Molecular Mechanism of Aflatoxin-Induced Hepatocellular Carcinoma Derived from a Bioinformatics Analysis



Abstract
:1. Introduction
2. Results
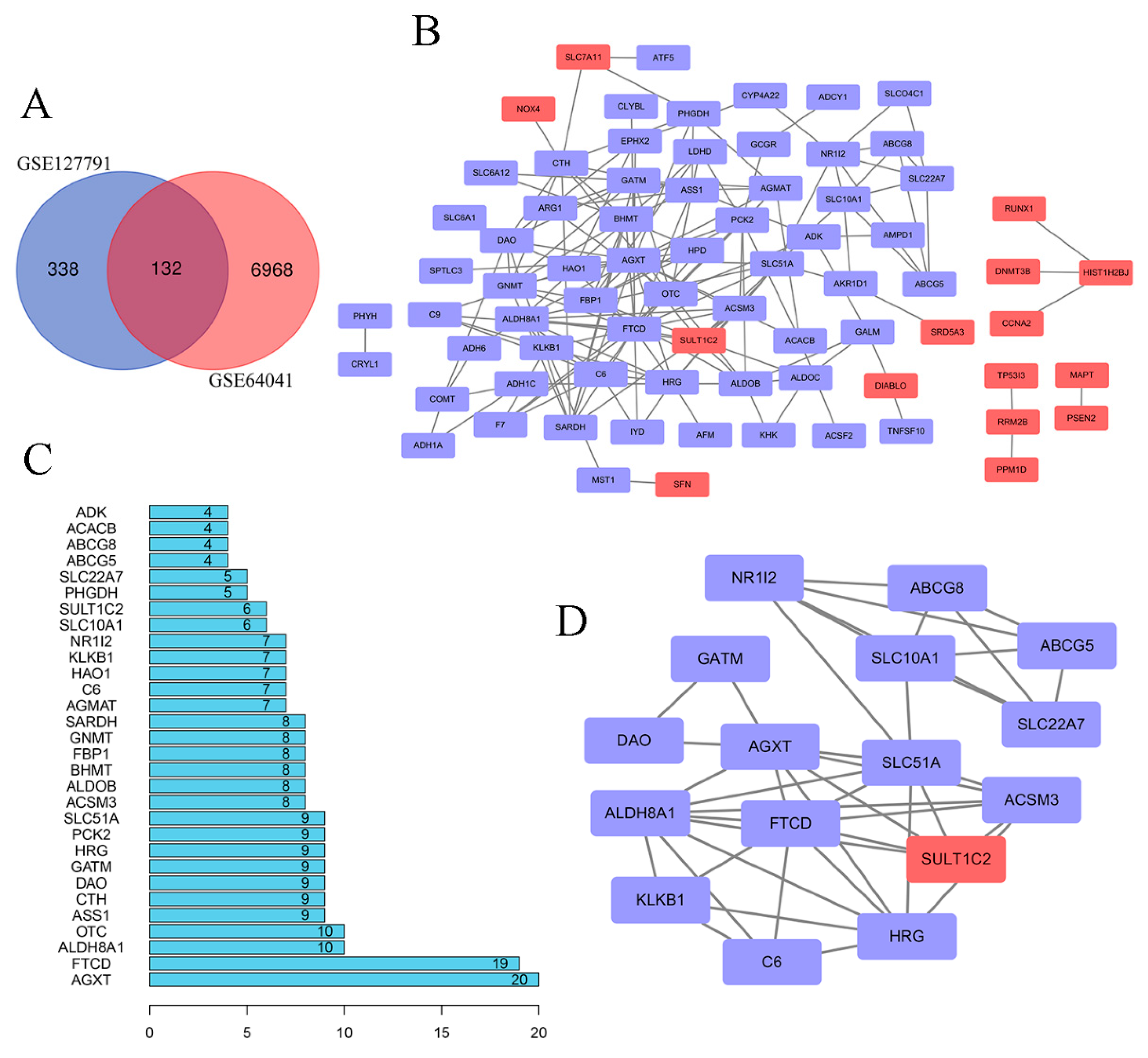
2.1. Screening and Identification of Differentially Expressed Genes (DEGs)
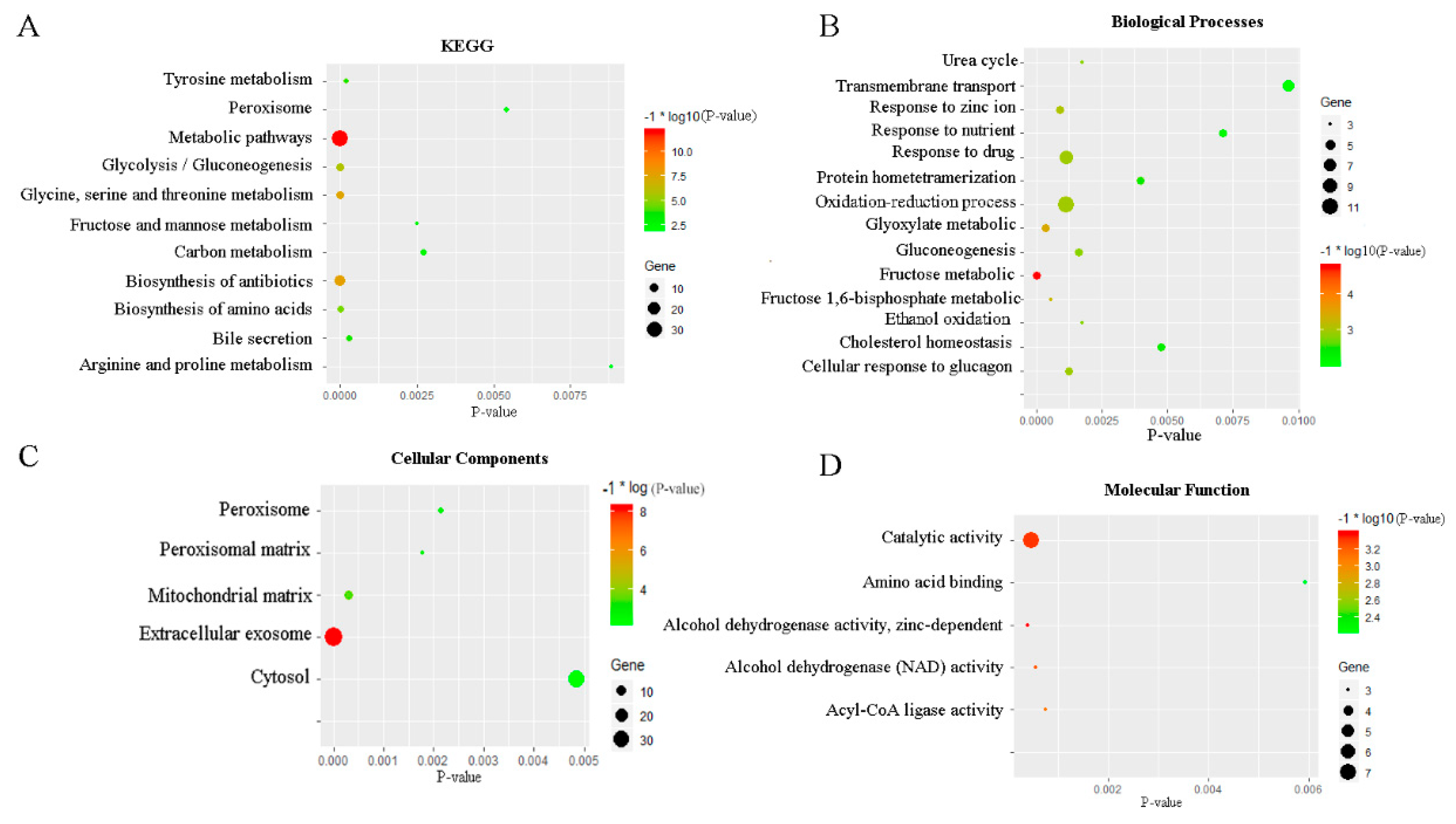
2.2. Enrichment Analysis of GO and KEGG of Differentially Expressed Genes (DEGs)
2.3. Hierarchical Clustering of Hub Genes
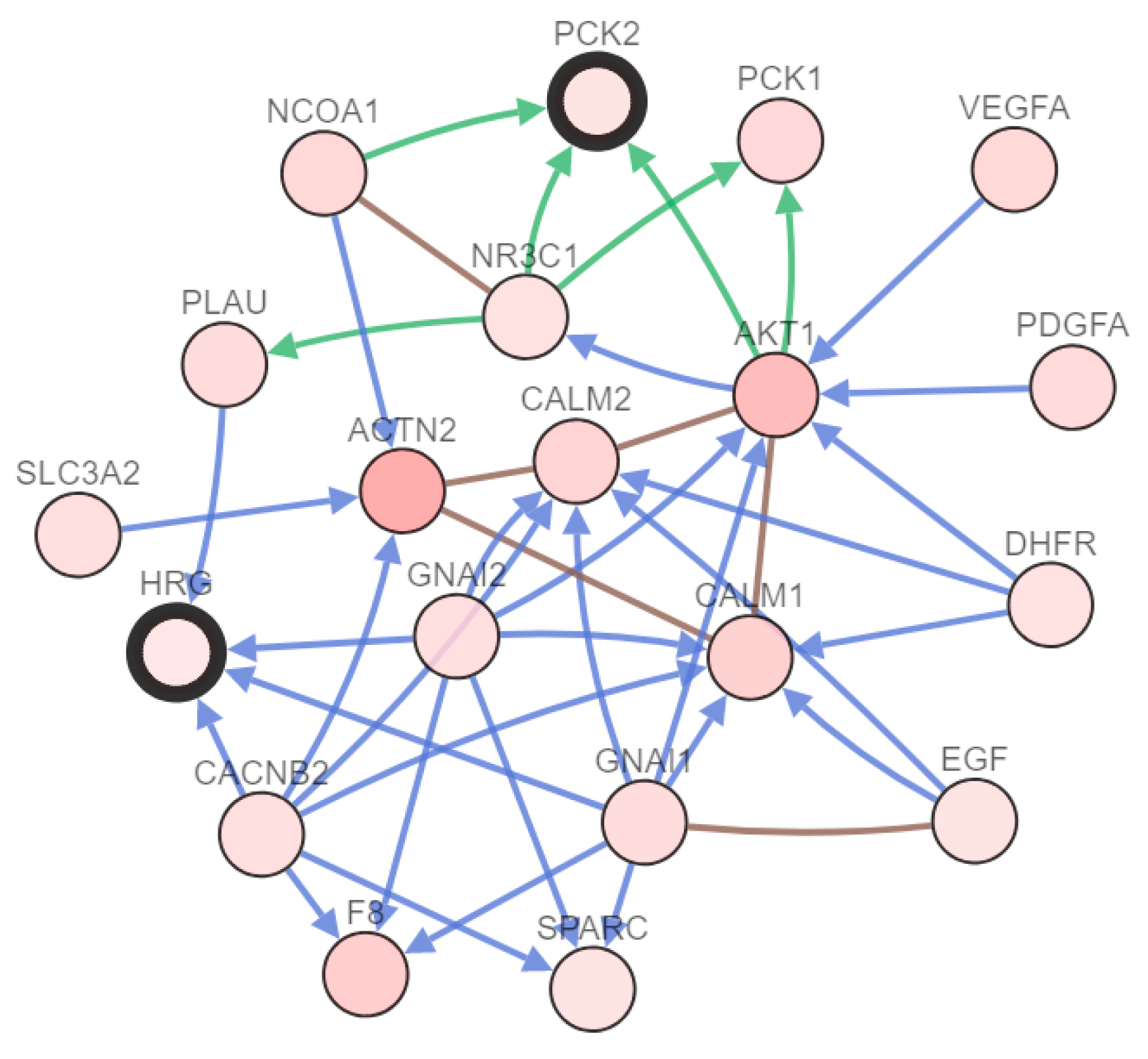
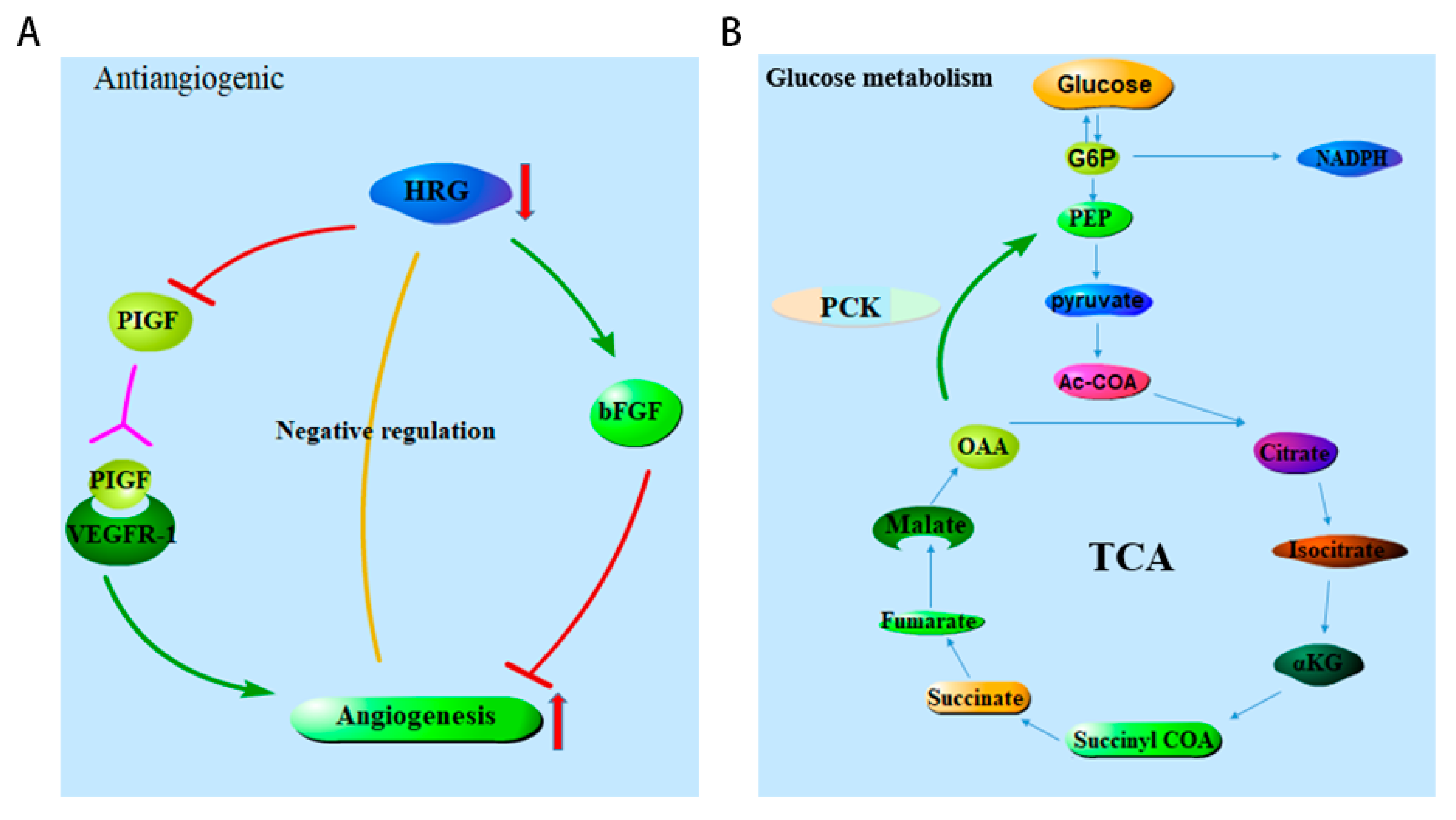
2.4. HRG and PCK2 Were Key Genes in the Hub Gene Dataset
2.5. Decreased Expression of HRG and PCK2 in HCC
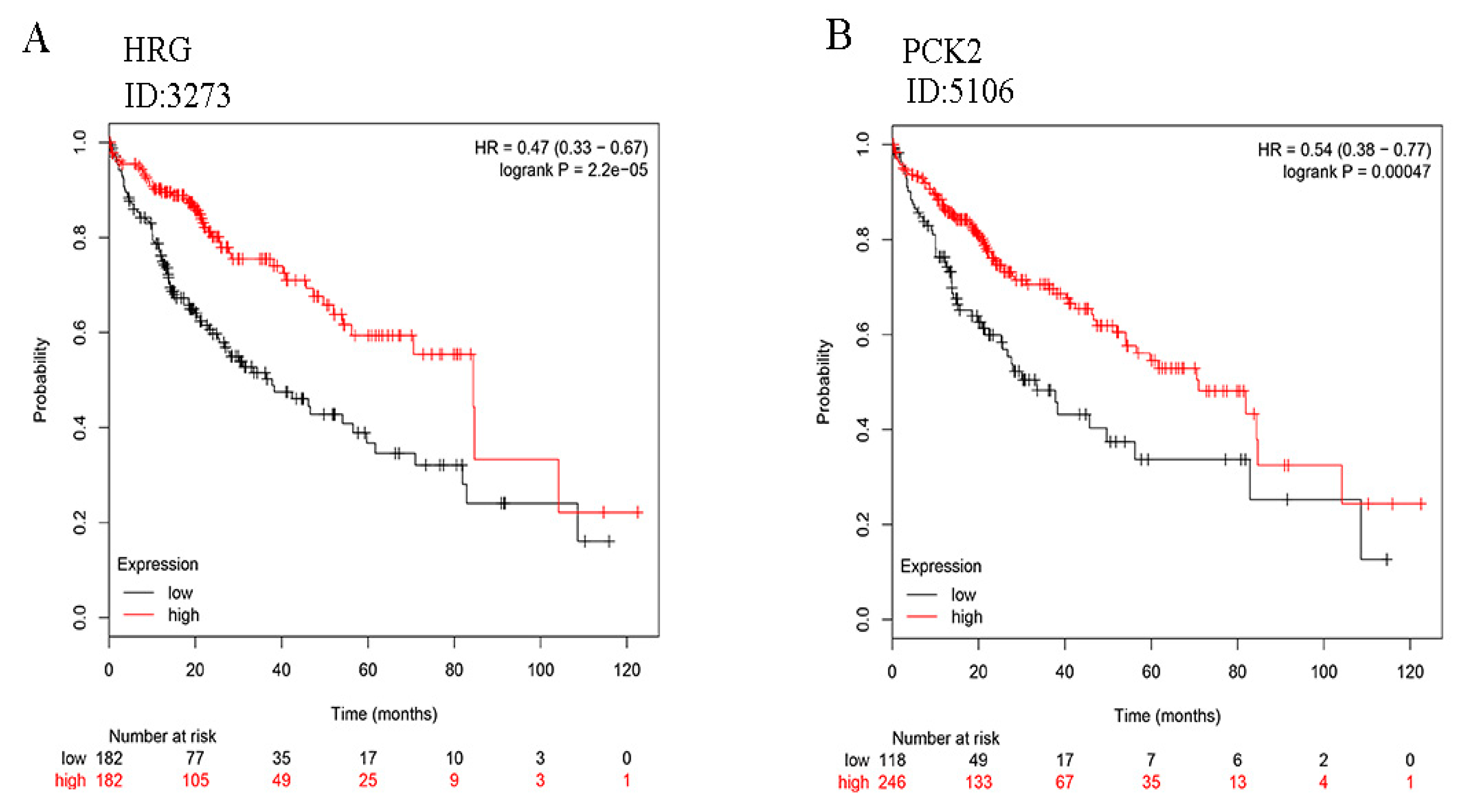
2.6. Survival Rate Analysis of HRG and PCK2 Expression of HCC Patients
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Screening of Datasets
5.2. Screening of DEGs
5.3. Protein-Protein Interaction (PPI) Analysis
5.4. KEGG Pathway and GO Term Enrichment Analysis
5.5. Screening and Analysis of Hub Genes
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Dietrich, P.; Hellerbrand, C.; Bosserhoff, A. The Delta Subunit of Rod-Specific Photoreceptor cGMP Phosphodiesterase (PDE6D) Contributes to Hepatocellular Carcinoma Progression. Cancers 2019, 11, 398. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, R.; Nelson, D.R. Review article: The management of hepatocellular carcinoma. Aliment. Pharmacol. Ther. 2010, 31, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 60, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Kolawole, O.; Meneely, J.; Greer, B.; Chevallier, O.; Jones, D.S.; Connolly, L.; Elliott, C. Comparative In Vitro