Potential of Matrix Metalloproteinase Inhibitors for the Treatment of Local Tissue Damage Induced by a Type P-I Snake Venom Metalloproteinase


Abstract
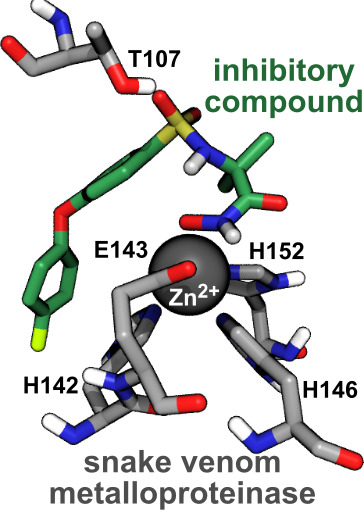
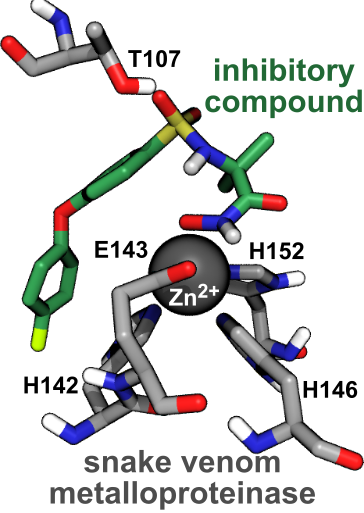
:
1. Introduction
2. Results and Discussion
2.1. Inhibition of Proteolytic Activity
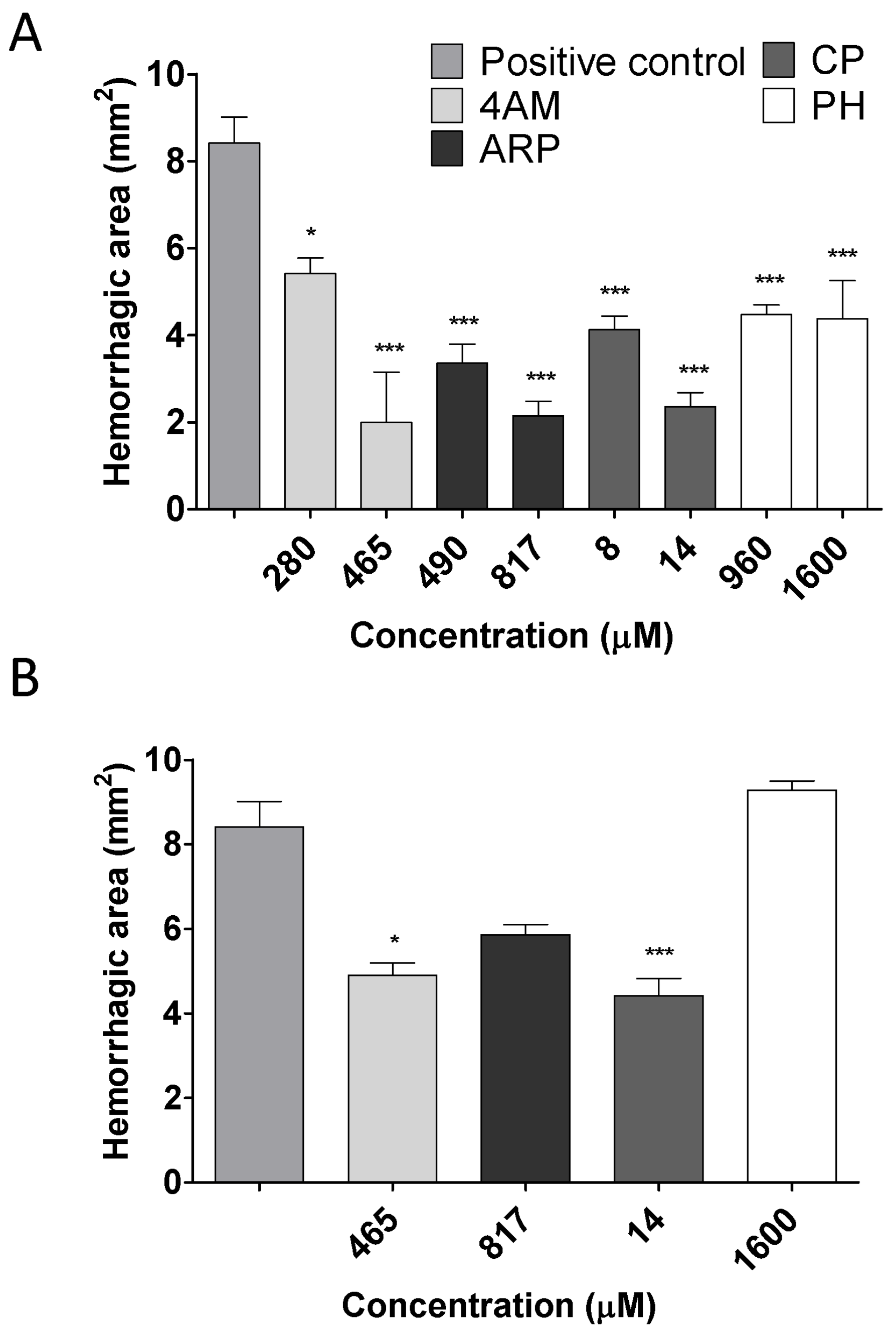
2.2. Inhibition of Hemorrhagic Activity
2.3. Inhibition of Edema-Forming Activity
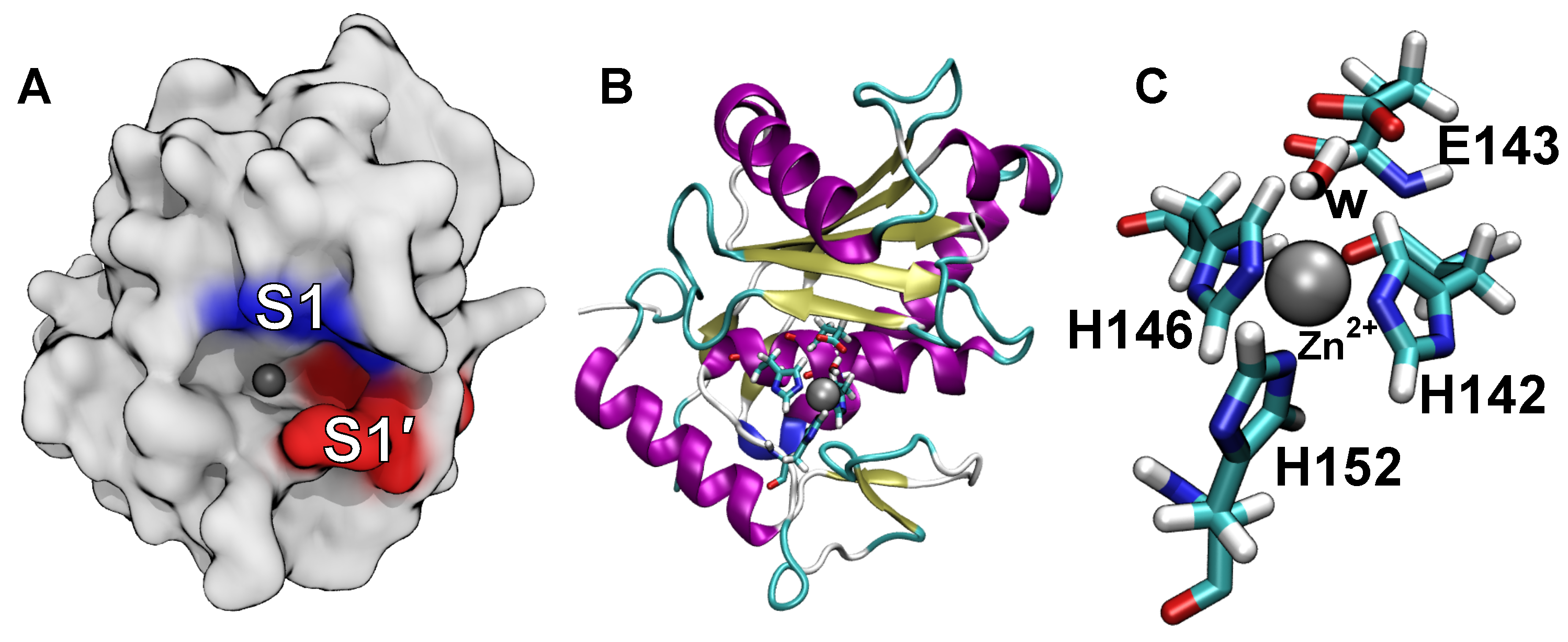
