The Role of Gut Dysbiosis in the Bone–Vascular Axis in Chronic Kidney Disease




{kind=link}
Abstract
:1. Introduction
2. Bone–Vascular Axis
3. Gut Microbial Ecosystem in Health and CKD
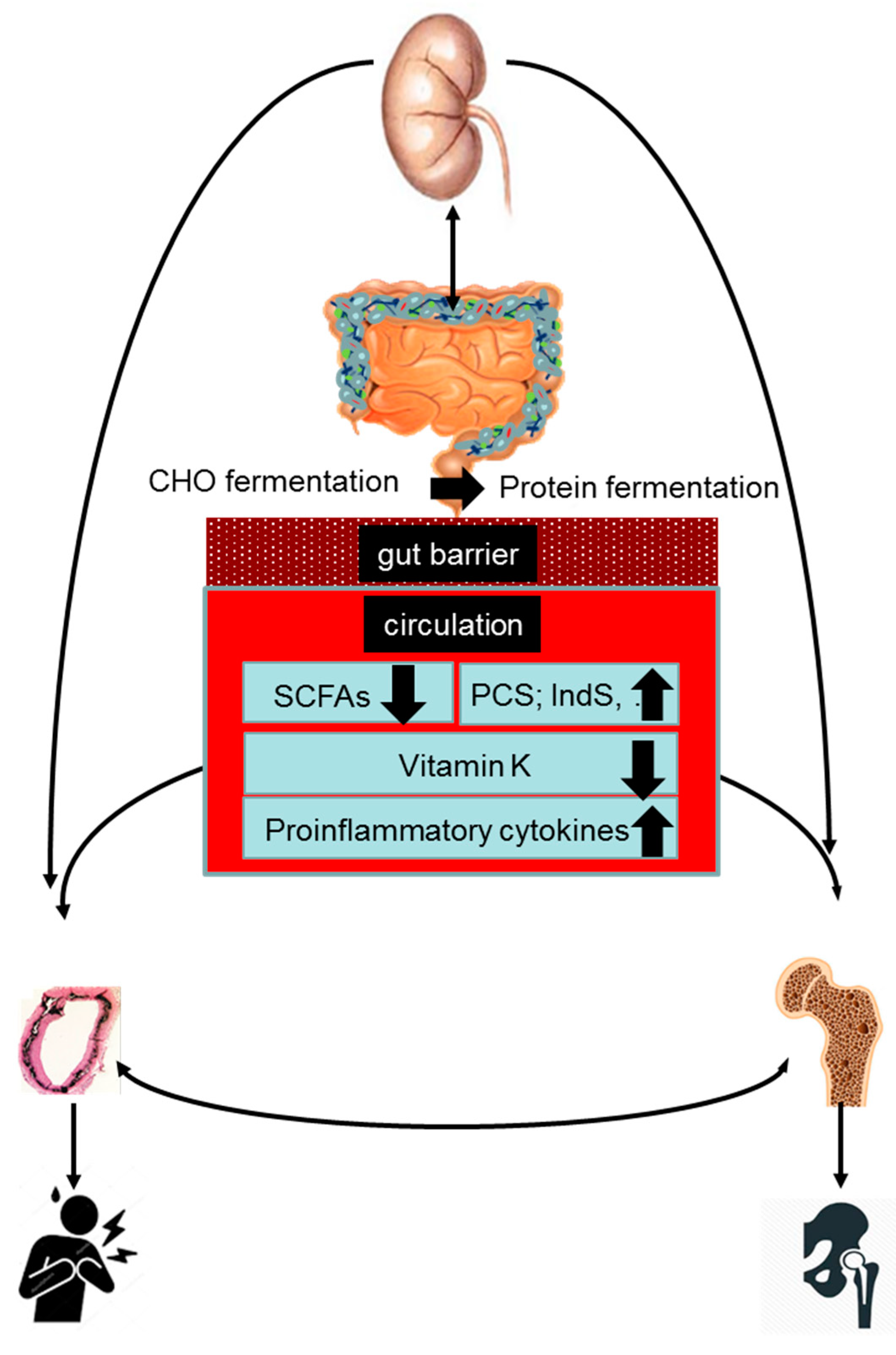
4. Gut–Bone–Vascular Axis in CKD
5. Role of Increased Protein Fermentation in the Bone–Vascular Axis
6. Role of Decreased Carbohydrate Fermentation in the Bone–Vascular Axis
7. Role of Vitamin K Deficiency in the Bone–Vascular Axis
8. Role of Inflammation in the Bone–Vascular Axis
9. Therapeutic Options
Author Contributions
Funding
Conflicts of Interest
References
- Moe, S.; Drueke, T.; Cunningham, J.; Goodman, W.; Martin, K.; Olgaard, K.; Ott, S.; Sprague, S.; Lameire, N.; Eknoyan, G. Definition, evaluation, and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006, 69, 1945–1953. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Opdebeeck, B.; David, K.; D’Haese, P.C. Bone-Vascular Axis in Chronic Kidney Disease. Adv. Chronic. Kidney Dis. 2019, 26, 472–483. [Google Scholar] [CrossRef]
- Stein, M.S.; Packham, D.K.; Ebeling, P.R.; Wark, J.D.; Becker, G.J. Prevalence and risk factors for osteopenia in dialysis patients. Am. J. Kidney Dis. 1996, 28, 515–522. [Google Scholar] [CrossRef]
- Rix, M.; Andreassen, H.; Eskildsen, P.; Langdahl, B.; Olgaard, K. Bone mineral density and biochemical markers of bone turnover in patients with predialysis chronic renal failure. Kidney Int. 1999, 56, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Urena, P.; Bernard-Poenaru, O.; Ostertag, A.; Baudoin, C.; Cohen-Solal, M.; Cantor, T.; de Vernejoul, M.C. Bone mineral density, biochemical markers and skeletal fractures in haemodialysis patients. Nephrol. Dial. Transplant. 2003, 18, 2325–2331. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Claes, K.; Meijers, B.; Laurent, M.R.; Bammens, B.; Naesens, M.; Sprangers, B.; Pottel, H.; Cavalier, E.; Kuypers, D. Bone mineral density, bone turnover markers, and incident fractures in de novo kidney transplant recipients. Kidney Int. 2019, 95, 1461–1470. [Google Scholar] [CrossRef]
- Chen, H.; Lips, P.; Vervloet, M.G.; van Schoor, N.M.; de Jongh, R.T. Association of renal function with bone mineral density and fracture risk in the Longitudinal Aging Study Amsterdam. Osteoporos. Int 2018, 29, 2129–2138. [Google Scholar] [CrossRef] [Green Version]
- Klawansky, S.; Komaroff, E.; Cavanaugh, P.F., Jr.; Mitchell, D.Y.; Gordon, M.J.; Connelly, J.E.; Ross, S.D. Relationship between age, renal function and bone mineral density in the US population. Osteoporos. Int. 2003, 14, 570–576. [Google Scholar] [CrossRef]
- Ishani, A.; Blackwell, T.; Jamal, S.A.; Cummings, S.R.; Ensrud, K.E. The effect of raloxifene treatment in postmenopausal women with CKD. J. Am. Soc. Nephrol. 2008, 19, 1430–1438. [Google Scholar] [CrossRef] [Green Version]
- Ketteler, M.; Block, G.A.; Evenepoel, P.; Fukagawa, M.; Herzog, C.A.; McCann, L.; Moe, S.M.; Shroff, R.; Tonelli, M.A.; Toussaint, N.D.; et al. Executive summary of the 2017 KDIGO Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Guideline Update: What’s changed and why it matters. Kidney Int. 2017, 92, 26–36. [Google Scholar] [CrossRef] [Green Version]
- Malluche, H.H.; Porter, D.S.; Monier-Faugere, M.C.; Mawad, H.; Pienkowski, D. Differences in bone quality in low- and high-turnover renal osteodystrophy. J. Am. Soc. Nephrol. 2012, 23, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Jadoul, M.; Albert, J.M.; Akiba, T.; Akizawa, T.; Arab, L.; Bragg-Gresham, J.L.; Mason, N.; Prutz, K.G.; Young, E.W.; Pisoni, R.L. Incidence and risk factors for hip or other bone fractures among hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study. Kidney Int. 2006, 70, 1358–1366. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, G.M.; Naves, D.M.; Cannata Andia, J.B. Bone metabolism, vascular calcifications and mortality: Associations beyond mere coincidence. J. Nephrol. 2005, 18, 458–463. [Google Scholar]
- Tentori, F.; McCullough, K.; Kilpatrick, R.D.; Bradbury, B.D.; Robinson, B.M.; Kerr, P.G.; Pisoni, R.L. High rates of death and hospitalization follow bone fracture among hemodialysis patients. Kidney Int. 2014, 85, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Naves, M.; Diaz-Lopez, J.B.; Gomez, C.; Rodriguez-Rebollar, A.; Rodriguez-Garcia, M.; Cannata-Andia, J.B. The effect of vertebral fracture as a risk factor for osteoporotic fracture and mortality in a Spanish population. Osteoporos. Int. 2003, 14, 520–524. [Google Scholar] [CrossRef]
- Vervloet, M.; Cozzolino, M. Vascular calcification in chronic kidney disease: Different bricks in the wall? Kidney Int. 2017, 91, 808–817. [Google Scholar] [CrossRef]
