Centipede Venom Peptides Acting on Ion Channels



Abstract
:1. Introduction
2. Centipede Toxins as an Abundant Source of Drug Leads
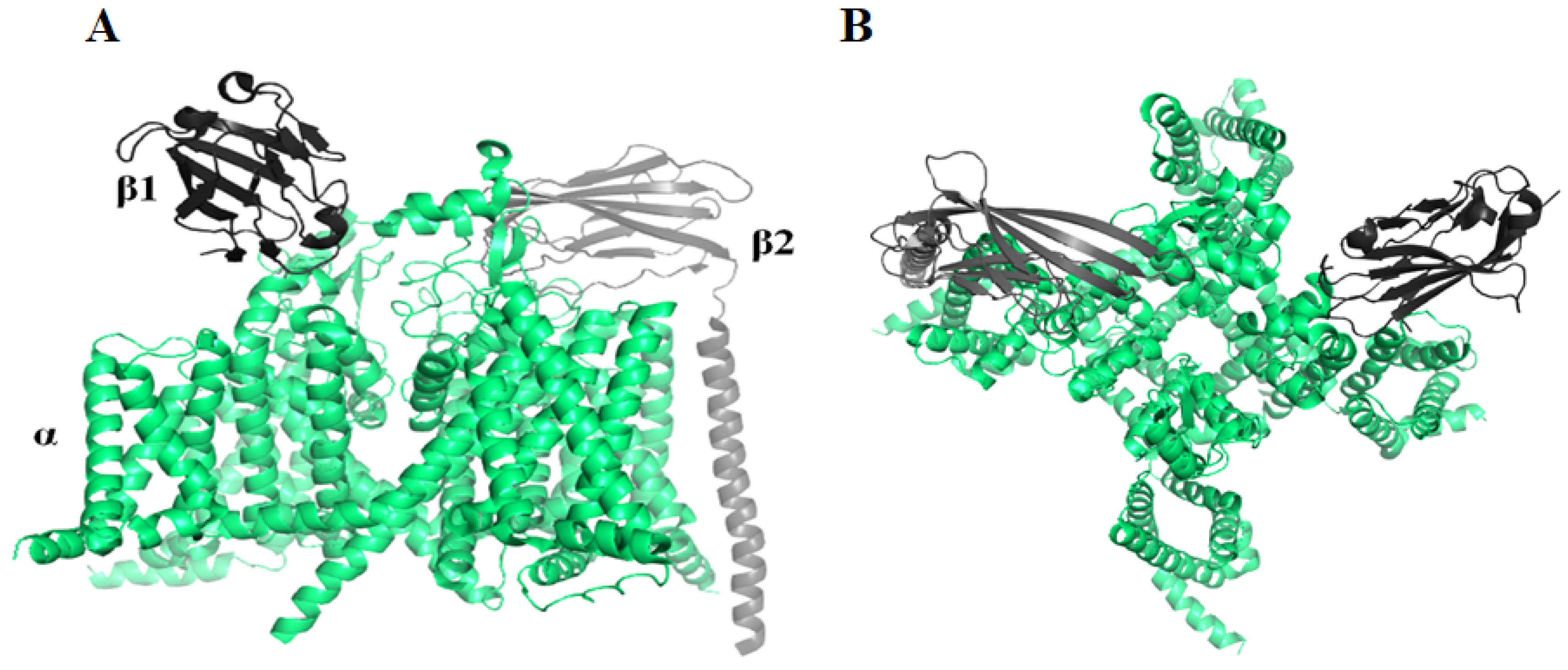
2.1. Voltage-Gated Sodium Channel (Nav) Blocker
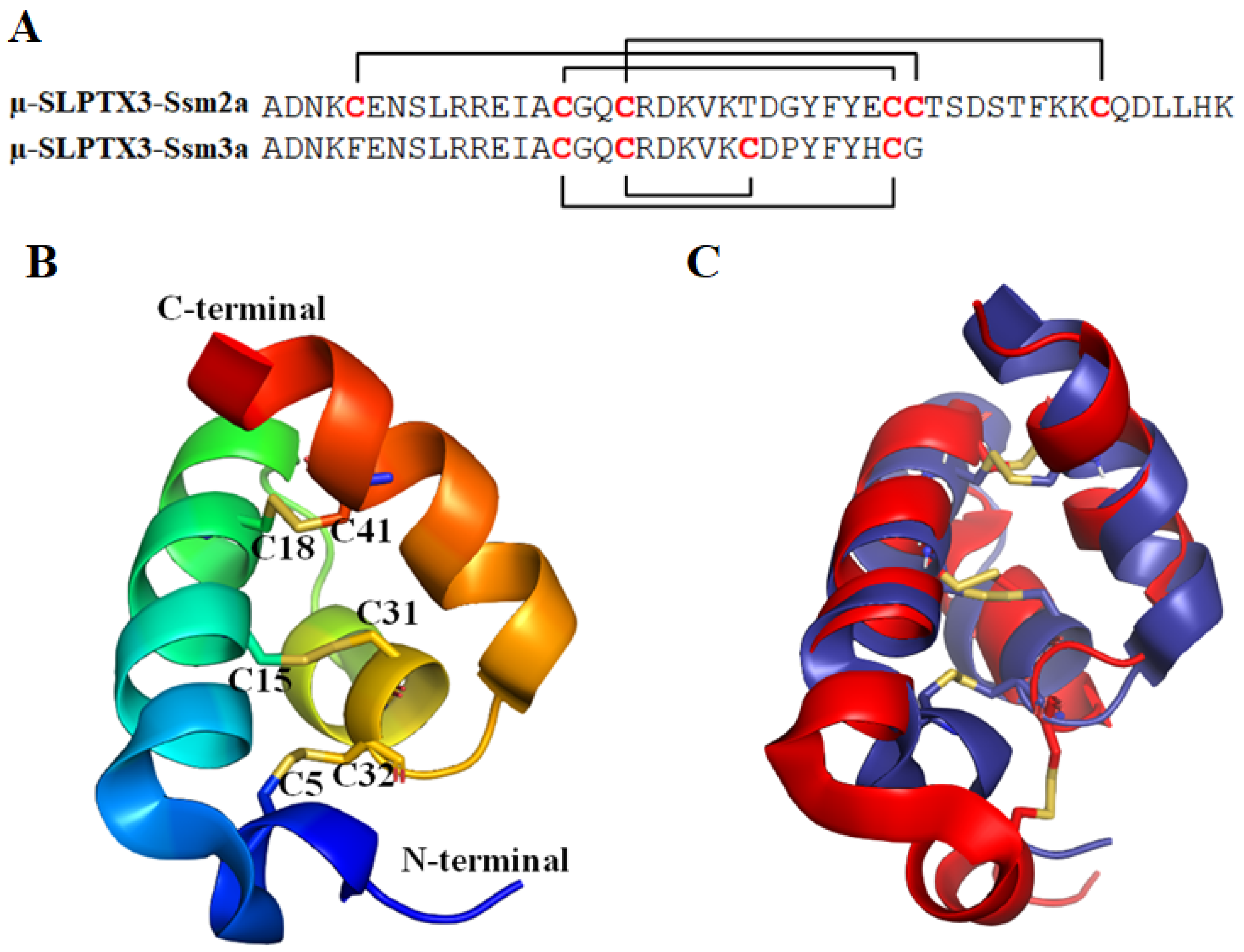
2.1.1. μ-SLPTX3-Ssm2a
2.1.2. μ-SLPTX3-Ssm3a
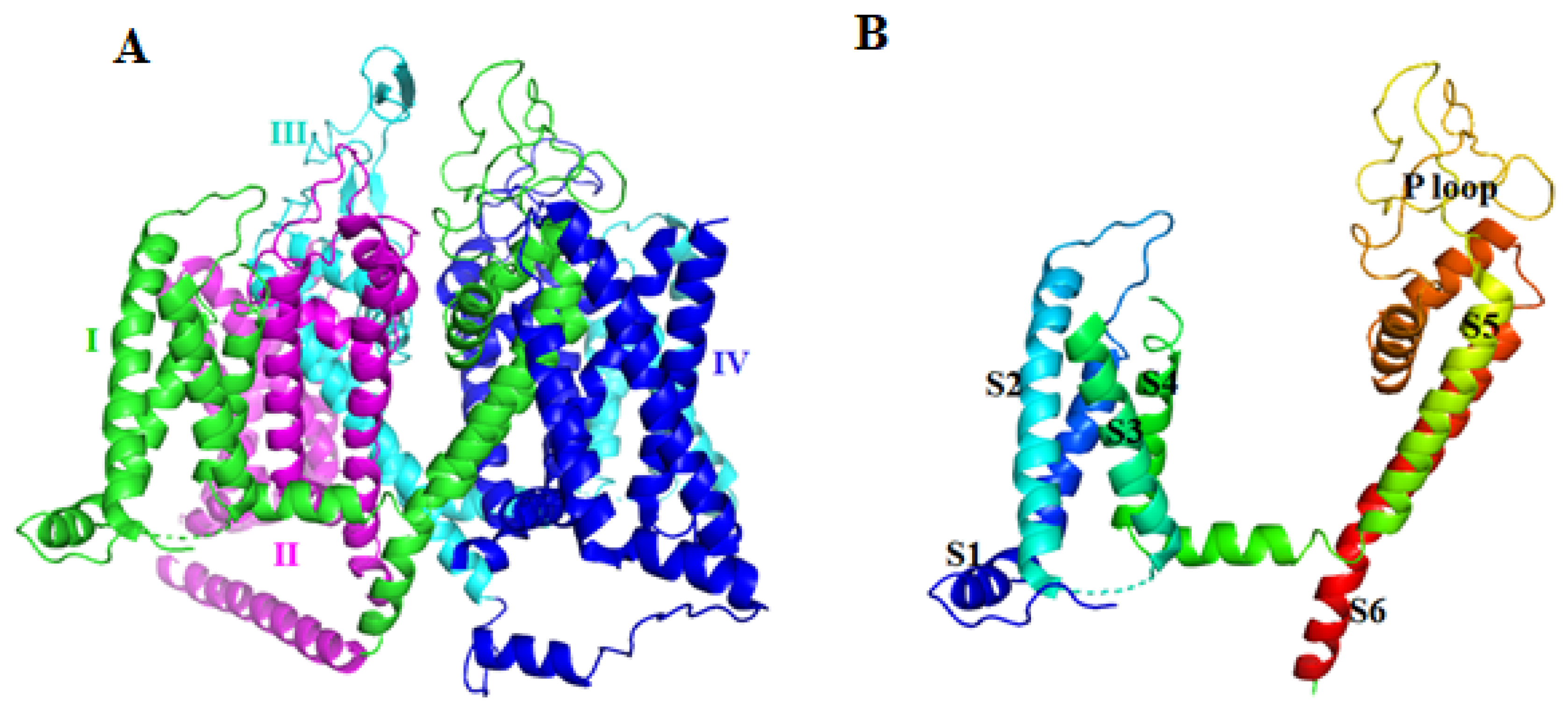
2.2. Voltage-Gated Potassium Channel (Kv) Inhibitor
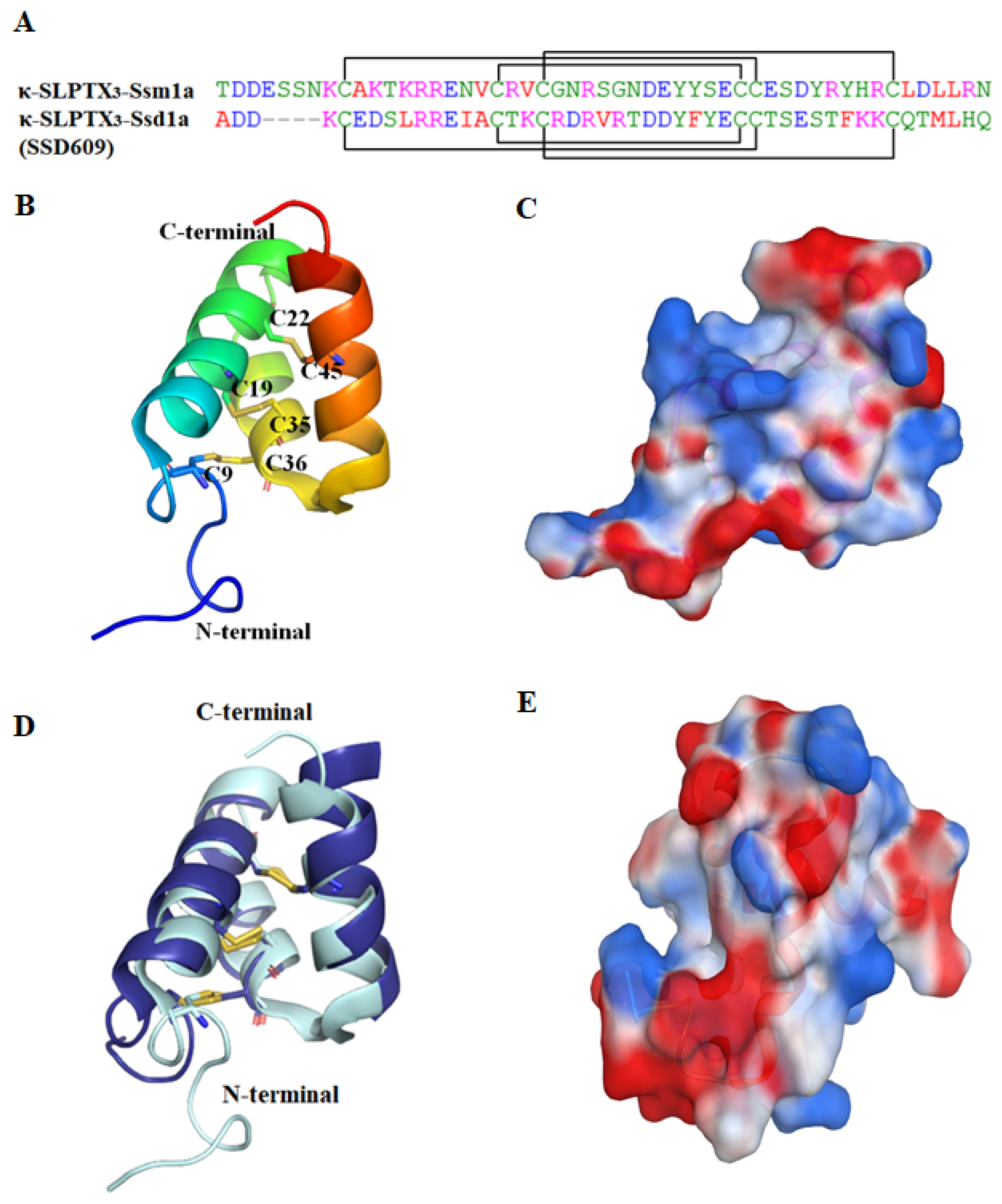
2.2.1. κ-SLPTX3-Ssm1a
2.2.2. SSD609
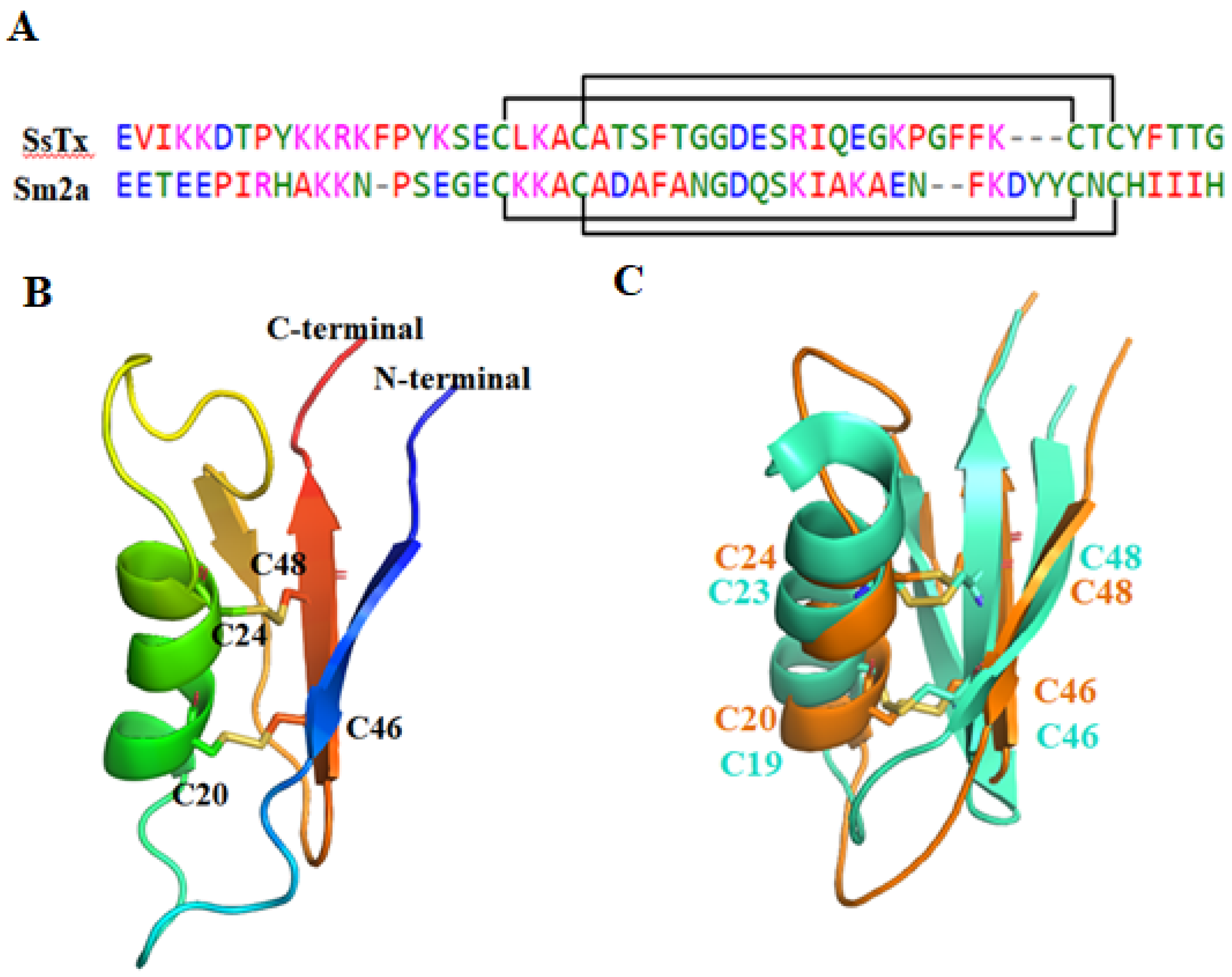
2.2.3. SsTx
2.2.4. κ-SLPTX7-Ssm2a
2.2.5. κ-SLPTX11-Ssm3a
2.2.6. κ-SLPTX15-Ssd2a
2.2.7. SsmTx-1
2.3. Voltage-Gated Calcium Channel (Cav) Modulator
2.3.1. ω-SLPTX5-Ssm1a
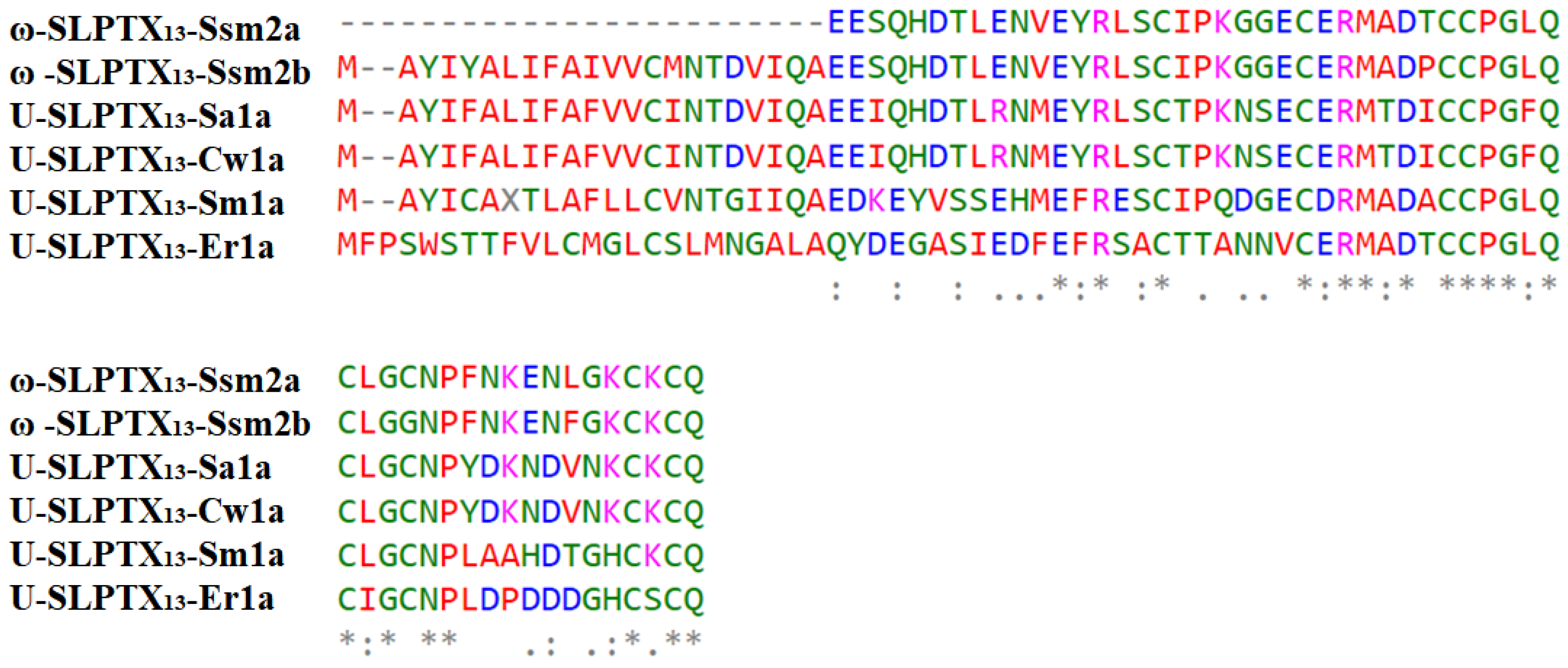
2.3.2. ω-SLPTX13-Ssm2a
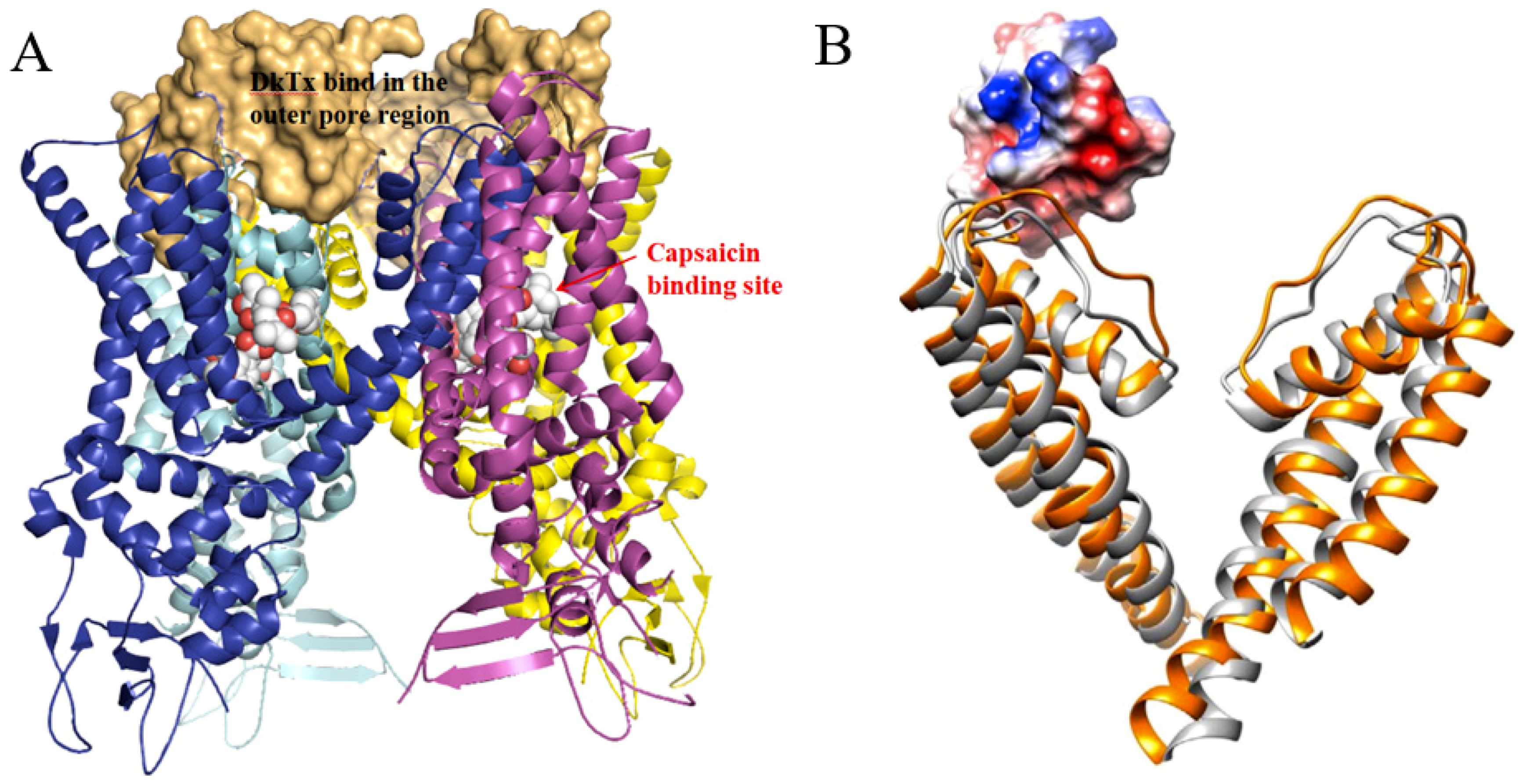
2.4. TRPV1 Activator
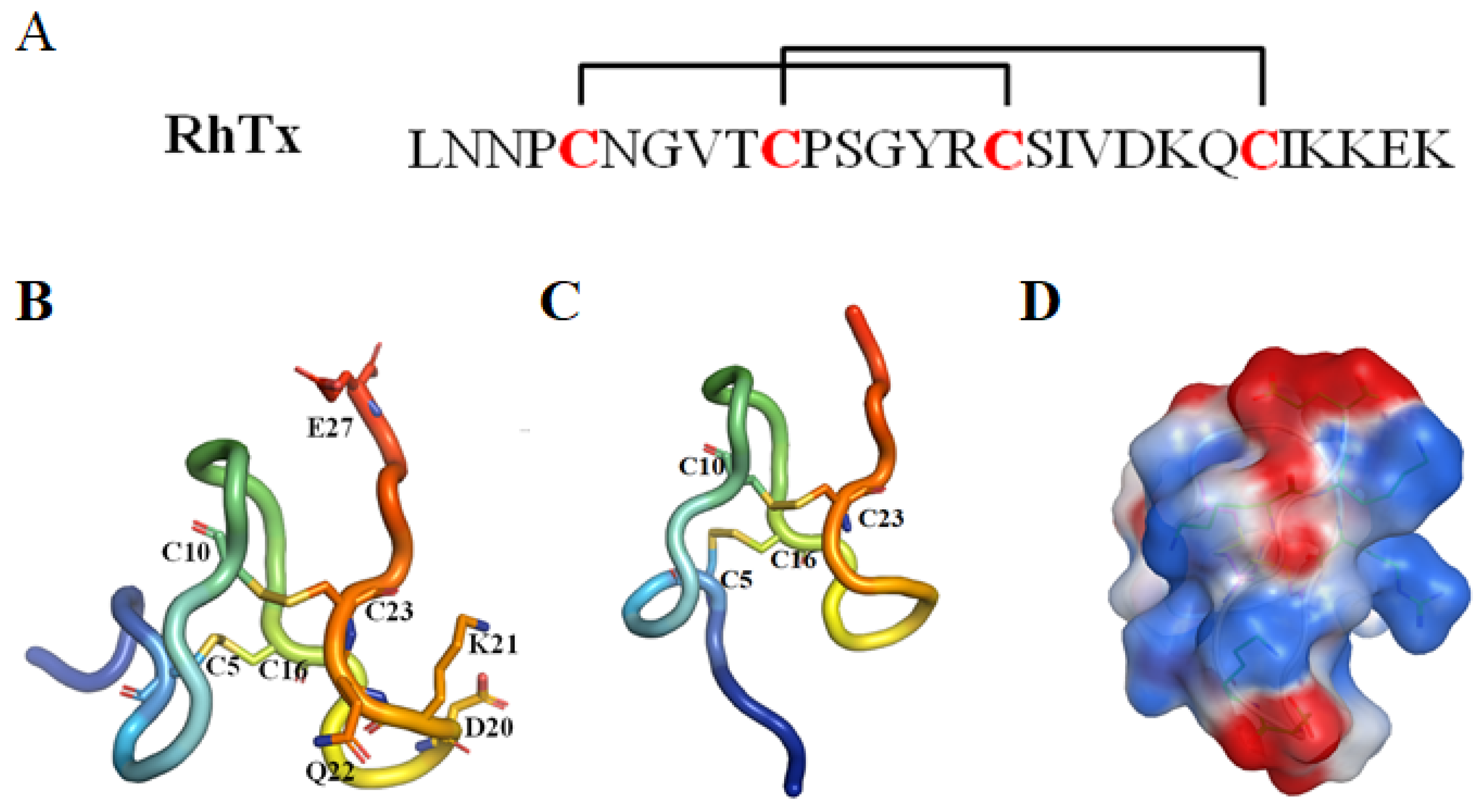
RhTx
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Undheim, E.A.; Jones, A.; Clauser, K.R.; Holland, J.W.; Pineda, S.S.; King, G.F.; Fry, B.G. Clawing through evolution: Toxin diversification and convergence in the ancient lineage Chilopoda (centipedes). Mol. Biol. Evol. 2014, 31, 2124–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siriwut, W.; Edgecombe, G.D.; Sutcharit, C.; Tongkerd, P.; Panha, S. A taxonomic review of the centipede genus Scolopendra Linnaeus, 1758 (Scolopendromorpha, Scolopendridae) in mainland Southeast Asia, with description of a new species from Laos. Zookeys 2016, 1–124. [Google Scholar] [CrossRef] [Green Version]
- Undheim, E.A.; King, G.F. On the venom system of centipedes (Chilopoda), a neglected group of venomous animals. Toxicon 2011, 57, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Edgecombe, G.D.; Giribet, G. Evolutionary biology of centipedes (Myriapoda: Chilopoda). Annu. Rev. Entomol. 2007, 52, 151–170. [Google Scholar] [CrossRef] [Green Version]
- Veraldi, S.; Cuka, E.; Gaiani, F. Scolopendra bites: A report of two cases and review of the literature. Int. J. Dermatol. 2014, 53, 869–872. [Google Scholar] [CrossRef]
- Rates, B.; Bemquerer, M.P.; Richardson, M.; Borges, M.H.; Morales, R.A.; De Lima, M.E.; Pimenta, A.M. Venomic analyses of Scolopendra viridicornis nigra and Scolopendra angulata (Centipede, Scolopendromorpha): Shedding light on venoms from a neglected group. Toxicon 2007, 49, 810–826. [Google Scholar] [CrossRef]
- Undheim, E.A.; Fry, B.G.; King, G.F. Centipede venom: Recent discoveries and current state of knowledge. Toxins (Basel) 2015, 7, 679–704. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Lan, X.; Li, T.; Xiang, Y.; Zhao, F.; Zhang, Y.; Lee, W.H. Proteotranscriptomic analysis and discovery of the profile and diversity of toxin-like proteins in centipede. Mol. Cell. Proteomics 2018, 17, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Sonoda, Y.; Hada, N.; Kaneda, T.; Suzuki, T.; Ohshio, T.; Takeda, T.; Kasahara, T. A synthetic glycosphingolipid-induced antiproliferative effect in melanoma cells is associated with suppression of FAK, Akt, and Erk activation. Biol. Pharm. Bull 2008, 31, 1279–1283. [Google Scholar] [CrossRef] [Green Version]