Assessment of a Range of Commercial Feed Additives with Multiple Mycotoxin Binding Claims. Toxins 2019, 11, 659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkekoglu, P.; Oral, D.; Chao, M.W.; Kocer-Gumusel, B. Hepatocellular Carcinoma and Possible Chemical and Biological Causes: A Review. J. Environ. Pathol. Toxicol. 2017, 36, 171. [Google Scholar] [CrossRef] [PubMed]
- Jin, X. Molecular biology of aflatoxin biosynthesis. J. Hyg. Res. 2003, 32, 628. [Google Scholar]
- Ismail, A.; Goncalves, B.L.; de Neeff, D.V.; Ponzilacqua, B.; Coppa, C.; Hintzsche, H.; Sajid, M.; Cruz, A.G.; Corassin, C.H.; Oliveira, C.A.F. Aflatoxin in foodstuffs: Occurrence and recent advances in decontamination. Food Res. Int. 2018, 113, 74–85. [Google Scholar] [CrossRef]
- Wogan, G.N.; Kensler, T.W.; Groopman, J.D. Present and future directions of translational research on aflatoxin and hepatocellular carcinoma. A review. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2012, 29, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Baan, R.; Grosse, Y.; Straif, K.; Secretan, B.; El, G.F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L. A review of human carcinogens—Part F: Chemical agents and related occupations. Lancet Oncol. 2009, 10, 1143–1144. [Google Scholar] [CrossRef]
- Kamdem, L.K.; Ingolf, M.; Ute, G.D.A.; Jürgen, B.L.; Leszek, W. Dominant contribution of P450 3A4 to the hepatic carcinogenic activation of aflatoxin B1. Chem. Res. Toxicol. 2006, 19, 577. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.P.; Turner, P.C. The toxicology of aflatoxins as a basis for public health decisions. Mutagenesis 2002, 17, 471. [Google Scholar] [CrossRef] [PubMed]
