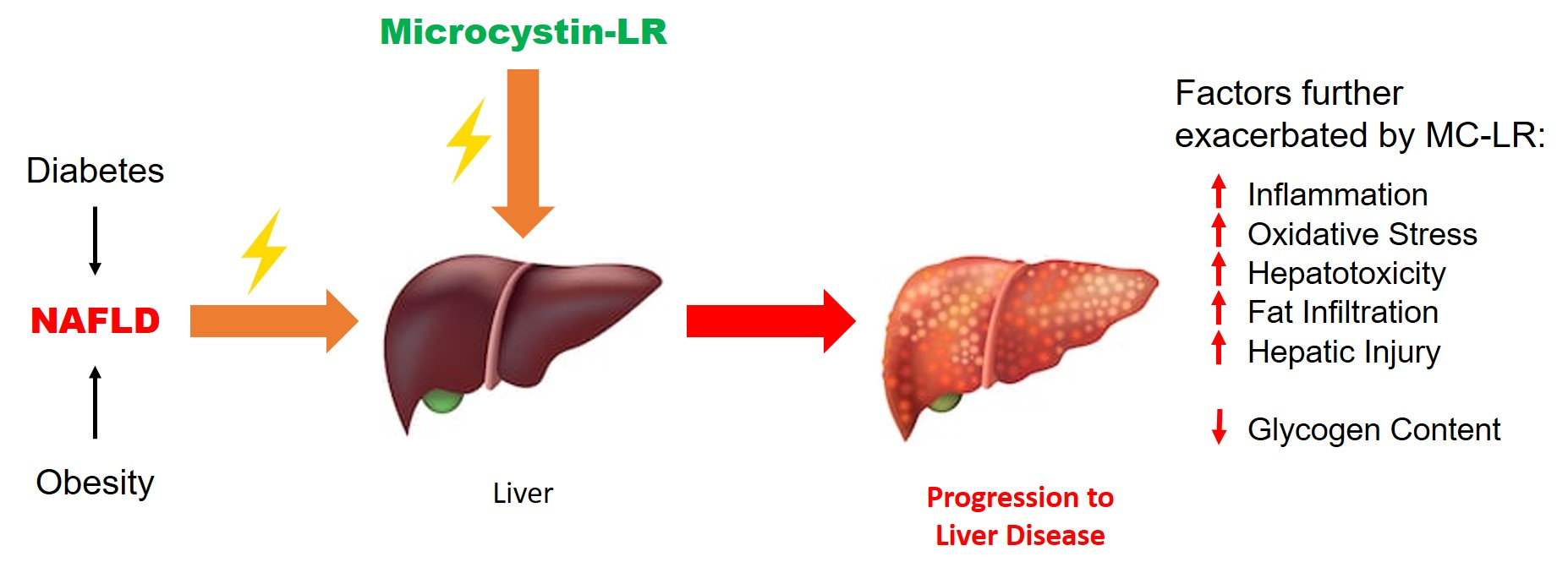
2.2. Detection of MC-LR in Plasma and Urine
2.3. Blood Biochemistry
2.4. Liver Histology
2.5. Genetic Analysis Hepatotoxicity and Oxidative Stress
2.6. Phosphoproteomic Analysis
3. Discussion
Limitations
4. Materials and Methods
4.1. Mice
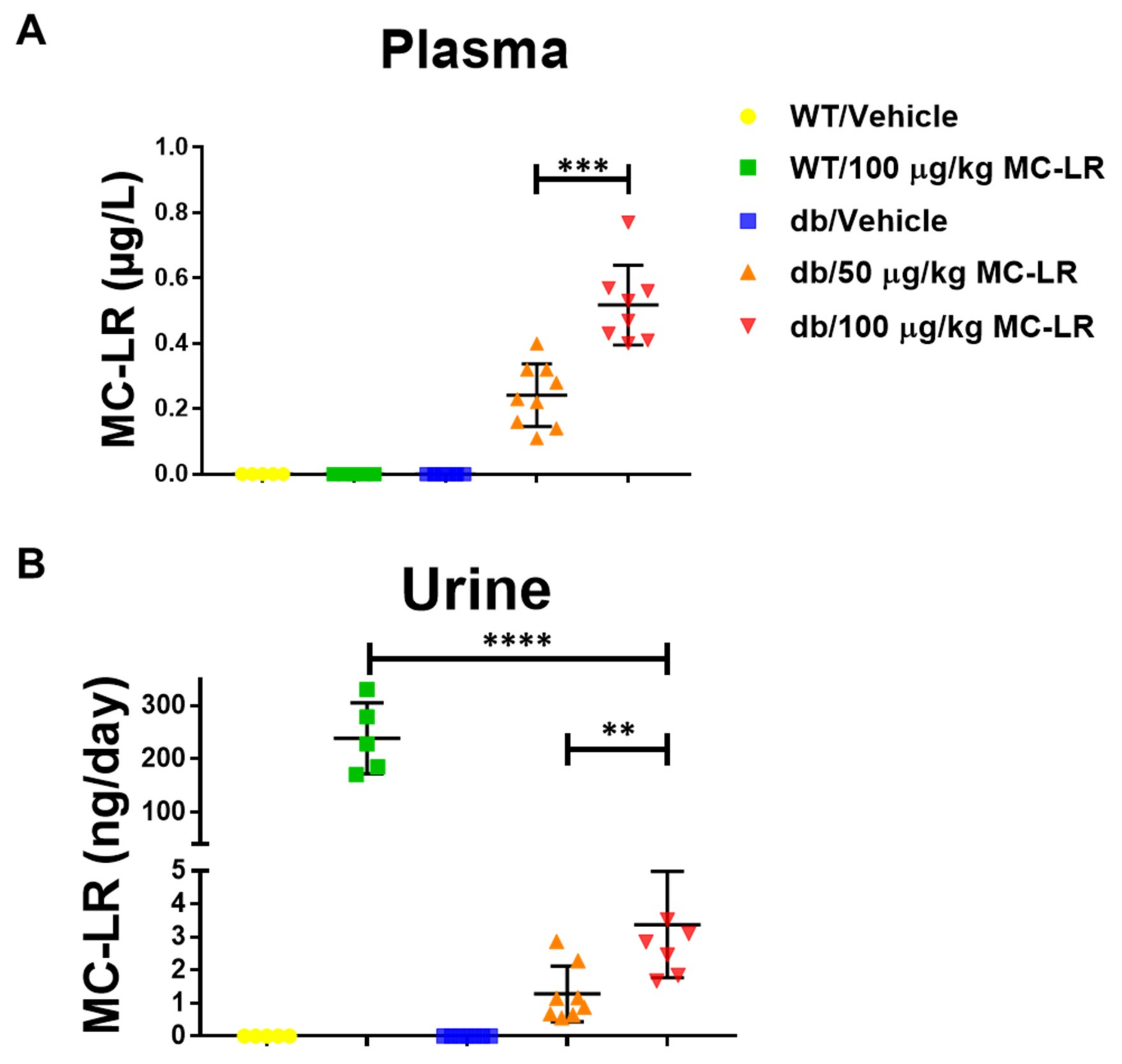
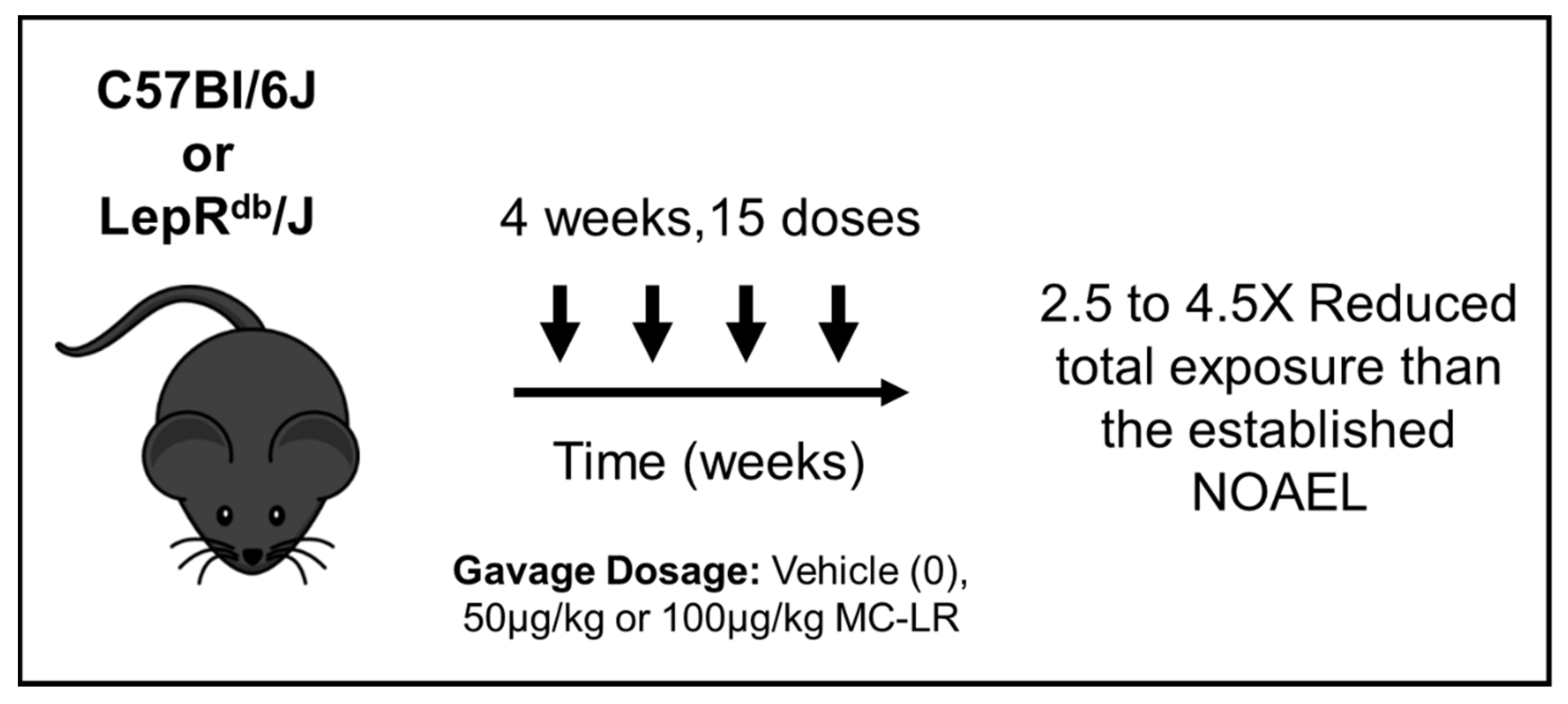
4.2. Exposure and Experimental Design
4.3. Histological Studies
4.4. Blood Chemistry
4.5. MC-LR Determination in Plasma and Urine
4.6. Genetic Analysis of Hepatotoxicity and Oxidative Stress
4.7. Proteomic Analyses of TiO2 Enriched Phosphopeptides
4.8. Proteomic Data Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fawell, J.K.; Mitchell, R.E.; Everett, D.J.; Hill, R.E. The toxicity of cyanobacterial toxins in the mouse: I Microcystin-LR. Hum. Exp. Toxicol. 1999, 18, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Sedan, D.; Laguens, M.; Copparoni, G.; Aranda, J.O.; Giannuzzi, L.; Marra, C.A.; Andrinolo, D. Hepatic and intestine alterations in mice after prolonged exposure to low oral doses of Microcystin-LR. Toxicon 2015, 104, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Vasas, G.; Farkas, O.; Borics, G.; Felföldi, T.; Sramkó, G.; Batta, G.; Bácsi, I.; Gonda, S. Appearance of Planktothrix rubescens Bloom with [D-Asp3, Mdha7]MC–RR in Gravel Pit Pond of a Shallow Lake-Dominated Area. Toxins 2013, 5, 2434–2455. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, Z.A. First report of toxic Cylindrospermopsis raciborskii and Raphidiopsis mediterranea (Cyanoprokaryota) in Egyptian fresh waters. FEMS Microbiol. Ecol. 2007, 59, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Mantzouki, E.; Lürling, M.; Fastner, J.; Domis, L.D.S.; Wilk-Wo’zniak, E.; Koreiviene, J.; Seelen, L.; Teurlincx, S.; Verstijnen, Y.; Krztoń, W.; et al. Temperature Effects Explain Continental Scale Distribution of Cyanobacterial Toxins. Toxins 2018, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Li, G.; Chen, J.; Lin, J.; Zeng, C.; Chen, J.; Deng, J.; Xie, P. Prolonged exposure to low-dose microcystin induces nonalcoholic steatohepatitis in mice: A systems toxicology study. Arch. Toxicol. 