- Rong, M.; Yang, S.; Wen, B.; Mo, G.; Kang, D.; Liu, J.; Lin, Z.; Jiang, W.; Li, B.; Du, C.; et al. Peptidomics combined with cDNA library unravel the diversity of centipede venom. J. Proteomics 2015, 114, 28–37. [Google Scholar] [CrossRef]
- Hakim, M.A.; Yang, S.; Lai, R. Centipede venoms and their components: Resources for potential therapeutic applications. Toxins (Basel) 2015, 7, 4832–4851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.M.; Khan, N.A.; Sagathevan, K.; Anwar, A.; Siddiqui, R. Biologically active metabolite(s) from haemolymph of red-headed centipede Scolopendra subspinipes possess broad spectrum antibacterial activity. AMB Express 2019, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, R.W. Insects and other arthropods used as drugs in Korean traditional medicine. J. Ethnopharmacol. 1999, 65, 207–216. [Google Scholar] [CrossRef]
- Malta, M.B.; Lira, M.S.; Soares, S.L.; Rocha, G.C.; Knysak, I.; Martins, R.; Guizze, S.P.; Santoro, M.L.; Barbaro, K.C. Toxic activities of Brazilian centipede venoms. Toxicon 2008, 52, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Fung, H.T.; Lam, S.K.; Wong, O.F. Centipede bite victims: A review of patients presenting to two emergency departments in Hong Kong. Hong Kong Med. J. 2011, 17, 381–385. [Google Scholar] [PubMed]
- Balit, C.R.; Harvey, M.S.; Waldock, J.M.; Isbister, G.K. Prospective study of centipede bites in Australia. J. Toxicol. Clin. Toxicol. 2004, 42, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, A.; Biceroglu, S.; Yakut, N.; Bilir, C.; Akdemir, R.; Akilli, A. Acute myocardial infarction in a young man caused by centipede sting. Emerg. Med. J. 2006, 23, e30. [Google Scholar] [CrossRef]
- Dunbar, J.P.; Sulpice, R.; Dugon, M.M. The kiss of (cell) death: Can venom-induced immune response contribute to dermal necrosis following arthropod envenomations? Clin. Toxicol. (Phila) 2019, 57, 677–685. [Google Scholar] [CrossRef]
- Ozsarac, M.; Karcioglu, O.; Ayrik, C.; Somuncu, F.; Gumrukcu, S. Acute coronary ischemia following centipede envenomation: Case report and review of the literature. Wilderness Environ. Med. 2004, 15, 109–112. [Google Scholar] [CrossRef]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [Google Scholar] [CrossRef]
- Robinson, S.D.; Undheim, E.A.B.; Ueberheide, B.; King, G.F. Venom peptides as therapeutics: Advances, challenges and the future of venom-peptide discovery. Expert Rev. Proteomics 2017, 14, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Cushman, D.W.; Ondetti, M.A. Design of angiotensin converting enzyme inhibitors. Nat. Med. 1999, 5, 1110–1113. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.P.; Keating, G.M. Eptifibatide: A review of its use in patients with acute coronary syndromes and/or undergoing percutaneous coronary intervention. Drugs 2005, 65, 2009–2035. [Google Scholar] [CrossRef] [PubMed]