- Szymański, P.; Olszewska, P.; Mikiciuk-Olasik, E.; Różalski, A.; Maszewska, A.; Markiewicz, Ł.; Cuchra, M.; Majsterek, I. Novel tetrahydroacridine and cyclopentaquinoline derivatives with fluorobenzoic acid moiety induce cell cycle arrest and apoptosis in lung cancer cells by activation of DNA damage signaling. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39, 1010428317695011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulik, L.; El-Serag, H.B. Epidemiology and Management of Hepatocellular Carcinoma. Gastroenterology 2019, 156, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, Y.; Zhu, Y.; Yu, Z.; Shao, M.; Luo, Y. Identification and Characterization of Cadmium-Related Genes in Liver Carcinoma. Biol. Trace Elem. Res. 2017, 182, 238–247. [Google Scholar] [CrossRef]
- Falzone, L.; Lupo, G.; La Rosa, G.R.M.; Crimi, S.; Anfuso, C.D.; Salemi, R.; Rapisarda, E.; Libra, M.; Candido, S. Identification of Novel MicroRNAs and Their Diagnostic and Prognostic Significance in Oral Cancer. Cancers 2019, 11, 610. [Google Scholar] [CrossRef] [Green Version]
- Moreno, E.C.; Pascual, A.; Prieto-Cuadra, D.; Laza, V.F.; Molina-Cerrillo, J.; Ramos-Munoz, M.E.; Rodriguez-Serrano, E.M.; Soto, J.L.; Carrato, A.; Garcia-Bermejo, M.L.; et al. Novel Molecular Characterization of Colorectal Primary Tumors Based on miRNAs. Cancers 2019, 11, 346. [Google Scholar] [CrossRef] [Green Version]
- Nicole, L.; Werner, T.V.; Christina, B.; Patrick, T.; Manfred, G.; Andreas, K.; Hans-Peter, L.; Norbert, G.; Eckart, M. Combining miRNA and mRNA Expression Profiles in Wilms Tumor Subtypes. Int. J. Mol. Sci. 2016, 17, 475. [Google Scholar]