2017, 91, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Ueno, Y.; Nagat, S.; Suttajit, M.; Mebs, D.; Vasconcelos, V. Immunochemical Survey of Microcystins in Environmental Water in Various Countries. In Mycotoxins and Phycotoxins: Developments in Chemistry, Toxicology and Food Safety; Alaken Inc.: Fort Collins, CO, USA, 1998; pp. 449–453. [Google Scholar]
- Rinehart, K.L.; Harada, K.; Namikoshi, M.; Chen, C.; Harvis, C.A.; Munro, M.H.G.; Blunt, J.W.; Mulligan, P.E.; Beasley, V.R. Nodularin, microcystin, and the configuration of Adda. J. Am. Chem. Soc. 1988, 110, 8557–8558. [Google Scholar] [CrossRef]
- Chen, L.; Xie, P. Mechanisms of microcystin-induced cytotoxicity and apoptosis. Mini Rev. Med. Chem. 2016, 16, 1018–1031. [Google Scholar] [CrossRef]
- Yoshizawa, S.; Matsushima, R.; Watanabe, M.F.; Harada, K.I.; Ichihara, A.; Carmichael, W.W.; Fujiki, H. Inhibition of protein phosphatases by microcystis and nodularin associated with hepatotoxicity. J. Cancer Res. Clin. Oncol. 1990, 116, 609–614. [Google Scholar] [CrossRef]
- Ding, W.X.; Shen, H.M.; Zhu, H.G.; Ong, C.N. Studies on Oxidative Damage Induced by Cyanobacteria Extract in Primary Cultured Rat Hepatocytes. Environ. Res. 1998, 78, 12–18. [Google Scholar] [CrossRef]
- Solter, P.F.; Wollenberg, G.K.; Huang, X.; Chu, F.S.; Runnegar, M.T. Prolonged Sublethal Exposure to the Protein Phosphatase Inhibitor Microcystin-LR Results in Multiple Dose-Dependent Hepatotoxic Effects. Toxicol. Sci. 1998, 44, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Codd, G.A. Mechanisms of action and health effects associated with cyanobacterial toxins. Toxicol. Lett. 1996, 88, 21. [Google Scholar] [CrossRef]
- Milutinović, A.; Živin, M.; Zorc-Pleskovič, R.; Sedmak, B.; Šuput, D. Nephrotoxic effects of chronic administration of microcystins-LR and-YR. Toxicon 2003, 42, 281–288. [Google Scholar] [CrossRef]