- Perumal Samy, R.; Stiles, B.G.; Franco, O.L.; Sethi, G.; Lim, L.H.K. Animal venoms as antimicrobial agents. Biochem. Pharmacol. 2017, 134, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Miljanich, G.P. Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Shanmughapriya, S.; Mok, M.C.Y.; Dong, Z.; Tomar, D.; Carvalho, E.; Rajan, S.; Junop, M.S.; Madesh, M.; Stathopulos, P.B. Structural insights into mitochondrial calcium uniporter regulation by divalent cations. Cell Chem. Biol. 2016, 23, 1157–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peigneur, S.; Tytgat, J. Toxins in drug discovery and pharmacology. Toxins (Basel) 2018, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Undheim, E.A.; Jenner, R.A.; King, G.F. Centipede venoms as a source of drug leads. Expert Opin. Drug Discov. 2016, 11, 1139–1149. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.W.; Serrano, S.M. Approaching the golden age of natural product pharmaceuticals from venom libraries: An overview of toxins and toxin-derivatives currently involved in therapeutic or diagnostic applications. Curr. Pharm. Des. 2007, 13, 2927–2934. [Google Scholar] [CrossRef] [Green Version]
- Tarcha, E.J.; Olsen, C.M.; Probst, P.; Peckham, D.; Munoz-Elias, E.J.; Kruger, J.G.; Iadonato, S.P. Safety and pharmacodynamics of dalazatide, a Kv1.3 channel inhibitor, in the treatment of plaque psoriasis: A randomized phase 1b trial. PLoS ONE 2017, 12, e0180762. [Google Scholar] [CrossRef]
- Saez, N.J.; Senff, S.; Jensen, J.E.; Er, S.Y.; Herzig, V.; Rash, L.D.; King, G.F. Spider-venom peptides as therapeutics. Toxins (Basel) 2010, 2, 2851–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, T.S.; Teichert, R.W.; Olivera, B.M.; Bulaj, G. Conus venoms - a rich source of peptide-based therapeutics. Curr. Pharm. Des. 2008, 14, 2462–2479. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.; Davis, J.L.; Rash, L.D.; Anangi, R.; Mobli, M.; Alewood, P.F.; Lewis, R.J.; King, G.F. Venomics: A new paradigm for natural products-based drug discovery. Amino Acids 2011, 40, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.K.; Lewis, R.J.; Alewood, D.; Drinkwater, R.; Palant, E.; Patterson, M.; Yaksh, T.L.; McCumber, D.; Smith, M.T. Anti-allodynic efficacy of the chi-conopeptide, Xen2174, in rats with neuropathic pain. Pain 2005, 118, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Trammel, J.; Wasiewski, W.W.; Ancrod Stroke Program (ASP) Study Team. Ancrod for acute ischemic stroke: A new dosing regimen derived from analysis of prior ancrod stroke studies. J. Stroke Cerebrovasc. Dis. 2009, 18, 23–27. [Google Scholar] [CrossRef]
- Mamelak, A.N.; Jacoby, D.B. Targeted delivery of antitumoral therapy to glioma and other malignancies with synthetic chlorotoxin (TM-601). Expert Opin. Drug Deliv. 2007, 4, 175–186. [Google Scholar] [CrossRef]
- Harvey, A.L. Toxins and drug discovery. Toxicon 2014, 92, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Dibas, A.; Millar, C.; Al-Farra, A.; Yorio, T. Neuroprotective effects of psalmotoxin-1, an acid-sensing ion channel (ASIC) inhibitor, in ischemia reperfusion in mouse eyes. Curr. Eye Res. 2018, 43, 921–933. [Google Scholar] [CrossRef]
- Liu, Z.C.; Zhang, R.; Zhao, F.; Chen, Z.M.; Liu, H.W.; Wang, Y.J.; Jiang, P.; Zhang, Y.; Wu, Y.; Ding, J.P.; et al. Venomic and transcriptomic analysis of centipede Scolopendra subspinipes dehaani. J. Proteome Res. 2012, 11, 6197–6212. [Google Scholar] [CrossRef]