- Guo, Y.; Bao, Y.; Ma, M.; Yang, W. Identification of Key Candidate Genes and Pathways in Colorectal Cancer by Integrated Bioinformatical Analysis. Int. J. Mol. Sci. 2017, 18, 722. [Google Scholar] [CrossRef] [Green Version]
- Fischer, C.; Mazzone, M.; Jonckx, B.; Carmeliet, P. FLT1 and its ligands VEGFB and PlGF: Drug targets for anti-angiogenic therapy? Nat. Rev. Cancer 2008, 8, 942–956. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.K.; Larsson, H.; Dixelius, J.; Johansson, I.; Lee, C.; Oellig, C.; Bjork, I.; Claesson-Welsh, L. A fragment of histidine-rich glycoprotein is a potent inhibitor of tumor vascularization. Cancer Res. 2004, 64, 599–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, I.K.; Patel, K.K.; Davis, D.S.; Parish, C.R.; Hulett, M.D. Histidine-rich glycoprotein: The Swiss Army knife of mammalian plasma. Blood 2011, 117, 2093–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlotte, R.; Massimiliano, M.; Sònia, T.; Damya, L.; Irja, J.; Cathy, C.; Mario Leonardo, S.; Inmaculada, S.; Xiujuan, L.; Ellen, K. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar]
- Bartneck, M.; Fech, V.; Ehling, J.; Govaere, O.; Theresa, W.K.; Hittatiya, K.; Vucur, M.; Gautheron, J.; Luedde, T.; Trautwein, C. Histidine-rich glycoprotein promotes macrophage activation and inflammation in chronic liver disease. Hepatology 2015, 63, 1310. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, J. Geometric Interpretation of Gene Coexpression Network Analysis. PLoS Comput. Biol. 2008, 4, e1000117. [Google Scholar]