- Budoff, M.J.; Rader, D.J.; Reilly, M.P.; Mohler III, E.R.; Lash, J.; Yang, W.; Rosen, L.; Glenn, M.; Teal, V.; Feldman, H.I. Relationship of estimated GFR and coronary artery calcification in the CRIC (Chronic Renal Insufficiency Cohort) Study. Am. J. Kidney Dis. 2011, 58, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Neven, E.; De Schutter, T.M.; De Broe, M.E.; D’Haese, P.C. Cell biological and physicochemical aspects of arterial calcification. Kidney Int. 2011, 79, 1166–1177. [Google Scholar] [CrossRef] [Green Version]
- Schlieper, G. Vascular calcification in chronic kidney disease: Not all arteries are created equal. Kidney Int. 2014, 85, 501–503. [Google Scholar] [CrossRef] [Green Version]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, W.C.; Adams, A.L. Breast arterial calcification in chronic kidney disease: Absence of smooth muscle apoptosis and osteogenic transdifferentiation. Kidney Int. 2014, 85, 668–676. [Google Scholar] [CrossRef] [Green Version]
- Okuno, S.; Ishimura, E.; Kitatani, K.; Fujino, Y.; Kohno, K.; Maeno, Y.; Maekawa, K.; Yamakawa, T.; Imanishi, Y.; Inaba, M.; et al. Presence of abdominal aortic calcification is significantly associated with all-cause and cardiovascular mortality in maintenance hemodialysis patients. Am. J. Kidney Dis. 2007, 49, 417–425. [Google Scholar] [CrossRef]
- Claes, K.J.; Heye, S.; Bammens, B.; Kuypers, D.R.; Meijers, B.; Naesens, M.; Vanrenterghem, Y.; Evenepoel, P. Aortic calcifications and arterial stiffness as predictors of cardiovascular events in incident renal transplant recipients. Transpl. Int 2013, 26, 973–981. [Google Scholar] [CrossRef]
- Chen, Z.; Qureshi, A.R.; Ripsweden, J.; Wennberg, L.; Heimburger, O.; Lindholm, B.; Barany, P.; Haarhaus, M.; Brismar, T.B.; Stenvinkel, P. Vertebral bone density associates with coronary artery calcification and is an independent predictor of poor outcome in end-stage renal disease patients. Bone 2016, 92, 50–57. [Google Scholar] [CrossRef]
- Viaene, L.; Behets, G.J.; Heye, S.; Claes, K.; Monbaliu, D.; Pirenne, J.; D’Haese, P.C.; Evenepoel, P. Inflammation and the bone-vascular axis in end-stage renal disease. Osteoporos. Int. 2016, 27, 489–497. [Google Scholar] [CrossRef]
- Naves, M.; Rodriguez-Garcia, M.; Diaz-Lopez, J.B.; Gomez-Alonso, C.; Cannata-Andia, J.B. Progression of vascular calcifications is associated with greater bone loss and increased bone fractures. Osteoporos. Int. 2008, 19, 1161–1166. [Google Scholar] [CrossRef]
- Adragao, T.; Herberth, J.; Monier-Faugere, M.C.; Branscum, A.J.; Ferreira, A.; Frazao, J.M.; Dias, C.J.; Malluche, H.H. Low bone volume--a risk factor for coronary calcifications in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 450–455. [Google Scholar] [CrossRef] [Green Version]
- Cejka, D.; Weber, M.; Diarra, D.; Reiter, T.; Kainberger, F.; Haas, M. Inverse association between bone microarchitecture assessed by HR-pQCT and coronary artery calcification in patients with end-stage renal disease. Bone 2014, 64, 33–38. [Google Scholar] [CrossRef]
- Barreto, D.V.; Barreto, F.C.; Carvalho, A.B.; Cuppari, L.; Cendoroglo, M.; Draibe, S.A.; Moyses, R.M.; Neves, K.R.; Jorgetti, V.; Blair, A.; et al. Coronary calcification in hemodialysis patients: The contribution of traditional and uremia-related risk factors. Kidney Int. 2005, 67, 1576–1582. [Google Scholar] [CrossRef] [Green Version]
- Schulz, E.; Arfai, K.; Liu, X.; Sayre, J.; Gilsanz, V. Aortic calcification and the risk of osteoporosis and fractures. J. Clin. Endocrinol. Metab. 2004, 89, 4246–4253. [Google Scholar] [CrossRef]
- Tanko, L.B.; Christiansen, C.; Cox, D.A.; Geiger, M.J.; McNabb, M.A.; Cummings, S.R. Relationship between osteoporosis and cardiovascular disease in postmenopausal women. J. Bone Miner. Res. 2005, 20, 1912–1920. [Google Scholar] [CrossRef] [Green Version]
- Hyder, J.A.; Allison, M.A.; Wong, N.; Papa, A.; Lang, T.F.; Sirlin, C.; Gapstur, S.M.; Ouyang, P.; Carr, J.J.; Criqui, M.H. Association of coronary artery and aortic calcium with lumbar bone density: The MESA Abdominal Aortic Calcium Study. Am. J. Epidemiol. 2009, 169, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Lampropoulos, C.E.; Papaioannou, I.; D’Cruz, D.P. Osteoporosis—A risk factor for cardiovascular disease? Nat. Rev. Rheumatol. 2012, 8, 587–598. [Google Scholar] [CrossRef]
- Flipon, E.; Liabeuf, S.; Fardellone, P.; Mentaverri, R.; Ryckelynck, T.; Grados, F.; Kamel, S.; Massy, Z.A.; Dargent-Molina, P.; Brazier, M. Is vascular calcification associated with bone mineral density and osteoporotic fractures in ambulatory, elderly women? Osteoporos. Int. 2011. [Google Scholar] [CrossRef]
- Persy, V.; D’Haese, P. Vascular calcification and bone disease: The calcification paradox. Trends Mol. Med. 2009, 15, 405–416. [Google Scholar] [CrossRef]