- Gonzalez-Morales, L.; Pedraza-Escalona, M.; Diego-Garcia, E.; Restano-Cassulini, R.; Batista, C.V.; Gutierrez Mdel, C.; Possani, L.D. Proteomic characterization of the venom and transcriptomic analysis of the venomous gland from the Mexican centipede Scolopendra viridis. J. Proteomics 2014, 111, 224–237. [Google Scholar] [CrossRef]
- Jimenez-Vargas, J.M.; Possani, L.D.; Luna-Ramirez, K. Arthropod toxins acting on neuronal potassium channels. Neuropharmacology 2017, 127, 139–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xiao, Y.; Kang, D.; Liu, J.; Li, Y.; Undheim, E.A.; Klint, J.K.; Rong, M.; Lai, R.; King, G.F. Discovery of a selective Nav1.7 inhibitor from centipede venom with analgesic efficacy exceeding morphine in rodent pain models. Proc. Natl. Acad. Sci. USA 2013, 110, 17534–17539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Liu, Z.; Xiao, Y.; Li, Y.; Rong, M.; Liang, S.; Zhang, Z.; Yu, H.; King, G.F.; Lai, R. Chemical punch packed in venoms makes centipedes excellent predators. Mol. Cell. Proteomics 2012, 11, 640–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, P.; Wu, F.; Wen, M.; Yang, X.; Wang, C.; Li, Y.; He, S.; Zhang, L.; Zhang, Y.; Tian, C. A distinct three-helix centipede toxin SSD609 inhibits Iks channels by interacting with the KCNE1 auxiliary subunit. Sci. Rep. 2015, 5, 13399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Li, B.; Wang, S.; Wu, F.; Wang, X.; Liang, P.; Ombati, R.; Chen, J.; Lu, X.; Cui, J.; et al. Centipedes subdue giant prey by blocking KCNQ channels. Proc. Natl. Acad. Sci. USA 2018, 115, 1646–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, C.; Li, J.; Shao, Z.; Mwangi, J.; Xu, R.; Tian, H.; Mo, G.; Lai, R.; Yang, S. Centipede KCNQ inhibitor SsTx also targets Kv1.3. Toxins (Basel) 2019, 11, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Li, J.; Zhang, F.; Liu, Z. Isolation and characterization of SsmTx-I, a specific Kv2.1 blocker from the venom of the centipede Scolopendra Subspinipes Mutilans L. Koch. J. Pept. Sci. 2014, 20, 159–164. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.; Yang, M.; Wu, C.; Zou, Z.; Tang, J.; Yang, X. Centipede venom peptide SsmTX-I with two intramolecular disulfide bonds shows analgesic activities in animal models. J. Pept. Sci. 2017, 23, 384–391. [Google Scholar] [CrossRef]
- Yang, S.; Yang, F.; Wei, N.; Hong, J.; Li, B.; Luo, L.; Rong, M.; Yarov-Yarovoy, V.; Zheng, J.; Wang, K.; et al. A pain-inducing centipede toxin targets the heat activation machinery of nociceptor TRPV1. Nat. Commun. 2015, 6, 8297. [Google Scholar] [CrossRef] [Green Version]
- Ombati, R.; Luo, L.; Yang, S.; Lai, R. Centipede envenomation: Clinical importance and the underlying molecular mechanisms. Toxicon 2018, 154, 60–68. [Google Scholar] [CrossRef]