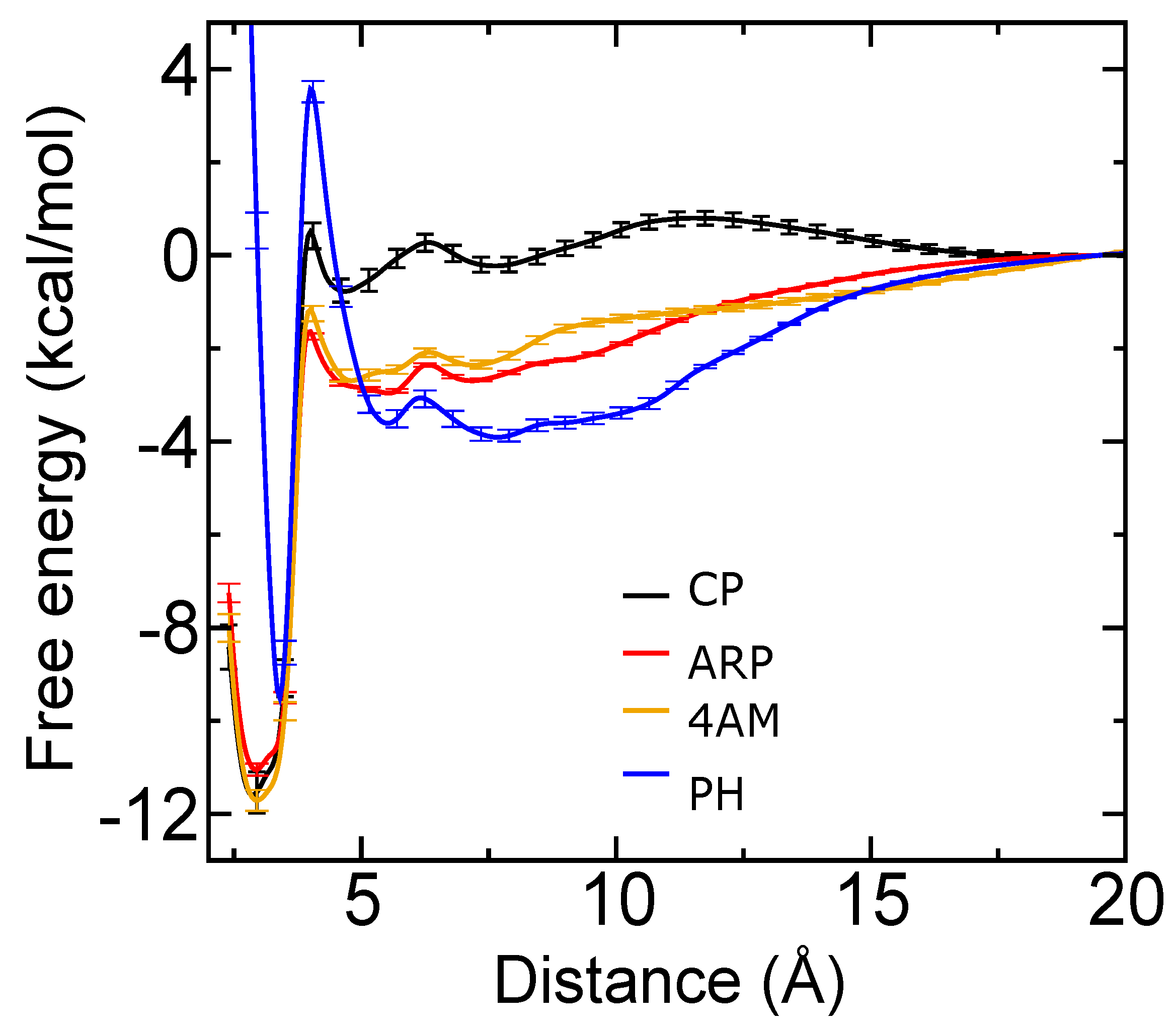
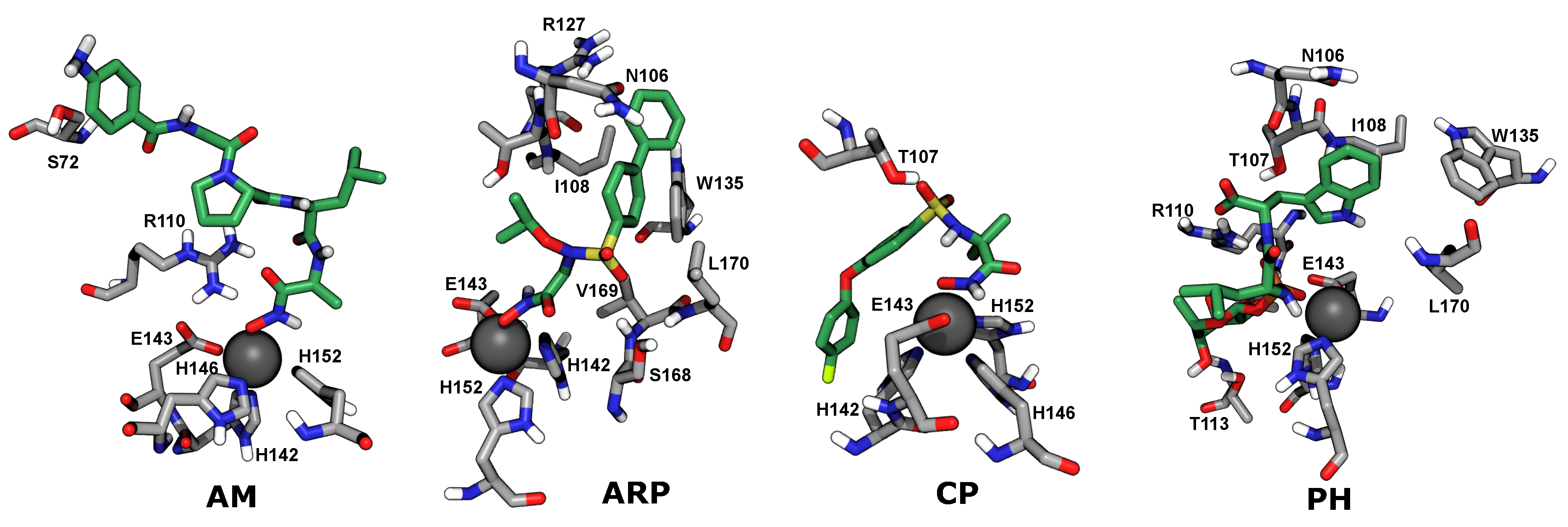
2.4. Computational Analysis of Inhibitor–BaP1 Interactions
3. Conclusions
4. Materials and Methods
4.1. Venom
4.2. Chemicals and Reagents
4.3. Animals
4.4. Purification of Metalloproteinase Batx-I
4.5. Inhibition of Proteolytic Activity
4.6. Inhibition of Hemorrhagic Activity
4.7. Inhibition of Edema-Forming Activity
4.8. Molecular Models
4.9. Molecular Dynamics
4.10. Free Energy Calculations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABF | Adaptive biasing force |
| Colvars | Collective variables |
| eABF | Extended adaptive biasing force |
| meta-eABF | Extended adaptive biasing force with sampling enhanced by metadynamics |
| MMPs | Matrix metalloproteinases |
| SVMPs | Snake venom metalloproteinases |
References
- Chippaux, J.P. Snakebite envenomation turns again into a neglected tropical disease! J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 38. [Google Scholar] [CrossRef]
- Chippaux, J.P. Incidence and mortality due to snakebite in the Americas. PLoS Negl. Trop. Dis. 2017, 11, e0005662. [Google Scholar] [CrossRef]
- Gutiérrez, J.M.; Calvete, J.J.; Habib, A.G.; Harrison, R.A.; Williams, D.J.; Warrell, D.A. Snakebite envenoming. Nat. Rev. Dis. Primers 2017, 3, 17063. [Google Scholar] [CrossRef]
- Informe Final de Accidente ofídico Colombia año 2018. Available online: https://www.ins.gov.co/buscador-eventos/Informesdeevento/ACCIDENTE%20OF%C3%8DDICO_2018.pdf (accessed on 16 December 2019).