- London, G.M.; Marty, C.; Marchais, S.J.; Guerin, A.P.; Metivier, F.; de Vernejoul, M.C. Arterial Calcifications and Bone Histomorphometry in End-Stage Renal Disease. J. Am. Soc. Nephrol. 2004, 15, 1943–1951. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, M.; Gomez-Alonso, C.; Naves-Diaz, M.; Diaz-Lopez, J.B.; Diaz-Corte, C.; Cannata-Andia, J.B. Vascular calcifications, vertebral fractures and mortality in haemodialysis patients. Nephrol. Dial. Transplant. 2009, 24, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Meijers, B.; Evenepoel, P.; Anders, H.J. Intestinal microbiome and fitness in kidney disease. Nat. Rev. Nephrol. 2019, 15, 531–545. [Google Scholar] [CrossRef]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; Desantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Xie, S.; Lv, D.; Wang, P.; He, H.; Zhang, T.; Zhou, Y.; Lin, Q.; Zhou, H.; Jiang, J.; et al. Alteration of the gut microbiota in Chinese population with chronic kidney disease. Sci. Rep. 2017, 7, 2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poesen, R.; Windey, K.; Neven, E.; Kuypers, D.; De Preter, V.; Augustijns, P.; D’Haese, P.; Evenepoel, P.; Verbeke, K.; Meijers, B. The Influence of CKD on Colonic Microbial Metabolism. J. Am. Soc. Nephrol. 2016, 27, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Evidence for impaired assimilation of protein in chronic renal failure. Kidney Int. 2003, 64, 2196–2203. [Google Scholar] [CrossRef] [PubMed]
- Evenepoel, P.; Meijers, B.K.I.; Bammens, B.R.M.; Verbeke, K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. 2009, 76, S12–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; Desantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Xie, S.; Lv, D.; Zhang, Y.; Deng, J.; Zeng, L.; Chen, Y. A reduction in the butyrate producing species Roseburia spp. and Faecalibacterium prausnitzii is associated with chronic kidney disease progression. Antonie Leeuwenhoek 2016, 109, 1389–1396. [Google Scholar] [CrossRef]
- Poesen, R.; Ramezani, A.; Claes, K.; Augustijns, P.; Kuypers, D.; Barrows, I.R.; Muralidharan, J.; Evenepoel, P.; Meijers, B.; Raj, D.S. Associations of Soluble CD14 and Endotoxin with Mortality, Cardiovascular Disease, and Progression of Kidney Disease among Patients with CKD. Clin. J. Am. Soc. Nephrol. 2015, 10, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, C.W.; Harrison, L.E.; Eldehni, M.T.; Jefferies, H.J.; Szeto, C.C.; John, S.G.; Sigrist, M.K.; Burton, J.O.; Hothi, D.; Korsheed, S.; et al. Circulating endotoxemia: A novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 133–141. [Google Scholar] [CrossRef]
- Andersen, K.; Kesper, M.S.; Marschner, J.A.; Konrad, L.; Ryu, M.; Kumar, V.S.; Kulkarni, O.P.; Mulay, S.R.; Romoli, S.; Demleitner, J.; et al. Intestinal Dysbiosis, Barrier Dysfunction, and Bacterial Translocation Account for CKD-Related Systemic Inflammation. J. Am. Soc. Nephrol. 2017, 28, 76–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M.; Wang, Z.; Liu, H.; Jiang, H.; Wang, M.; Liang, S.; Shi, K.; Feng, J. Probiotic Bifidobacterium animalis subsp. lactis Bi-07 alleviates bacterial translocation and ameliorates microinflammation in experimental uraemia. Nephrology (Carlton.) 2014, 19, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, M.; Magnusson, K.E.; Sundqvist, T.; Denneberg, T. Impaired intestinal barrier function measured by differently sized polyethylene glycols in patients with chronic renal failure. Gut 1991, 32, 754–759. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.J.; Andersen, K.; Stecher, B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013, 83, 1010–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D.; Yuan, J.; Nazertehrani, S.; Ni, Z.; Liu, S. Chronic kidney disease causes disruption of gastric and small intestinal epithelial tight junction. Am. J. Nephrol. 2013, 38, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am. J. Nephrol. 2013, 37, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach Knudsen, K.E.; Laerke, H.N.; Hedemann, M.S.; Nielsen, T.S.; Ingerslev, A.K.; Gundelund Nielsen, D.S.; Theil, P.K.; Purup, S.; Hald, S.; Schioldan, A.G.; et al. Impact of Diet-Modulated Butyrate Production on Intestinal Barrier Function and Inflammation. Nutrients 2018, 10, 499. [Google Scholar] [CrossRef] [Green Version]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host. Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Hatayama, H.; Iwashita, J.; Kuwajima, A.; Abe, T. The short chain fatty acid, butyrate, stimulates MUC2 mucin production in the human colon cancer cell line, LS174T. Biochem. Biophys. Res. Commun. 2007, 356, 599–603. [Google Scholar] [CrossRef]