- Pahan, K.; Gu, F.; Gruenberg, J.; Sheikh, F.G.; Namboodiri, A.M.S.; Singh, I. Inhibitors of Protein Phosphatase 1 and 2A Differentially Regulate the Expression of Inducible Nitric-oxide Synthase in Rat Astrocytes and Macrophages. J. Biol. Chem. 1998, 273, 12219–12226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, R.E.; Solter, P.F. Characterization of Sublethal Microcystin-LR Exposure in Mice. Veter. Pathol. 2002, 39, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Valério, E.; Vasconcelos, V.; Campos, A. New insights on the mode of action of microcystins in animal cells –A review. Mini. Rev. Med. Chem. 2016, 16, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Dzierlenga, A.; Arman, T.; Toth, E.; Li, H.; Lynch, K.D.; Tian, D.-D.; Goedken, M.; Paine, M.F.; Cherrington, N. Nonalcoholic fatty liver disease alters microcystin-LR toxicokinetics and acute toxicity. Toxicon 2019, 162, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lone, Y.; Bhide, M.; Koiri, R.K. Microcystin-LR Induced Immunotoxicity in Mammals. J. Toxicol. 2016, 2016, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Campos, A.; Vasconcelos, V. Molecular Mechanisms of Microcystin Toxicity in Animal Cells. Int. J. Mol. Sci. 2010, 11, 268–287. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.G.; Codd, G.A. Cyanobacterial toxins and human health. Rev. Med. Microbiol. 1994, 5, 256–264. [Google Scholar] [CrossRef]
- Takahashi, S.; Kaya, K. Quail spleen is enlarged by microcystin RR as a blue-green algal hepatotoxin. Nat. Toxins 1993, 1, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhao, X.; Liu, Y.; Shi, Q.; Hua, Z.; Shen, P. Analysis of immunomodulating nitric oxide, iNOS and cytokines mRNA in mouse macrophages induced by microcystin-LR. Toxicology 2004, 197, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Soares, R.M.; Yuan, M.; Servaites, J.C.; Delgado, A.; Magalhães, V.F.; Hilborn, E.D.; Carmichael, W.W.; Azevedo, S.M.F.O. Sublethal exposure from microcystins to renal insufficiency patients in Rio de Janeiro, Brazil. Environ. Toxicol. 2006, 21, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xie, P.; Tang, R.; Zhang, X.; Li, L.; Li, D. In vivo studies on the toxic effects of microcystins on mitochondrial electron transport chain and ion regulation in liver and heart of rabbit. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2008, 148, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Lone, Y.; Koiri, R.K.; Bhide, M. An overview of the toxic effect of potential human carcinogen Microcystin-LR on testis. Toxicol. Rep. 2015, 2, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLellan, N.L.; Manderville, R.A. Toxic mechanisms of microcystins in mammals. Toxicol. Res. 2017, 6, 391–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Kimono, D.; Albadrani, M.; Seth, R.K.; Busbee, P.; Alghetaa, H.; Porter, D.E.; Scott, G.I.; Brooks, B.; Nagarkatti, M.; et al. Environmental microcystin targets the microbiome and increases the risk of intestinal inflammatory pathology via NOX2 in underlying murine model of Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 8742. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xie, P.; Li, L.; Xu, J. First Identification of the Hepatotoxic Microcystins in the Serum of a Chronically Exposed Human Population Together with Indication of Hepatocellular Damage. Toxicol. Sci. 2009, 108, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, D.R.; Ernst, B.; Day, B.W. Human Consumer Death and Algal Supplement Consumption: A Post Mortem Assessment of Potential Microcystin-Intoxication Via Microcystin Immunoistochemical (MCICH) Analyses. In Proceedings of the 7th International Conference on Toxic Cyanobacteria, Rio de Janeiro State, Brazil, 5–10 August 2007. [Google Scholar]
- D’Anglada, L.V.; Joyce, M.D.; Jamie, S.; Belinda, H. Health Effects Support Document for the Cyanobacterial Toxin Cylindrospermopsin; US Environmental Protection Agency, Office of Water, Health and Ecological Central Division: Washington, DC, USA, 2015.
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Le, M.H.; Devaki, P.; Ha, N.B.; Jun, D.W.; Te, H.S.; Cheung, R.C.; Nguyen, M.H. Prevalence of non-alcoholic fatty liver disease and risk factors for advanced fibrosis and mortality in the United States. PLoS ONE 2017, 12, e0173499. [Google Scholar] [CrossRef]
- Brown, G.T.; Kleiner, D.E. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism 2016, 65, 1080. [Google Scholar] [CrossRef] [PubMed]