- Otero-Patiño, R. Epidemiological, clinical and therapeutic aspects of Bothrops asper bites. Toxicon 2009, 54, 998–1011. [Google Scholar] [CrossRef]
- Gutiérrez, J.M.; Rucavado, A.; Chaves, F.; Díaz, C.; Escalante, T. Experimental pathology of local tissue damage induced by Bothrops asper snake venom. Toxicon 2009, 54, 958–975. [Google Scholar] [CrossRef]
- Georgieva, D.; Arni, R.K.; Betzel, C. Proteome analysis of snake venom toxins: Pharmacological insights. Exp. Rev. Proteomic 2008, 5, 787–797. [Google Scholar] [CrossRef]
- Bode, W.; Gomis-Rüth, F.X.; Stöckler, W. Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (HEXXHXXGXXH and Met-turn) and topologies and should be grouped into a common family, the ‘metzincins’. FEBS Lett. 1993, 331, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.W.; Serrano, S.M. Insights into and speculations about snake venom metalloproteinase (SVMP) synthesis, folding and disulfide bond formation and their contribution to venom complexity. FEBS J. 2008, 275, 3016–3030. [Google Scholar] [CrossRef]
- Lingott, T.; Schleberger, C.; Gutiérrez, J.M.; Merfort, I. High-resolution crystal structure of the snake venom metalloproteinase BaP1 complexed with a peptidomimetic: Insight into inhibitor binding. Biochemistry 2009, 48, 6166–6174. [Google Scholar] [CrossRef]
- Alape-Girón, A.; Sanz, L.; Escolano, J.; Flores-Diaz, M.; Madrigal, M.; Sasa, M.; Calvete, J.J. Snake venomics of the lancehead pitviper Bothrops asper: Geographic, individual, and ontogenetic variations. J. Proteome Res. 2008, 7, 3556–3571. [Google Scholar] [CrossRef]
- Berger, A.; Schechter, I. Mapping the active site of papain with the aid of peptide substrates and inhibitors. Proc. R. Soc. Lond. B. (Biol. Sci.) 1970, 257, 249–264. [Google Scholar] [CrossRef]
- Gupta, S.P.; Patil, V.M. Specificity of binding with matrix metalloproteinases. In Matrix Metalloproteinase Inhibitors; Springer: Berlin, Germany, 2012; pp. 35–56. [Google Scholar]
- Gutiérrez, J.; Escalante, T.; Rucavado, A.; Herrera, C. Hemorrhage caused by snake venom metalloproteinases: A journey of discovery and understanding. Toxins 2016, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, J.M.; Rucavado, A. Snake venom metalloproteinases: Their role in the pathogenesis of local tissue damage. Biochimie 2000, 82, 841–850. [Google Scholar] [CrossRef]
- Gutiérrez, J.M.; León, G.; Rojas, G.; Lomonte, B.; Rucavado, A.; Chaves, F. Neutralization of local tissue damage induced by Bothrops asper (terciopelo) snake venom. Toxicon 1998, 36, 1529–1538. [Google Scholar] [CrossRef]
- Lancet. Snakebite—Emerging from the shadows of neglect. Lancet 2019, 393, 2175. [Google Scholar] [CrossRef]
- Rasmussen, H.S. Batimastat and Marimastat in Cancer. In Antiangiogenic Agents in Cancer Therapy; Springer: Berlin, Germany, 1999; pp. 399–405. [Google Scholar]
- Escalante, T.; Franceschi, A.; Rucavado, A.; Gutiérrez, J.M. Effectiveness of batimastat, a synthetic inhibitor of matrix metalloproteinases, in neutralizing local tissue damage induced by BaP1, a hemorrhagic metalloproteinase from the venom of the snake Bothrops asper. Biochem. Pharmacol. 2000, 60, 269–274. [Google Scholar] [CrossRef]