- Schilderink, R.; Verseijden, C.; Seppen, J.; Muncan, V.; van den Brink, G.R.; Lambers, T.T.; van Tol, E.A.; de Jonge, W.J. The SCFA butyrate stimulates the epithelial production of retinoic acid via inhibition of epithelial HDAC. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G1138–G1146. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, C.M. Diet, gut microbiome, and bone health. Curr. Osteoporos. Rep. 2015, 13, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.J.; Guss, J.D.; Luna, M.; Goldring, S.R. Links Between the Microbiome and Bone. J. Bone Miner. Res. 2016, 31, 1638–1646. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, M.M.; Jones, R.M.; Schett, G.; Pacifici, R. The gut-bone axis: How bacterial metabolites bridge the distance. J. Clin. Investig. 2019, 129, 3018–3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, F.H.; Fak, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Backhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [Green Version]
- Jovanovich, A.; Isakova, T.; Stubbs, J. Microbiome and Cardiovascular Disease in CKD. Clin. J. Am. Soc. Nephrol. 2018, 13, 1598–1604. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Claus, D.; Geypens, B.; Hiele, M.; Geboes, K.; Rutgeerts, P.; Ghoos, Y. Amount and fate of egg protein escaping assimilation in the small intestine of humans. AJP-Gastrointest. Liver Physiol. 1999, 277, G935–G943. [Google Scholar] [CrossRef] [Green Version]
- Meijers, B.; Farre, R.; Dejongh, S.; Vicario, M.; Evenepoel, P. Intestinal Barrier Function in Chronic Kidney Disease. Toxins (Basel) 2018, 10, 298. [Google Scholar] [CrossRef] [Green Version]
- Poesen, R.; Evenepoel, P.; de Loor, H.; Kuypers, D.; Augustijns, P.; Meijers, B. Metabolism, Protein Binding, and Renal Clearance of Microbiota-Derived p-Cresol in Patients with CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 1136–1144. [Google Scholar] [CrossRef] [Green Version]
- Poesen, R.; Viaene, L.; Verbeke, K.; Claes, K.; Bammens, B.; Sprangers, B.; Naesens, M.; Vanrenterghem, Y.; Kuypers, D.; Evenepoel, P.; et al. Renal clearance and intestinal generation of p-cresyl sulfate and indoxyl sulfate in CKD. Clin. J. Am. Soc. Nephrol. 2013, 8, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins (Basel) 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-kappaB activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Muteliefu, G.; Enomoto, A.; Jiang, P.; Takahashi, M.; Niwa, T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol. Dial. Transplant. 2009, 24, 2051–2058. [Google Scholar] [CrossRef] [Green Version]
- Meijers, B.K.; Van, K.S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef]
- Buendia, P.; Montes de Oca, A.; Madueno, J.A.; Merino, A.; Martin-Malo, A.; Aljama, P.; Ramirez, R.; Rodriguez, M.; Carracedo, J. Endothelial microparticles mediate inflammation-induced vascular calcification. FASEB J. 2015, 29, 173–181. [Google Scholar] [CrossRef]
- Rodrigues, S.D.; Santos, S.S.; Meireles, T.; Romero, N.; Glorieux, G.; Pecoits-Filho, R.; Zhang, D.D.; Nakao, L.S. Uremic toxins promote accumulation of oxidized protein and increased sensitivity to hydrogen peroxide in endothelial cells by impairing the autophagic flux. Biochem. Biophys. Res. Commun. 2019. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Chen, J.; Shen, Z.; Gu, Y.; Xu, L.; Hu, J.; Zhang, X.; Ding, X. Indoxyl sulfate accelerates vascular smooth muscle cell calcification via microRNA-29b dependent regulation of Wnt/beta-catenin signaling. Toxicol. Lett. 2018, 284, 29–36. [Google Scholar] [CrossRef]
- Stockler-Pinto, M.B.; Soulage, C.O.; Borges, N.A.; Cardozo, L.F.M.F.; Dolenga, C.J.; Nakao, L.S.; Pecoits-Filho, R.; Fouque, D.; Mafra, D. From bench to the hemodialysis clinic: Protein-bound uremic toxins modulate NF-kappaB/Nrf2 expression. Int Urol. Nephrol. 2018, 50, 347–354. [Google Scholar] [CrossRef]
- Adijiang, A.; Goto, S.; Uramoto, S.; Nishijima, F.; Niwa, T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol. Dial. Transplant. 2008, 23, 1892–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adijiang, A.; Higuchi, Y.; Nishijima, F.; Shimizu, H.; Niwa, T. Indoxyl sulfate, a uremic toxin, promotes cell senescence in aorta of hypertensive rats. Biochem. Biophys. Res. Commun. 2010, 399, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Opdebeeck, B.; Maudsley, S.; Azmi, A.; De, M.A.; De, L.W.; Meijers, B.; Verhulst, A.; Evenepoel, P.; D’Haese, P.C.; Neven, E. Indoxyl Sulfate and p-Cresyl Sulfate Promote Vascular Calcification and Associate with Glucose Intolerance. J. Am. Soc. Nephrol. 2019, 30, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.I.; Claes, K.; Bammens, B.; de Loor, H.; Viaene, L.; Verbeke, K.; Kuypers, D.; Vanrenterghem, Y.; Evenepoel, P. p-Cresol and Cardiovascular Risk in Mild-to-Moderate Kidney Disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 1182–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; et al. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Viaene, L.; Thijs, L.; Jin, Y.; Liu, Y.; Gu, Y.; Meijers, B.; Claes, K.; Staessen, J.; Evenepoel, P. Heritability and Clinical Determinants of Serum Indoxyl Sulfate and p-Cresyl Sulfate, Candidate Biomarkers of the Human Microbiome Enterotype. PLoS ONE 2014, 9, e79682. [Google Scholar] [CrossRef] [Green Version]