- Fazel, Y.; Koenig, A.B.; Sayiner, M.; Goodman, Z.D.; Younossi, Z.M. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism 2016, 65, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Tiribelli, C.; Marchesini, G.; Bellentani, S. Prevalence of and risk factors for nonalcoholic fatty liver disease: The Dionysos nutrition and liver study. Hepatology 2005, 42, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Soejima, Y.; Fukusato, T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2012, 18, 2300–2308. [Google Scholar] [CrossRef] [PubMed]
- Albadrani, M.; Alhasson, F.; Dattaroy, D.; Chandrashekaran, V.; Seth, R.; Nagarkatti, M.; Nagarkatti, P.; Chatterjee, S. Microcystin exposure Exacerbates Non-alcoholic Fatty Liver Disease (NAFLD) via NOX2 Dependent Activation of miR21-induced Inflammatory Pathways. Free. Radic. Biol. Med. 2017, 112, 61. [Google Scholar] [CrossRef]
- Park, S.; Rich, J.; Hanses, F.; Lee, J.C. Defects in innate immunity predispose C57BL/6J-Leprdb/Leprdb mice to infection by Staphylococcus aureus. Infect. Immun. 2009, 77, 1008–1014. [Google Scholar] [CrossRef]
- Väremo, L.; Nielsen, J.; Nookaew, I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013, 41, 4378–4391. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2017, 46, D649–D655. [Google Scholar] [CrossRef]
- Lau, J.K.; Zhang, X.; Yu, J. Animal models of non-alcoholic fatty liver disease: Current perspectives and recent advances. J. Pathol. 2017, 241, 36–44. [Google Scholar] [CrossRef]
- Palagama, D.S.; Baliu-Rodriguez, D.; Lad, A.; Levison, B.S.; Kennedy, D.J.; Haller, S.T.; Westrick, J.; Hensley, K.; Isailovic, D. Development and applications of solid-phase extraction and liquid chromatography-mass spectrometry methods for quantification of microcystins in urine, plasma, and serum. J. Chromatogr. A 2018, 1573, 66–77. [Google Scholar] [CrossRef]
- Giannini, E.G.; Testa, R.; Savarino, V. Liver enzyme alteration: A guide for clinicians. Can. Med Assoc. J. 2005, 172, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Abbas, A.K.; Aster, J.C. Robbins and Cotran Pathologic Basis of Disease, 9th ed.; Elsevier Saunders: Philadelphia, PA, USA.