- Mossanen, J.C.; Tacke, F. Role of lymphocytes in liver cancer. Oncoimmunology 2013, 2, e26468. [Google Scholar] [CrossRef] [Green Version]
- Coulon, S.; Heindryckx, F.; Geerts, A.; Van Steenkiste, C.; Colle, I.; Van Vlierberghe, H. Angiogenesis in chronic liver disease and its complications. Liver Int. Off. J. Int. Assoc. Study Liver 2011, 31, 146–162. [Google Scholar] [CrossRef]
- Zhang, Q.; Jiang, K.; Li, Y.; Gao, D.; Sun, L.; Zhang, S.; Liu, T.; Guo, K.; Liu, Y. Histidine-rich glycoprotein function in hepatocellular carcinoma depends on its N-glycosylation status, and it regulates cell proliferation by inhibiting Erk1/2 phosphorylation. Oncotarget 2015, 6, 30222–30231. [Google Scholar] [CrossRef]
- Wang, S.F.; Wung, C.H.; Chen, M.S.; Chen, C.F.; Yin, P.H.; Yeh, T.S.; Chang, Y.L.; Chou, Y.C.; Hung, H.H.; Lee, H.C. Activated Integrated Stress Response Induced by Salubrinal Promotes Cisplatin Resistance in Human Gastric Cancer Cells via Enhanced xCT Expression and Glutathione Biosynthesis. Int. J. Mol. Sci. 2018, 19, 3389. [Google Scholar] [CrossRef] [Green Version]
- Balsa-Martinez, E.; Puigserver, P. Cancer Cells Hijack Gluconeogenic Enzymes to Fuel Cell Growth. Mol. Cell 2015, 60, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, E.E.; Sergushichev, A.; Griss, T.; Gingras, M.C.; Samborska, B.; Ntimbane, T.; Coelho, P.P.; Blagih, J.; Raissi, T.C.; Choiniere, L.; et al. Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth. Mol. Cell 2015, 60, 195–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leithner, K.; Hrzenjak, A.; Trotzmuller, M.; Moustafa, T.; Kofeler, H.C.; Wohlkoenig, C.; Stacher, E.; Lindenmann, J.; Harris, A.L.; Olschewski, A.; et al. PCK2 activation mediates an adaptive response to glucose depletion in lung cancer. Oncogene 2015, 34, 1044–1050. [Google Scholar] [CrossRef]
- Luo, S.; Li, Y.; Ma, R.; Liu, J.; Xu, P.; Zhang, H.; Tang, K.; Ma, J.; Liu, N.; Zhang, Y.; et al. Downregulation of PCK2 remodels tricarboxylic acid cycle in tumor-repopulating cells of melanoma. Oncogene 2017, 36, 3609–3617. [Google Scholar] [CrossRef]
- Liu, M.X.; Jin, L.; Sun, S.J.; Liu, P.; Feng, X.; Cheng, Z.L.; Liu, W.R.; Guan, K.L.; Shi, Y.H.; Yuan, H.X.; et al. Metabolic reprogramming by PCK1 promotes TCA cataplerosis, oxidative stress and apoptosis in liver cancer cells and suppresses hepatocellular carcinoma. Oncogene 2018, 37, 1637–1653. [Google Scholar] [CrossRef]
- Niranjan, B.; Bhat, N.; Avadhani, N. Preferential attack of mitochondrial DNA by aflatoxin B1 during hepatocarcinogenesis. Science 1982, 215, 73–75. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, W. Aflatoxin B1 impairs mitochondrial functions, activates ROS generation, induces apoptosis and involves Nrf2 signal pathway in primary broiler hepatocytes. Anim. Sci. J. 2016, 87, 1490–1500. [Google Scholar] [CrossRef]
- Grasmann, G.; Smolle, E.; Olschewski, H.; Leithner, K. Gluconeogenesis in cancer cells—Repurposing of a starvation-induced metabolic pathway? Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 24–36. [Google Scholar] [CrossRef]