- Rucavado, A.; Escalante, T.; Gutiérrez, J. Effect of the metalloproteinase inhibitor batimastat in the systemic toxicity induced by Bothrops asper snake venom: Understanding the role of metalloproteinases in envenomation. Toxicon 2004, 43, 417–424. [Google Scholar] [CrossRef]
- Arias, A.S.; Rucavado, A.; Gutiérrez, J.M. Peptidomimetic hydroxamate metalloproteinase inhibitors abrogate local and systemic toxicity induced by Echis ocellatus (saw-scaled) snake venom. Toxicon 2017, 132, 40–49. [Google Scholar] [CrossRef]
- Odake, S.; Morita, Y.; Morikawa, T.; Yoshida, N.; Hori, H.; Nagai, Y. Inhibition of matrix metalloproteinases by peptidyl hydroxamic acids. Biochem. Biophys. Res. Commun. 1994, 199, 1442–1446. [Google Scholar] [CrossRef]
- Rossello, A.; Nuti, E.; Orlandini, E.; Carelli, P.; Rapposelli, S.; Macchia, M.; Minutolo, F.; Carbonaro, L.; Albini, A.; Benelli, R.; et al. New N-arylsulfonyl-N-alkoxyaminoacetohydroxamic acids as selective inhibitors of gelatinase A (MMP-2). Bioorg. Med. Chem. 2004, 12, 2441–2450. [Google Scholar] [CrossRef]
- Rohde, L.E.; Ducharme, A.; Arroyo, L.H.; Aikawa, M.; Sukhova, G.H.; Lopez-Anaya, A.; McClure, K.F.; Mitchell, P.G.; Libby, P.; Lee, R.T. Matrix metalloproteinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice. Circulation 1999, 99, 3063–3070. [Google Scholar] [CrossRef] [Green Version]
- Umezawa, H. Structures and activities of protease inhibitors of microbial origin. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1976; Volume 45, pp. 678–695. [Google Scholar]
- Odake, S.; Okayama, T.; Obata, M.; Morikawa, T.; Hattori, S.; HORI, H.; Nagai, Y. Vertebrate collagenase inhibitor. II. Tetrapeptidyl hydroxamic acids. Chem. Pharm. Bull. 1991, 39, 1489–1494. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Saravanan, C.; Singh, S.K. Sulphonamides: Deserving class as MMP inhibitors? Eur. J. Med. Chem. 2013, 60, 89–100. [Google Scholar] [CrossRef]
- Selman, M.; Cisneros-Lira, J.; Gaxiola, M.; Ramirez, R.; Kudlacz, E.M.; Mitchell, P.G.; Pardo, A. Matrix metalloproteinases inhibition attenuates tobacco smoke-induced emphysema in guinea pigs. Chest 2003, 123, 1633–1641. [Google Scholar] [CrossRef]
- Van den Burg, B.; Eijsink, V. Thermolysin and related Bacillus metallopeptidases. In Handbook of Proteolytic Enzymes; Elsevier: Amsterdam, The Netherlands, 2004; pp. 374–387. [Google Scholar]
- Suda, H.; Aoyago, T.; Takeuchi, T.; Umezawa, H. A thermolysin inhibitor produced by actinomycetes: Phosphoramidon. J. Antibiot. 1973, 26, 621–623. [Google Scholar] [CrossRef] [Green Version]
- Bjarnason, J.B.; Fox, J.W. Hemorrhagic metalloproteinases from snake venoms. Pharmacol. Therapeut. 1994, 62, 325–372. [Google Scholar] [CrossRef]
- Patiño, A.C.; Pereañez, J.A.; Núñez, V.; Benjumea, D.M.; Fernandez, M.; Rucavado, A.; Sanz, L.; Calvete, J.J. Isolation and biological characterization of Batx-I, a weak hemorrhagic and fibrinogenolytic PI metalloproteinase from Colombian Bothrops atrox venom. Toxicon 2010, 56, 936–943. [Google Scholar] [CrossRef]
- Preciado, L.M.; Rey-Suarez, P.; Henao, I.C.; Pereañez, J.A. Betulinic, oleanolic and ursolic acids inhibit the enzymatic and biological effects induced by a PI snake venom metalloproteinase. Chem. Biol. Int. 2018, 279, 219–226. [Google Scholar] [CrossRef]
- Gutiérrez, J.; Romero, M.; Díaz, C.; Borkow, G.; Ovadia, M. Isolation and characterization of a metalloproteinase with weak hemorrhagic activity from the venom of the snake Bothrops asper (terciopelo). Toxicon 1995, 33, 19–29. [Google Scholar] [CrossRef]