- Kazama, J.J.; Iwasaki, Y.; Fukagawa, M. Uremic osteoporosis. Kidney Int. Suppl. (2011) 2013, 3, 446–450. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Iwasaki, Y.; Yamato, H.; Mori, Y.; Komaba, H.; Watanabe, H.; Maruyama, T.; Fukagawa, M. p-Cresyl sulfate induces osteoblast dysfunction through activating JNK and p38 MAPK pathways. Bone 2013, 56, 347–354. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kwak, K.A.; Gil, H.W.; Song, H.Y.; Hong, S.Y. Indoxyl sulfate promotes apoptosis in cultured osteoblast cells. BMC. Pharmacol. Toxicol. 2013, 14, 60. [Google Scholar] [CrossRef] [Green Version]
- Mozar, A.; Louvet, L.; Godin, C.; Mentaverri, R.; Brazier, M.; Kamel, S.; Massy, Z.A. Indoxyl sulphate inhibits osteoclast differentiation and function. Nephrol. Dial. Transplant. 2012, 27, 2176–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanza, D.; Perna, A.F.; Oliva, A.; Vanholder, R.; Pletinck, A.; Guastafierro, S.; Di, N.A.; Vigorito, C.; Capasso, G.; Jankowski, V.; et al. Impact of the uremic milieu on the osteogenic potential of mesenchymal stem cells. PLoS ONE 2015, 10, e0116468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, Y.; Kazama, J.J.; Yamato, H.; Fukagawa, M. Changes in chemical composition of cortical bone associated with bone fragility in rat model with chronic kidney disease. Bone 2011, 48, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Kazama, J.J.; Yamato, H.; Shimoda, H.; Fukagawa, M. Accumulated uremic toxins attenuate bone mechanical properties in rats with chronic kidney disease. Bone 2013, 57, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Nii-Kono, T.; Iwasaki, Y.; Uchida, M.; Fujieda, A.; Hosokawa, A.; Motojima, M.; Yamato, H.; Kurokawa, K.; Fukagawa, M. Indoxyl sulfate induces skeletal resistance to parathyroid hormone in cultured osteoblastic cells. Kidney Int. 2007. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Bover, J.; Urena, T.P. Parathyroid hormone metabolism and signaling in health and chronic kidney disease. Kidney Int. 2016, 90, 1184–1190. [Google Scholar] [CrossRef]
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012, 81, 640–650. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, X.; Zhang, H.; Liu, T.; Zhang, H.; Teng, J.; Ji, J.; Ding, X. Indoxyl Sulfate Enhance the Hypermethylation of Klotho and Promote the Process of Vascular Calcification in Chronic Kidney Disease. Int, J. Biol. Sci. 2016, 12, 1236–1246. [Google Scholar] [CrossRef] [Green Version]
- Mencke, R.; Hillebrands, J.L. The role of the anti-ageing protein Klotho in vascular physiology and pathophysiology. Ageing Res. Rev. 2017, 35, 124–146. [Google Scholar] [CrossRef]
- Kawaguchi, H.; Manabe, N.; Miyaura, C.; Chikuda, H.; Nakamura, K.; Kuro-o, M. Independent impairment of osteoblast and osteoclast differentiation in klotho mouse exhibiting low-turnover osteopenia. J. Clin. Investig. 1999, 104, 229–237. [Google Scholar] [CrossRef]
- Lindberg, K.; Olauson, H.; Amin, R.; Ponnusamy, A.; Goetz, R.; Taylor, R.F.; Mohammadi, M.; Canfield, A.; Kublickiene, K.; Larsson, T.E. Arterial klotho expression and FGF23 effects on vascular calcification and function. PLoS ONE 2013, 8, e60658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, Y.; Bivi, N.; Farrow, E.; Lezcano, V.; Plotkin, L.I.; White, K.E.; Bellido, T. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 2011, 49, 636–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komaba, H.; Kaludjerovic, J.; Hu, D.Z.; Nagano, K.; Amano, K.; Ide, N.; Sato, T.; Densmore, M.J.; Hanai, J.I.; Olauson, H.; et al. Klotho expression in osteocytes regulates bone metabolism and controls bone formation. Kidney Int. 2017, 92, 599–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Stumpff, F. A look at the smelly side of physiology: Transport of short chain fatty acids. Pflugers Arch. 2018, 470, 571–598. [Google Scholar] [CrossRef]