- Toivola, D.M.; Eriksson, J.E.; Brautigan, D.L. Identification of protein phosphatase 2A as the primary target for microcystin-LR in rat liver homogenates. FEBS Lett. 1994, 344, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Andrinolo, D.; Sedan, D.; Telese, L.; Aura, C.; Masera, S.; Giannuzzi, L.; Marra, C.A.; De Alaniz, M.J. Hepatic recovery after damage produced by sub-chronic intoxication with the cyanotoxin microcystin LR. Toxicon 2008, 51, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, J.; Xia, Z. Microcystin-LR exhibits immunomodulatory role in mouse primary hepatocytes through activation of the NF-kappaB and MAPK signaling pathways. Toxicol. Sci. 2013, 136, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Song, L.; Liu, J. Responses of antioxidant systems in the hepatocytes of common carp (Cyprinus carpio L.) to the toxicity of microcystin-LR. Toxicon 2003, 42, 85–89. [Google Scholar] [CrossRef]
- Svirčev, Z.; Baltić, V.; Gantar, M.; Juković, M.; Stojanović, D.; Baltic, M. Molecular Aspects of Microcystin-induced Hepatotoxicity and Hepatocarcinogenesis. J. Environ. Sci. Health Part C 2010, 28, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Puerto, M.; Pichardo, S.; Jos, A.; Prieto, A.I.; Sevilla, E.; Frías, J.E.; Cameán, A.M. Differential oxidative stress responses to pure Microcystin-LR and Microcystin-containing and non-containing cyanobacterial crude extracts on Caco-2 cells. Toxicon 2010, 55, 514–522. [Google Scholar] [CrossRef]
- Li, Y.; Han, X. Microcystin–LR causes cytotoxicity effects in rat testicular Sertoli cells. Environ. Toxicol. Pharmacol. 2012, 33, 318–326. [Google Scholar] [CrossRef]
- Ma, J.; Li, Y.; Duan, H.; Sivakumar, R.; Li, X. Chronic exposure of nanomolar MC-LR caused oxidative stress and inflammatory responses in HepG2 cells. Chemosphere 2018, 192, 305–317. [Google Scholar] [CrossRef]
- Sedan, D.; Giannuzzi, L.; Rosso, L.; Marra, C.A.; Andrinolo, D. Biomarkers of prolonged exposure to microcystin-LR in mice. Toxicon 2013, 68, 9–17. [Google Scholar] [CrossRef]
- Li, H.; Cai, Y.; Xie, P.; Li, G.; Hao, L.; Xiong, Q. Identification and Expression Profiles of IL-8 in Bighead Carp (Aristichthys nobilis) in Response to Microcystin-LR. Arch. Environ. Contam. Toxicol. 2013, 65, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, J.; Zhang, Q.; Xiang, Z.; Li, D.; Han, X. Microcystin-leucine arginine exhibits immunomodulatory roles in testicular cells resulting in orchitis. Environ. Pollut. 2017, 229, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, X.X.; Wu, B.; Yin, J.; Yu, Y.; Yang, L. Comprehensive insights into microcystin-LR effects on hepatic lipid metabolism using cross-omics technologies. J. Hazard Mater. 2016, 315, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Hillebrandt, S.; Goos, C.; Matern, S.; Lammert, F. Genome-wide analysis of hepatic fibrosis in inbred mice identifies the susceptibility locus Hfib1 on chromosome 15. Gastroenterology 2002, 123, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Wakil, A.E.; Rockey, D.C. Strain-specific differences in mouse hepatic wound healing are mediated by divergent T helper cytokine responses. Proc. Natl. Acad. Sci. USA 1997, 94, 10663–10668. [Google Scholar] [CrossRef] [Green Version]
- Sediment and Algae Color the Great Lakes. Available online: https://coastalscience.noaa.gov/news/sediment-and-algae-color-the-great-lakes-image-of-the-day/ (accessed on 6 August 2019).