- Geron, M.; Hazan, A.; Priel, A. Animal toxins providing insights into TRPV1 activation mechanism. Toxins (Basel) 2017, 9, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Shao, Y.; Chen, H.; Ming, X.; Wang, J.B.; Li, Z.Y.; Wei, J.F. A novel factor Xa-inhibiting peptide from centipedes venom. Int. J. Pept. Res. Ther. 2013, 19, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, K.; Kong, Y.; Zhai, L.; Wu, X.; Jia, P.; Liu, J.; Yu, H. Two novel antimicrobial peptides from centipede venoms. Toxicon 2010, 55, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Yan, W.; Du, K.; Ye, Y.; Cao, Q.; Ren, W. Construction and expression of an antimicrobial peptide scolopin 1 from the centipede venoms of Scolopendra subspinipes mutilans in Escherichia coli using SUMO fusion partner. Protein Expr. Purif. 2013, 92, 230–234. [Google Scholar] [CrossRef]
- Rivara, M.; Zuliani, V. Novel sodium channel antagonists in the treatment of neuropathic pain. Expert Opin. Investig. Drugs 2016, 25, 215–226. [Google Scholar] [CrossRef]
- Kushnarev, M.; Pirvulescu, I.P.; Candido, K.D.; Knezevic, N.N. Neuropathic pain: Preclinical and early clinical progress with voltage-gated sodium channel blockers. Expert Opin. Investig. Drugs 2020, 29, 259–271. [Google Scholar] [CrossRef]
- Xu, L.; Ding, X.; Wang, T.; Mou, S.; Sun, H.; Hou, T. Voltage-gated sodium channels: Structures, functions, and molecular modeling. Drug Discov. Today 2019, 24, 1389–1397. [Google Scholar] [CrossRef]
- Tikhonov, D.B.; Zhorov, B.S. Mechanism of sodium channel block by local anesthetics, antiarrhythmics, and anticonvulsants. J. Gen. Physiol. 2017, 149, 465–481. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Waxman, S.G. Diversity of composition and function of sodium channels in peripheral sensory neurons. Pain 2015, 156, 2406–2407. [Google Scholar]
- Ma, R.S.Y.; Kayani, K.; Whyte-Oshodi, D.; Whyte-Oshodi, A.; Nachiappan, N.; Gnanarajah, S.; Mohammed, R. Voltage gated sodium channels as therapeutic targets for chronic pain. J. Pain Res. 2019, 12, 2709–2722. [Google Scholar] [CrossRef] [Green Version]
- Leipold, E.; Hanson-Kahn, A.; Frick, M.; Gong, P.; Bernstein, J.A.; Voigt, M.; Katona, I.; Oliver Goral, R.; Altmuller, J.; Nurnberg, P.; et al. Cold-aggravated pain in humans caused by a hyperactive Nav1.9 channel mutant. Nat. Commun. 2015, 6, 10049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, E.C.; Luiz, A.P.; Wood, J.N. Nav1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin. Ther. Targets 2016, 20, 975–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dib-Hajj, S.D.; Black, J.A.; Waxman, S.G. Nav1.9: A sodium channel linked to human pain. Nat. Rev. Neurosci. 2015, 16, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Huang, J.; Waxman, S.G. Sodium channel Nav1.8: Emerging links to human disease. Neurology 2016, 86, 473–483. [Google Scholar] [CrossRef]
- Xu, H.; Li, T.; Rohou, A.; Arthur, C.P.; Tzakoniati, F.; Wong, E.; Estevez, A.; Kugel, C.; Franke, Y.; Chen, J.; et al. Structural basis of Nav1.7 inhibition by a gating-modifier spider toxin. Cell 2019, 176, 702–715. [Google Scholar] [CrossRef] [Green Version]
- Undheim, E.A.; Grimm, L.L.; Low, C.F.; Morgenstern, D.; Herzig, V.; Zobel-Thropp, P.; Pineda, S.S.; Habib, R.; Dziemborowicz, S.; Fry, B.G.; et al. Weaponization of a hormone: Convergent recruitment of hyperglycemic hormone into the venom of arthropod predators. Structure 2015, 23, 1283–1292. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Liu, D.; Wu, K.; Lei, J.; Yan, N. Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science 2019, 363, 1303–1308. [Google Scholar] [CrossRef]