- Tanya, B.; Troup, D.B.; Wilhite, S.E.; Pierre, L.; Carlos, E.; Kim, I.F.; Maxim, T.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M. NCBI GEO: Archive for functional genomics data sets—10 years on. Nucleic Acids Res. 2013, 39, 1005–1010. [Google Scholar]
- Tryndyak, V.; Borowa-Mazgaj, B.; Beland, F.A.; Pogribny, I.P. Gene expression and cytosine DNA methylation alterations in induced pluripotent stem-cell-derived human hepatocytes treated with low doses of chemical carcinogens. Archive Für Toxikologie 2019, 93, 3335–3344. [Google Scholar] [CrossRef] [PubMed]
- Makowska, Z.; Boldanova, T.; Adametz, D.; Quagliata, L.; Vogt, J.E.; Dill, M.T.; Matter, M.S.; Roth, V.; Terracciano, L.; Heim, M.H. Gene expression analysis of biopsy samples reveals critical limitations of transcriptome-based molecular classifications of hepatocellular carcinoma. J. Pathol. Clin. Res. 2016, 2, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, M.P.H.; Porter, M.A. Critical Truths About Power Laws. Science 2012, 335, 665–666. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.S.; Yang, S.K.; Lan, C.Y.; Chuang, Y.J. A systems biology approach to construct the gene regulatory network of systemic inflammation via microarray and databases mining. BMC Med. Genom. 2008, 1, 46. [Google Scholar] [CrossRef] [PubMed]
- Bi, D.; Ning, H.; Liu, S.; Que, X.; Ding, K. Chemistry, Gene expression patterns combined with network analysis identify hub genes associated with bladder cancer. Comput. Biol. Chem. 2015, 56, 71–83. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Da, W.H.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44. [Google Scholar]