- Bullerjahn, G.S.; McKay, R.M.; Davis, T.W.; Baker, D.B.; Boyer, G.L.; D’Anglada, L.V.; Doucette, G.J.; Ho, J.C.; Irwin, E.G.; Kling, C.L.; et al. Global solutions to regional problems: Collecting global expertise to address the problem of harmful cyanobacterial blooms. A Lake Erie case study. Harmful Algae 2016, 54, 223–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, T.L.; Utsumi, M. An overview of the accumulation of microcystins in aquatic ecosystems. J. Environ. Manag. 2018, 213, 520–529. [Google Scholar] [CrossRef]
- Svirčev, Z.; Drobac, D.; Tokodi, N.; Mijović, B.; Codd, G.A.; Meriluoto, J. Toxicology of microcystins with reference to cases of human intoxications and epidemiological investigations of exposures to cyanobacteria and cyanotoxins. Arch. Toxicol. 2017, 91, 621–650. [Google Scholar] [CrossRef]
- Stewart, I.; Webb, P.M.; Schluter, P.J.; Fleming, L.E.; Burns, J.W.; Gantar, M.; Backer, L.C.; Shaw, G.R. Epidemiology of recreational exposure to freshwater cyanobacteria—An international prospective cohort study. BMC Public Health 2006, 6, 93. [Google Scholar] [CrossRef]
- Su, R.C.; Blomquist, T.M.; Kleinhenz, A.L.; Khalaf, F.K.; Dube, P.; Lad, A.; Breidenbach, J.D.; Mohammed, C.J.; Zhang, S.; Baum, C.E.; et al. Exposure to the Harmful Algal Bloom (HAB) Toxin Microcystin-LR (MC-LR) Prolongs and Increases Severity of Dextran Sulfate Sodium (DSS)-Induced Colitis. Toxins 2019, 11, 371. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining clusters from a hierarchical cluster tree: The Dynamic Tree Cut package for R. Bioinformatics 2007, 24, 719–720. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote) omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, N.J.; Rosenspire, A.J.; Caruso, J.A.; Stemmer, P.M. Low level Hg2+ exposure modulates the B-cell cytoskeletal phosphoproteome. J. Proteom. 2018, 173, 107–114. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test | q < 0.10 | q < 0.30 |
|---|---|---|
| 50 µg/kg vs. Ctrl | 10 | 66 |
| 100 µg/kg vs. Ctrl | 93 | 459 |
| Linear regression | 25 | 368 |
| Pathway | Similar Pathways a | Mean t-Statistic | Sites | FDR |
|---|---|---|---|---|
| Vehicle vs. 50 μg/kg MC-LR | ||||
| Urogenital system development | 18 | −0.493 | 71 | 0.029 |
| Regulation of T cell proliferation | 9 | −0.730 | 35 | 0.029 |
| Appendage morphogenesis | 9 | −0.813 | 30 | 0.029 |
| Regulation of DNA-binding transcription factor activity | 7 | −0.368 | 102 | 0.036 |
| Regulation of striated muscle tissue development | 7 | −0.590 | 36 | 0.058 |
| Regulation of cellular response to oxidative stress | 6 | −0.677 | 30 | 0.046 |
| Energy derivation by oxidation of organic compounds | 6 | −0.460 | 92 | 0.029 |
| Positive regulation of cell cycle process | 6 | −0.464 | 86 | 0.029 |
| Defense response to other organism | 5 | −0.431 | 85 | 0.029 |
| Response to wounding | 4 | −0.367 | 95 | 0.056 |
| Cognition | 4 | −0.414 | 76 | 0.056 |
| Regulation of microtubule cytoskeleton organization | 4 | −0.345 | 102 | 0.046 |
| Monosaccharide metabolic process | 3 | −0.479 | 66 | 0.045 |
| Protein autophosphorylation | 3 | −0.346 | 95 | 0.068 |
| Lung alveolus development | 3 | −0.736 | 24 | 0.070 |
| Coronary vasculature development | 4 | −1.032 | 17 | 0.029 |
| Vehicle vs. 100 μg/kg MC-LR | ||||
| Toll-like receptor TLR6:TLR2 cascade | 17 | −1.073 | 32 | 0.0292 |
| Transport of mature mRNA derived from an intron-less transcript | 6 | −1.257 | 21 | 0.0292 |
| Cell cycle, mitotic | 5 | −0.555 | 132 | 0.0292 |
| Resolution of sister chromatid cohesion | 5 | −1.111 | 30 | 0.0292 |
| Mitotic prometaphase | 4 | −0.797 | 49 | 0.0426 |