- Loffek, S.; Ullrich, N.; Gorgens, A.; Murke, F.; Eilebrecht, M.; Menne, C.; Giebel, B.; Schadendorf, D.; Singer, B.; Helfrich, I. CEACAM1-4L Promotes Anchorage-Independent Growth in Melanoma. Front. Oncol. 2015, 5, 234. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, T.; Takeuchi, A.; Munesue, S.; Yamamoto, N.; Hayashi, K.; Kimura, H.; Miwa, S.; Inatani, H.; Shimozaki, S.; Kato, T.; et al. Anti-tumor effects of a nonsteroidal anti-inflammatory drug zaltoprofen on chondrosarcoma via activating peroxisome proliferator-activated receptor gamma and suppressing matrix metalloproteinase-2 expression. Can. Med. 2018, 7, 1944–1954. [Google Scholar] [CrossRef] [Green Version]
- Ulasov, I.; Thaci, B.; Sarvaiya, P.; Yi, R.; Guo, D.; Auffinger, B.; Pytel, P.; Zhang, L.; Kim, C.K.; Borovjagin, A.; et al. Inhibition of MMP 14 potentiates the therapeutic effect of temozolomide and radiation in gliomas. Can. Med. 2013, 2, 457–467. [Google Scholar] [CrossRef]
- Meng, H.; Xing, G.; Blanco, E.; Song, Y.; Zhao, L.; Sun, B.; Li, X.; Wang, P.C.; Korotcov, A.; Li, W.; et al. Gadolinium metallofullerenol nanoparticles inhibit cancer metastasis through matrix metalloproteinase inhibition: Imprisoning instead of poisoning cancer cells. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Le Quément, C.; Lagente, V.; Guénon, I.; Muzio, V.; Gillon, J.Y.; Boichot, E. Anti-inflammatory properties of MMP inhibitors in experimental models of chronic obstructive pulmonary disease and lung inflammation. In Matrix Metalloproteinases in Tissue Remodelling and Inflammation; Springer: Berlin, Germany, 2008; pp. 57–69. [Google Scholar]
- Aerts, J.; Vandenbroucke, R.; Dera, R.; Balusu, S.; Van Wonterghem, E.; Moons, L.; Libert, C.; Dehaen, W.; Arckens, L. Synthesis and validation of a hydroxypyrone-based, potent, and specific matrix metalloproteinase-12 inhibitor with anti-inflammatory activity in vitro and in vivo. Mediat. Inflamm. 2015, 2015. [Google Scholar] [CrossRef]
- Rucavado, A.; Escalante, T.; Teixeira, C.F.; Fernándes, C.M.; Díaz, C.; Gutiérrez, J.M. Increments in cytokines and matrix metalloproteinases in skeletal muscle after injection of tissue-damaging toxins from the venom of the snake Bothrops asper. Mediat. Inflamm. 2002, 11, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Nissinen, L.; Kähäri, V.M. Matrix metalloproteinases in inflammation. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2571–2580. [Google Scholar] [CrossRef]
- Manicone, A.M.; McGuire, J.K. Matrix metalloproteinases as modulators of inflammation. In Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 19, pp. 34–41. [Google Scholar]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Meth. 2017, 14, 71. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Zhang, H.; Chen, H.; Shao, X.; Chipot, C.; Cai, W. Zooming across the Free-Energy Landscape: Shaving Barriers, and Flooding Valleys. J. Chem. Theory Comput. 2018, 9, 4738–4745. [Google Scholar] [CrossRef]
- Rao, B.G. Recent developments in the design of specific matrix metalloproteinase inhibitors aided by structural and computational studies. Curr. Pharm. Des. 2005, 11, 295–322. [Google Scholar] [CrossRef]