- Zweig, A.S.; Karolchik, D.; Kuhn, R.M.; Haussler, D.; Kent, W.J. UCSC genome browser tutorial. Genom. 2008, 92, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Menyhart, O.; Nagy, A.; Gyorffy, B. Determining consistent prognostic biomarkers of overall survival and vascular invasion in hepatocellular carcinoma. R. Soc. Open Sci. 2018, 5, 181006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyorffy, B.; Lanczky, A.; Szallasi, Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr. Relat. Cancer 2012, 19, 197–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, P.; Zheng, H.; She, J.; Feng, N.; Zou, H.; Gu, J.; Yuan, Y.; Liu, X.; Liu, Z.; Bian, J. Molecular Mechanism of Aflatoxin-Induced Hepatocellular Carcinoma Derived from a Bioinformatics Analysis. Toxins 2020, 12, 203. https://doi.org/10.3390/toxins12030203
Cai P, Zheng H, She J, Feng N, Zou H, Gu J, Yuan Y, Liu X, Liu Z, Bian J. Molecular Mechanism of Aflatoxin-Induced Hepatocellular Carcinoma Derived from a Bioinformatics Analysis. Toxins. 2020; 12(3):203. https://doi.org/10.3390/toxins12030203
Chicago/Turabian StyleCai, Peirong, Hao Zheng, Jinjin She, Nannan Feng, Hui Zou, Jianhong Gu, Yan Yuan, Xuezhong Liu, Zongping Liu, and Jianchun Bian. 2020. "Molecular Mechanism of Aflatoxin-Induced Hepatocellular Carcinoma Derived from a Bioinformatics Analysis" Toxins 12, no. 3: 203. https://doi.org/10.3390/toxins12030203
APA StyleCai, P., Zheng, H., She, J., Feng, N., Zou, H., Gu, J., Yuan, Y., Liu, X., Liu, Z., & Bian, J. (2020). Molecular Mechanism of Aflatoxin-Induced Hepatocellular Carcinoma Derived from a Bioinformatics Analysis. Toxins, 12(3), 203. https://doi.org/10.3390/toxins12030203