- Wang, C.; Shan, B.; Wang, Q.; Xu, Q.; Zhang, H.; Lei, H. Fusion of Ssm6a with a protein scaffold retains selectivity on Nav 1.7 and improves its therapeutic potential against chronic pain. Chem. Biol. Drug Des. 2017, 89, 825–833. [Google Scholar] [CrossRef]
- Murray, J.K.; Ligutti, J.; Liu, D.; Zou, A.; Poppe, L.; Li, H.; Andrews, K.L.; Moyer, B.D.; McDonough, S.I.; Favreau, P.; et al. Engineering potent and selective analogues of GpTx-1, a tarantula venom peptide antagonist of the Nav1.7 sodium channel. J. Med. Chem. 2015, 58, 2299–2314. [Google Scholar] [CrossRef]
- Matthies, D.; Bae, C.; Toombes, G.E.; Fox, T.; Bartesaghi, A.; Subramaniam, S.; Swartz, K.J. Single-particle cryo-EM structure of a voltage-activated potassium channel in lipid nanodiscs. Elife 2018, 7, e37558. [Google Scholar] [CrossRef]
- Whicher, J.R.; MacKinnon, R. Structure of the voltage-gated K+ channel Eag1 reveals an alternative voltage sensing mechanism. Science 2016, 353, 664–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grizel, A.V.; Glukhov, G.S.; Sokolova, O.S. Mechanisms of activation of voltage-gated potassium channels. Acta Naturae 2014, 6, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lin, Z.; Mattmann, M.E.; Zou, B.; Terrenoire, C.; Zhang, H.; Wu, M.; McManus, O.B.; Kass, R.S.; Lindsley, C.W.; et al. Dynamic subunit stoichiometry confers a progressive continuum of pharmacological sensitivity by KCNQ potassium channels. Proc. Natl. Acad. Sci. USA 2013, 110, 8732–8737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajo, K.; Kubo, Y. KCNQ1 channel modulation by KCNE proteins via the voltage-sensing domain. J. Physiol. 2015, 593, 2617–2625. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Xiong, Q.; Sun, H.; Li, M. Desensitization of chemical activation by auxiliary subunits: Convergence of molecular determinants critical for augmenting KCNQ1 potassium channels. J. Biol. Chem. 2008, 283, 22649–22658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pusch, M.; Magrassi, R.; Wollnik, B.; Conti, F. Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys. J. 1998, 75, 785–792. [Google Scholar] [CrossRef] [Green Version]
- Dash, T.S.; Shafee, T.; Harvey, P.J.; Zhang, C.; Peigneur, S.; Deuis, J.R.; Vetter, I.; Tytgat, J.; Anderson, M.A.; Craik, D.J.; et al. A centipede toxin family defines an ancient class of CSalphabeta defensins. Structure 2019, 27, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, F.; Flockerzi, V.; Kahl, S.; Wegener, J.W. L-type Cav1.2 calcium channels: From in vitro findings to in vivo function. Physiol. Rev. 2014, 94, 303–326. [Google Scholar] [CrossRef] [Green Version]
- Andrade, A.; Brennecke, A.; Mallat, S.; Brown, J.; Gomez-Rivadeneira, J.; Czepiel, N.; Londrigan, L. Genetic associations between voltage-gated calcium channels and psychiatric disorders. Int. J. Mol. Sci. 2019, 20, 3537. [Google Scholar] [CrossRef] [Green Version]