- Cheng, X.C.; Wang, Q.; Fang, H.; Xu, W.F. Role of sulfonamide group in matrix metalloproteinase inhibitors. Curr. Med. Chem. 2008, 15, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, J.; Gené, J.; Rojas, G.; Cerdas, L. Neutralization of proteolytic and hemorrhagic activities of Costa Rican snake venoms by a polyvalent antivenom. Toxicon 1985, 23, 887–893. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2015, 12, 405–413. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hopkins, C.W.; Le Grand, S.; Walker, R.C.; Roitberg, A.E. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. [Google Scholar] [CrossRef]
- Darden, T.A.; York, D.M.; Pedersen, L.G. Particle mesh Ewald: An N log N method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Fiorin, G.; Klein, M.L.; Hénin, J. Using collective variables to drive molecular dynamics simulations. Math. Probl. Eng. 2013, 111, 3345–3362. [Google Scholar] [CrossRef]
- Darve, E.; Pohorille, A. Calculating Free Energies Using Average Force. J. Chem. Phys. 2001, 115, 9169–9183. [Google Scholar] [CrossRef] [Green Version]
- Comer, J.; Roux, B.; Chipot, C. Achieving Ergodic Sampling Using Replica-exchange Free-energy Calculations. Mol. Simul. 2014, 40, 218–228. [Google Scholar] [CrossRef]
- Fu, H.; Shao, X.; Chipot, C.; Cai, W. Extended adaptive biasing force algorithm. An on-the-fly implementation for accurate free-energy calculations. J. Chem. Theory Comput. 2016, 12, 3506–3513. [Google Scholar] [CrossRef]
- Bussi, G.; Laio, A.; Parrinello, M. Equilibrium free energies from nonequilibrium metadynamics. Phys. Rev. Lett. 2006, 96, 090601. [Google Scholar] [CrossRef] [Green Version]
- Limongelli, V.; Bonomi, M.; Parrinello, M. Funnel metadynamics as accurate binding free-energy method. Proc. Natl. Acad. Sci. USA 2013, 110, 6358–6363. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC (M) | CI (95%) (M) |
|---|---|---|
| 4AM | 408.3 | 328.8–507.1 |
| ARP | 55.4 | 43.1–71.1 |
| CP | 8.9 | 7.6–10.5 |
| PH | 793.2 | 450.6–1396.0 |
| Compound | IC (M) | CI (95%) (M) | IC (M) | CI (95%) (M) |
|---|---|---|---|---|
| 4AM | 87.1 | 72.6–104.4 | 92.9 | 79.3–108.8 |
| ARP | 36.5 | 29.5–45.2 | 163.8 | 156.1–171.8 |
| CP | 11.6 | 9.3–11.3 | 2.5 | 3.1–3.0 |
| PH | 358.3 | 327.0–392.0 | 321.1 | 212.7–484.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Preciado, L.M.; Pereañez, J.A.; Comer, J. Potential of Matrix Metalloproteinase Inhibitors for the Treatment of Local Tissue Damage Induced by a Type P-I Snake Venom Metalloproteinase. Toxins 2020, 12, 8. https://doi.org/10.3390/toxins12010008
Preciado LM, Pereañez JA, Comer J. Potential of Matrix Metalloproteinase Inhibitors for the Treatment of Local Tissue Damage Induced by a Type P-I Snake Venom Metalloproteinase. Toxins. 2020; 12(1):8. https://doi.org/10.3390/toxins12010008
Chicago/Turabian StylePreciado, Lina María, Jaime Andrés Pereañez, and Jeffrey Comer. 2020. "Potential of Matrix Metalloproteinase Inhibitors for the Treatment of Local Tissue Damage Induced by a Type P-I Snake Venom Metalloproteinase" Toxins 12, no. 1: 8. https://doi.org/10.3390/toxins12010008
APA StylePreciado, L. M., Pereañez, J. A., & Comer, J. (2020). Potential of Matrix Metalloproteinase Inhibitors for the Treatment of Local Tissue Damage Induced by a Type P-I Snake Venom Metalloproteinase. Toxins, 12(1), 8. https://doi.org/10.3390/toxins12010008