| Cytokine signaling in immune system | 3 | −0.580 | 144 | 0.0200 |
| Carbohydrate metabolism | 3 | −0.717 | 105 | 0.0200 |
| L1CAM interactions | 2 | −0.846 | 55 | 0.0292 |
| Process | Mean t-Statistic | Sites | FDR |
|---|---|---|---|
| Vehicle vs. 50 μg/kg MC-LR | |||
| Cluster 3 | |||
| Renal system development | −0.506 | 68 | 0.029 |
| Regulation of stem cell proliferation | −0.838 | 31 | 0.029 |
| Regulation of epithelial cell proliferation | −0.414 | 111 | 0.029 |
| Regulation of mononuclear cell proliferation | −0.689 | 37 | 0.029 |
| Regulation of leukocyte proliferation | −0.689 | 37 | 0.029 |
| Regulation of T cell proliferation | −0.730 | 35 | 0.029 |
| Positive regulation of cell cycle process | −0.464 | 86 | 0.029 |
| Regulation of lymphocyte proliferation | −0.689 | 37 | 0.029 |
| Urogenital system development | −0.493 | 71 | 0.029 |
| Kidney development | −0.566 | 62 | 0.029 |
| Coronary vasculature development | −1.032 | 17 | 0.029 |
| Cluster 4 | |||
| Defense response to another organism | −0.431 | 85 | 0.029 |
| Cluster 6 | |||
| Generation of precursor metabolites and energy | −0.375 | 133 | 0.029 |
| Vehicle vs. 100 μg/kg MC-LR | |||
| Cluster 1 | |||
| Pre-mRNA splicing | 0.246 | 223 | 0.0601 |
| mRNA splicing | 0.246 | 223 | 0.0601 |
| Cluster 2 | |||
| Post-translational protein modification | −0.31193 | 326 | 0.080192 |
| Axon guidance | −0.30646 | 203 | 0.18749 |
| Cluster 3 | |||
| Recruitment of mitotic centrosome proteins and complexes | −0.78878 | 24 | 0.14638 |
| Cell-cell junction organization | −0.56824 | 45 | 0.15862 |
| Centrosome maturation | −0.78878 | 24 | 0.14638 |
| Toll-like receptor 4 (TLR4) cascade | −0.86804 | 41 | 0.042602 |
| Signaling by interleukins | −0.57231 | 103 | 0.033413 |
| Innate immune system | −0.37959 | 287 | 0.044106 |
| Cytokine signaling in immune system | −0.58043 | 144 | 0.020048 |
| Cluster 4 | |||
| Cellular senescence | −0.65136 | 49 | 0.082148 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lad, A.; Su, R.C.; Breidenbach, J.D.; Stemmer, P.M.; Carruthers, N.J.; Sanchez, N.K.; Khalaf, F.K.; Zhang, S.; Kleinhenz, A.L.; Dube, P.; et al. Chronic Low Dose Oral Exposure to Microcystin-LR Exacerbates Hepatic Injury in a Murine Model of Non-Alcoholic Fatty Liver Disease. Toxins 2019, 11, 486. https://doi.org/10.3390/toxins11090486
Lad A, Su RC, Breidenbach JD, Stemmer PM, Carruthers NJ, Sanchez NK, Khalaf FK, Zhang S, Kleinhenz AL, Dube P, et al. Chronic Low Dose Oral Exposure to Microcystin-LR Exacerbates Hepatic Injury in a Murine Model of Non-Alcoholic Fatty Liver Disease. Toxins. 2019; 11(9):486. https://doi.org/10.3390/toxins11090486
Chicago/Turabian StyleLad, Apurva, Robin C. Su, Joshua D. Breidenbach, Paul M. Stemmer, Nicholas J. Carruthers, Nayeli K. Sanchez, Fatimah K. Khalaf, Shungang Zhang, Andrew L. Kleinhenz, Prabhatchandra Dube, and et al. 2019. "Chronic Low Dose Oral Exposure to Microcystin-LR Exacerbates Hepatic Injury in a Murine Model of Non-Alcoholic Fatty Liver Disease" Toxins 11, no. 9: 486. https://doi.org/10.3390/toxins11090486
APA StyleLad, A., Su, R. C., Breidenbach, J. D., Stemmer, P. M., Carruthers, N. J., Sanchez, N. K., Khalaf, F. K., Zhang, S., Kleinhenz, A. L., Dube, P., Mohammed, C. J., Westrick, J. A., Crawford, E. L., Palagama, D., Baliu-Rodriguez, D., Isailovic, D., Levison, B., Modyanov, N., Gohara, A. F., ... Kennedy, D. J. (2019). Chronic Low Dose Oral Exposure to Microcystin-LR Exacerbates Hepatic Injury in a Murine Model of Non-Alcoholic Fatty Liver Disease. Toxins, 11(9), 486. https://doi.org/10.3390/toxins11090486