- Striessnig, J.; Ortner, N.J.; Pinggera, A. Pharmacology of L-type calcium channels: Novel drugs for old targets? Curr. Mol. Pharmacol. 2015, 8, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.R.; Vetter, I.; Lewis, R.J. Venom peptides as a rich source of cav2.2 channel blockers. Toxins (Basel) 2013, 5, 286–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A.; Lenaeus, M.J.; Gamal El-Din, T.M. Structure and pharmacology of voltage-gated sodium and calcium channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 133–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Ortiz, W.; Cardozo, T.J. An Improved method for modeling voltage-gated ion channels at atomic accuracy applied to human Cav channels. Cell Rep. 2018, 23, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Decher, N.; Kumar, P.; Sachse, F.B.; Beggs, A.H.; Sanguinetti, M.C.; Keating, M.T. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 8089–8096. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.; Montagut-Bordas, C.; Dickenson, A.H. Calcium channel modulation as a target in chronic pain control. Br. J. Pharmacol. 2018, 175, 2173–2184. [Google Scholar] [CrossRef]
- Jiang, D.; Gamal El-Din, T.M.; Ing, C.; Lu, P.; Pomes, R.; Zheng, N.; Catterall, W.A. Structural basis for gating pore current in periodic paralysis. Nature 2018, 557, 590–594. [Google Scholar] [CrossRef]
- Zheng, J. Molecular mechanism of TRP channels. Compr. Physiol. 2013, 3, 221–242. [Google Scholar]
- Gavva, N.R.; Treanor, J.J.; Garami, A.; Fang, L.; Surapaneni, S.; Akrami, A.; Alvarez, F.; Bak, A.; Darling, M.; Gore, A.; et al. Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain 2008, 136, 202–210. [Google Scholar] [CrossRef]
- Cao, E.; Liao, M.; Cheng, Y.; Julius, D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 2013, 504, 113–118. [Google Scholar] [CrossRef]
- Levine, J.D.; Alessandri-Haber, N. TRP channels: Targets for the relief of pain. Biochim. Biophys. Acta 2007, 1772, 989–1003. [Google Scholar] [CrossRef] [Green Version]
- Kawamata, T.; Niiyama, Y.; Yamamoto, J.; Furuse, S. Reduction of bone cancer pain by CB1 activation and TRPV1 inhibition. J. Anesth. 2010, 24, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Jardin, I.; Lopez, J.J.; Diez, R.; Sanchez-Collado, J.; Cantonero, C.; Albarran, L.; Woodard, G.E.; Redondo, P.C.; Salido, G.M.; Smani, T.; et al. TRPs in pain sensation. Front. Physiol. 2017, 8, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghilardi, J.R.; Rohrich, H.; Lindsay, T.H.; Sevcik, M.A.; Schwei, M.J.; Kubota, K.; Halvorson, K.G.; Poblete, J.; Chaplan, S.R.; Dubin, A.E.; et al. Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. J. Neurosci. 2005, 25, 3126–3131. [Google Scholar] [CrossRef]
- Lee, B.H.; Zheng, J. Proton block of proton-activated TRPV1 current. J. Gen. Physiol. 2015, 146, 147–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordt, S.E.; Tominaga, M.; Julius, D. Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc. Natl. Acad. Sci. USA 2000, 97, 8134–8139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Ma, L.; Cao, X.; Wang, K.; Zheng, J. Divalent cations activate TRPV1 through promoting conformational change of the extracellular region. J. Gen. Physiol. 2014, 143, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Ahern, G.P.; Brooks, I.M.; Miyares, R.L.; Wang, X.B. Extracellular cations sensitize and gate capsaicin receptor TRPV1 modulating pain signaling. J. Neurosci. 2005, 25, 5109–5116. [Google Scholar] [CrossRef]