- Mishima, E.; Fukuda, S.; Mukawa, C.; Yuri, A.; Kanemitsu, Y.; Matsumoto, Y.; Akiyama, Y.; Fukuda, N.N.; Tsukamoto, H.; Asaji, K.; et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. 2017, 92, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Terpstra, M.L.; Sinnige, M.J.; Hugenholtz, F.; Peters-Sengers, H.; Remmerswaal, E.B.; Geerlings, S.E.; Bemelman, F.J. Butyrate production in patients with end-stage renal disease. Int, J. Nephrol. Renovasc. Dis. 2019, 12, 87–101. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Lv, D.; Jiang, S.; Jiang, J.; Liang, M.; Hou, F.; Chen, Y. Quantitative reduction in short-chain fatty acids, especially butyrate, contributes to the progression of chronic kidney disease. Clin. Sci. (Lond.) 2019, 133, 1857–1870. [Google Scholar] [CrossRef]
- Jadoon, A.; Mathew, A.V.; Byun, J.; Gadegbeku, C.A.; Gipson, D.S.; Afshinnia, F.; Pennathur, S. Gut Microbial Product Predicts Cardiovascular Risk in Chronic Kidney Disease Patients. Am. J. Nephrol. 2018, 48, 269–277. [Google Scholar] [CrossRef]
- Lucas, S.; Omata, Y.; Hofmann, J.; Bottcher, M.; Iljazovic, A.; Sarter, K.; Albrecht, O.; Schulz, O.; Krishnacoumar, B.; Kronke, G.; et al. Short-chain fatty acids regulate systemic bone mass and protect from pathological bone loss. Nat. Commun. 2018, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- Katono, T.; Kawato, T.; Tanabe, N.; Suzuki, N.; Iida, T.; Morozumi, A.; Ochiai, K.; Maeno, M. Sodium butyrate stimulates mineralized nodule formation and osteoprotegerin expression by human osteoblasts. Arch. Oral Biol. 2008, 53, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Montalvany-Antonucci, C.C.; Duffles, L.F.; de Arruda, J.A.A.; Zicker, M.C.; de Oliveira, S.; Macari, S.; Garlet, G.P.; Madeira, M.F.M.; Fukada, S.Y.; Andrade, I.; et al. Short-chain fatty acids and FFAR2 as suppressors of bone resorption. Bone 2019, 125, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.M.; Yu, M.; Darby, T.M.; Vaccaro, C.; Li, J.Y.; Owens, J.A.; Hsu, E.; Adams, J.; Weitzmann, M.N.; Jones, R.M.; et al. The Microbial Metabolite Butyrate Stimulates Bone Formation via T Regulatory Cell-Mediated Regulation of WNT10B Expression. Immunity 2018, 49, 1116–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, T.M.; Westendorf, J.J. Histone deacetylase inhibitors promote osteoblast maturation. J. Bone Miner. Res. 2005, 20, 2254–2263. [Google Scholar] [CrossRef]
- Yan, J.; Herzog, J.W.; Tsang, K.; Brennan, C.A.; Bower, M.A.; Garrett, W.S.; Sartor, B.R.; Aliprantis, A.O.; Charles, J.F. Gut microbiota induce IGF-1 and promote bone formation and growth. Proc. Natl. Acad. Sci. USA 2016, 113, E7554–E7563. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Viaene, L.; Meijers, B. Calcium balance in chronic kidney disease: Walking the tightrope. Kidney Int. 2012, 81, 1057–1059. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Jia, Y.; Yang, S.; Zhao, N.; Hu, Y.; Hong, J.; Gao, S.; Zhao, R. Sodium butyrate protects against high-fat diet-induced oxidative stress in rat liver by promoting expression of nuclear factor E2-related factor 2. Br. J. Nutr. 2019, 122, 400–410. [Google Scholar] [CrossRef]
- Wu, J.; Jiang, Z.; Zhang, H.; Liang, W.; Huang, W.; Zhang, H.; Li, Y.; Wang, Z.; Wang, J.; Jia, Y.; et al. Sodium butyrate attenuates diabetes-induced aortic endothelial dysfunction via P300-mediated transcriptional activation of Nrf2. Free Radic. Biol. Med. 2018, 124, 454–465. [Google Scholar] [CrossRef]
- Yaku, K.; Enami, Y.; Kurajyo, C.; Matsui-Yuasa, I.; Konishi, Y.; Kojima-Yuasa, A. The enhancement of phase 2 enzyme activities by sodium butyrate in normal intestinal epithelial cells is associated with Nrf2 and p53. Mol. Cell Biochem. 2012, 370, 7–14. [Google Scholar] [CrossRef]
- Guo, W.; Liu, J.; Sun, J.; Gong, Q.; Ma, H.; Kan, X.; Cao, Y.; Wang, J.; Fu, S. Butyrate alleviates oxidative stress by regulating NRF2 nuclear accumulation and H3K9/14 acetylation via GPR109A in bovine mammary epithelial cells and mammary glands. Free Radic. Biol. Med. 2020. [Google Scholar] [CrossRef]
- Ranganna, K.; Mathew, O.P.; Yatsu, F.M.; Yousefipour, Z.; Hayes, B.E.; Milton, S.G. Involvement of glutathione/glutathione S-transferase antioxidant system in butyrate-inhibited vascular smooth muscle cell proliferation. FEBS J. 2007, 274, 5962–5978. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Enaka, M.; Muragaki, Y. Activation of KEAP1/NRF2/P62 signaling alleviates high phosphate-induced calcification of vascular smooth muscle cells by suppressing reactive oxygen species production. Sci. Rep. 2019, 9, 10366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groenen-van Dooren, M.M.; Ronden, J.E.; Soute, B.A.; Vermeer, C. Bioavailability of phylloquinone and menaquinones after oral and colorectal administration in vitamin K-deficient rats. Biochem. Pharmacol. 1995, 50, 797–801. [Google Scholar] [CrossRef]
- Komai, M.; Shirakawa, H.; Kimura, S. Newly developed model for vitamin K deficiency in germfree mice. Int, J. Vitam. Nutr. Res. 1988, 58, 55–59. [Google Scholar]
- Allison, P.M.; Mummah-Schendel, L.L.; Kindberg, C.G.; Harms, C.S.; Bang, N.U.; Suttie, J.W. Effects of a vitamin K-deficient diet and antibiotics in normal human volunteers. J. Lab. Clin. Med. 1987, 110, 180–188. [Google Scholar] [PubMed]
- Frick, P.G.; Riedler, G.; Brogli, H. Dose response and minimal daily requirement for vitamin K in man. J. Appl. Physiol 1967, 23, 387–389. [Google Scholar] [CrossRef]
- Guss, J.D.; Taylor, E.; Rouse, Z.; Roubert, S.; Higgins, C.H.; Thomas, C.J.; Baker, S.P.; Vashishth, D.; Donnelly, E.; Shea, M.K.; et al. The microbial metagenome and bone tissue composition in mice with microbiome-induced reductions in bone strength. Bone 2019, 127, 146–154. [Google Scholar] [CrossRef]
- Krueger, T.; Westenfeld, R.; Ketteler, M.; Schurgers, L.J.; Floege, J. Vitamin K deficiency in CKD patients: A modifiable risk factor for vascular calcification? Kidney Int. 2009, 76, 18–22. [Google Scholar] [CrossRef] [Green Version]
- Holden, R.M.; Morton, A.R.; Garland, J.S.; Pavlov, A.; Day, A.G.; Booth, S.L. Vitamins K and D status in stages 3-5 chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 590–597. [Google Scholar] [CrossRef]
- Cranenburg, E.C.; Schurgers, L.J.; Uiterwijk, H.H.; Beulens, J.W.; Dalmeijer, G.W.; Westerhuis, R.; Magdeleyns, E.J.; Herfs, M.; Vermeer, C.; Laverman, G.D. Vitamin K intake and status are low in hemodialysis patients. Kidney Int. 2012, 82, 605–610. [Google Scholar] [CrossRef] [Green Version]
- Schlieper, G.; Westenfeld, R.; Kruger, T.; Cranenburg, E.C.; Magdeleyns, E.J.; Brandenburg, V.M.; Djuric, Z.; Damjanovic, T.; Ketteler, M.; Vermeer, C.; et al. Circulating nonphosphorylated carboxylated matrix gla protein predicts survival in ESRD. J. Am. Soc. Nephrol. 2011, 22, 387–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boxma, P.Y.; van den Berg, E.; Geleijnse, J.M.; Laverman, G.D.; Schurgers, L.J.; Vermeer, C.; Kema, I.P.; Muskiet, F.A.; Navis, G.; Bakker, S.J.; et al. Vitamin k intake and plasma desphospho-uncarboxylated matrix Gla-protein levels in kidney transplant recipients. PLoS ONE 2012, 7, e47991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evenepoel, P.; Claes, K.; Meijers, B.; Laurent, M.; Bammens, B.; Naesens, M.; Sprangers, B.; Pottel, H.; Cavalier, E.; Kuypers, D. Poor Vitamin K Status Is Associated With Low Bone Mineral Density and Increased Fracture Risk in End-Stage Renal Disease. J. Bone Miner. Res. 2019, 34, 262–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansz, T.T.; Neradova, A.; van Ballegooijen, A.J.; Verhaar, M.C.; Vervloet, M.G.; Schurgers, L.J.; van Jaarsveld, B.C. The role of kidney transplantation and phosphate binder use in vitamin K status. PLoS ONE 2018, 13, e0203157. [Google Scholar] [CrossRef] [PubMed]
- Kaesler, N.; Magdeleyns, E.; Herfs, M.; Schettgen, T.; Brandenburg, V.; Fliser, D.; Vermeer, C.; Floege, J.; Schlieper, G.; Kruger, T. Impaired vitamin K recycling in uremia is rescued by vitamin K supplementation. Kidney Int. 2014, 86, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Delanaye, P.; Krzesinski, J.M.; Warling, X.; Moonen, M.; Smelten, N.; Medart, L.; Pottel, H.; Cavalier, E. Dephosphorylated-uncarboxylated Matrix Gla protein concentration is predictive of vitamin K status and is correlated with vascular calcification in a cohort of hemodialysis patients. BMC. Nephrol. 2014, 15, 145. [Google Scholar] [CrossRef] [Green Version]
- Fain, M.E.; Kapuku, G.K.; Paulson, W.D.; Williams, C.F.; Raed, A.; Dong, Y.; Knapen, M.H.J.; Vermeer, C.; Pollock, N.K. Inactive Matrix Gla Protein, Arterial Stiffness, and Endothelial Function in African American Hemodialysis Patients. Am. J. Hypertens. 2018, 31, 735–741. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Barreto, D.V.; Barreto, F.C.; Liabeuf, S.; Renard, C.; Magdeleyns, E.J.; Vermeer, C.; Choukroun, G.; Massy, Z.A. The circulating inactive form of matrix gla protein is a surrogate marker for vascular calcification in chronic kidney disease: A preliminary report. Clin. J. Am. Soc. Nephrol. 2010, 5, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Fusaro, M.; Noale, M.; Viola, V.; Galli, F.; Tripepi, G.; Vajente, N.; Plebani, M.; Zaninotto, M.; Guglielmi, G.; Miotto, D.; et al. Vitamin K, vertebral fractures, vascular calcifications, and mortality: VItamin K Italian (VIKI) dialysis study. J. Bone Miner. Res. 2012, 27, 2271–2278. [Google Scholar] [CrossRef]