- Bohlen, C.J.; Priel, A.; Zhou, S.; King, D.; Siemens, J.; Julius, D. A bivalent tarantula toxin activates the capsaicin receptor, TRPV1, by targeting the outer pore domain. Cell 2010, 141, 834–845. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Toxin (Other Name) | Number of Residues | Disulfide/Cysteine Numbers (Cysteine Pairs) | Bioactivity |
|---|---|---|---|
| μ-SLPTX3-Ssm2a (µ-SLPTX-Ssm6a) | 46 | 3/6 (C5–C32, C15–C31, C18–C41) | Nav1.7, IC50 = 25.4 nM; Nav1.1, IC50 = 4.1 µM; Nav1.2, IC50 = 813 nM; Nav1.6, IC50 = 15.2 µM. No effect on Nav1.3, Nav1.4, Nav1.5, Nav1.8 and hERG [42]. |
| μ-SLPTX3-Ssm3a (μ-SLPTX-Ssm1a) | 32 | 2/4 (-) | Specifically inhibited TTX-S Nav channel current in rat DRGs, IC50 = ~9 nM [11,43]. No effect on TTX-R Nav. |
| κ-SLPTX3-Ssm1a (κ-SLPTX-Ssm1a) | 51 | 3/6 (C9–C36, C19–C35, C22–C45) | Inhibited Kv current in DRG neurons, IC50 = ~44.2 nM [43]. |
| SSD609 (κ-SLPTX3- Ssd1a) | 47 | 3/6 (C5–C32, C15–C31, C18–C41) | Inhibited the channel conductance of Iks, IC50 = 652.7 nM [39,44]. |
| SsTx (μ-SLPTX15-Ssm1a) | 53 | 2/4 (C20–C46, C24–C53) | KV7.4, IC50 = 2.5 µM; Kv7.1, IC50 = 2.8 µM; Kv7.2, IC50 = 2.7 µM; Kv7.5, IC50 = 2.7 µM; Kv1.3, IC50 = 5.26 µM; no inhibition of TRPV1, TRPV2, Kv2.1, Kv4.1, hERG, Nav or Cav in DRG neurons [45,46]. |
| κ-SLPTX7-Ssm2a (κ-SLPTX7-Ssm2a) | 31 | 3/6 (-) | KV, IC50 = ~570 nM [43]. |
| κ-SLPTX11-Ssm3a | 68 | 2/4 (-) | Inhibited Kv current amplitude by 25% at a concentration of 200 nM, did not fully inhibit peak Kv currents even at concentrations up to 5 µM [43]. |
| κ-SLPTX15-Ssd2a | 72 | -/6 (-) | Irreversibly blocked KV currents, IC50 = ~10 nM [39]. |
| SsmTx-1 | 36 | 2/4 (C8–C19, C13–C26) | Kv, IC50 = 200 nM; Kv2.1, IC50 = 41.7 nM. No effect on Nav [47,48]. |
| ω-SLPTX5-Ssm1a | 86 | 3/7 (-) | Cav activator, 1 μM ω-SLPTX5-Ssm1a increased CaV current in DRG neurons by 70%; 10 μM toxin increased Cav current by 120% [7,43]. |
| ω-SLPTX13-Ssm2a | 54 | 4/8 (-) | Cav, IC50 = 1590 nM [43]. |
| RhTx (τ-SLPTX4-Sm1a) | 27 | 2/4 (C5–C16, C10–C23) | TRPV1 activator, EC50 = 521.5 nM. No effect on other TRPV channels [28,49,50,51]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, Y.; Qiu, P.; Yu, R. Centipede Venom Peptides Acting on Ion Channels. Toxins 2020, 12, 230. https://doi.org/10.3390/toxins12040230
Chu Y, Qiu P, Yu R. Centipede Venom Peptides Acting on Ion Channels. Toxins. 2020; 12(4):230. https://doi.org/10.3390/toxins12040230
Chicago/Turabian StyleChu, YanYan, PeiJu Qiu, and RiLei Yu. 2020. "Centipede Venom Peptides Acting on Ion Channels" Toxins 12, no. 4: 230. https://doi.org/10.3390/toxins12040230
APA StyleChu, Y., Qiu, P., & Yu, R. (2020). Centipede Venom Peptides Acting on Ion Channels. Toxins, 12(4), 230. https://doi.org/10.3390/toxins12040230