- Zoch, M.L.; Clemens, T.L.; Riddle, R.C. New insights into the biology of osteocalcin. Bone 2016, 82, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Azuma, K.; Shiba, S.; Hasegawa, T.; Ikeda, K.; Urano, T.; Horie-Inoue, K.; Ouchi, Y.; Amizuka, N.; Inoue, S. Osteoblast-Specific gamma-Glutamyl Carboxylase-Deficient Mice Display Enhanced Bone Formation with Aberrant Mineralization. J. Bone Miner. Res. 2015, 30, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Maruyama-Nagao, A.; Sakuraba, K.; Kawai, S. Level of serum undercarboxylated osteocalcin correlates with bone quality assessed by calcaneal quantitative ultrasound sonometry in young Japanese females. Exp. Ther. Med. 2017, 13, 1937–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabb, M.M.; Sun, A.; Zhou, C.; Grun, F.; Errandi, J.; Romero, K.; Pham, H.; Inoue, S.; Mallick, S.; Lin, M.; et al. Vitamin K2 regulation of bone homeostasis is mediated by the steroid and xenobiotic receptor SXR. J. Biol. Chem. 2003, 278, 43919–43927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shea, M.K.; Booth, S.L.; Massaro, J.M.; Jacques, P.F.; D’Agostino, R.B., Sr.; Dawson-Hughes, B.; Ordovas, J.M.; O’Donnell, C.J.; Kathiresan, S.; Keaney, J.F.; et al. Vitamin K and vitamin D status: Associations with inflammatory markers in the Framingham Offspring Study. Am. J. Epidemiol. 2008, 167, 313–320. [Google Scholar] [CrossRef] [Green Version]
- Stenvinkel, P.; Wanner, C.; Metzger, T.; Heimburger, O.; Mallamaci, F.; Tripepi, G.; Malatino, L.; Zoccali, C. Inflammation and outcome in end-stage renal failure: Does female gender constitute a survival advantage? Kidney Int. 2002, 62, 1791–1798. [Google Scholar] [CrossRef] [Green Version]
- Kotanko, P.; Carter, M.; Levin, N.W. Intestinal bacterial microflora--a potential source of chronic inflammation in patients with chronic kidney disease. Nephrol. Dial. Transplant. 2006, 21, 2057–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D. CKD impairs barrier function and alters microbial flora of the intestine: A major link to inflammation and uremic toxicity. Curr. Opin. Nephrol. Hypertens. 2012, 21, 587–592. [Google Scholar] [CrossRef] [Green Version]
- Lau, W.L.; Kalantar-Zadeh, K.; Vaziri, N.D. The Gut as a Source of Inflammation in Chronic Kidney Disease. Nephron 2015, 130, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, A.; Krieg, R.; Massey, H.D.; Carl, D.; Ghosh, S.; Gehr, T.W.B.; Ghosh, S.S. Sodium butyrate ameliorates insulin resistance and renal failure in CKD rats by modulating intestinal permeability and mucin expression. Nephrol. Dial. Transplant. 2019, 34, 783–794. [Google Scholar] [CrossRef]
- Yang, J.; Lim, S.Y.; Ko, Y.S.; Lee, H.Y.; Oh, S.W.; Kim, M.G.; Cho, W.Y.; Jo, S.K. Intestinal barrier disruption and dysregulated mucosal immunity contribute to kidney fibrosis in chronic kidney disease. Nephrol. Dial. Transplant. 2019, 34, 419–428. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Dure-Smith, B.; Miller, R.; Mirahmadi, M.K. Pathology of gastrointestinal tract in chronic hemodialysis patients: An autopsy study of 78 cases. Am. J. Gastroenterol. 1985, 80, 608–611. [Google Scholar] [PubMed]
- Schepers, E.; Meert, N.; Glorieux, G.; Goeman, J.; Van der Eycken, J.; Vanholder, R. P-cresylsulphate, the main in vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol. Dial. Transplant. 2007, 22, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Katsuki, S.; Chen, M.; Decano, J.L.; Halu, A.; Lee, L.H.; Pestana, D.V.S.; Kum, A.S.T.; Kuromoto, R.K.; Golden, W.S.; et al. Uremic Toxin Indoxyl Sulfate Promotes Proinflammatory Macrophage Activation Via the Interplay of OATP2B1 and Dll4-Notch Signaling. Circulation 2019, 139, 78–96. [Google Scholar] [CrossRef] [PubMed]
- Viaene, L.; Evenepoel, P.; Meijers, B.; Vanderschueren, D.; Overbergh, L.; Mathieu, C. Uremia Suppresses Immune Signal-Induced CYP27B1 Expression in Human Monocytes. Am. J. Nephrol. 2012, 36, 497–508. [Google Scholar] [CrossRef]
- Hsu, H.J.; Yen, C.H.; Wu, I.W.; Hsu, K.H.; Chen, C.K.; Sun, C.Y.; Chou, C.C.; Chen, C.Y.; Tsai, C.J.; Wu, M.S.; et al. The association of uremic toxins and inflammation in hemodialysis patients. PLoS ONE 2014, 9, e102691. [Google Scholar] [CrossRef]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: A study with relevance to inflammatory bowel disease. World, J. Gastroenterol. 2007, 13, 2826–2832. [Google Scholar] [CrossRef]
- Meijer, K.; de Vos, P.; Priebe, M.G. Butyrate and other short-chain fatty acids as modulators of immunity: What relevance for health? Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 715–721. [Google Scholar] [CrossRef]
- Koh, A.; De, V.F.; Kovatcheva-Datchary, P.; Backhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Hjortnaes, J.; Butcher, J.; Figueiredo, J.L.; Riccio, M.; Kohler, R.H.; Kozloff, K.M.; Weissleder, R.; Aikawa, E. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: A role for inflammation. Eur. Heart, J. 2010, 31, 1975–1984. [Google Scholar] [CrossRef]
- Khosla, S. The bone and beyond: A shift in calcium. Nat. Med. 2011, 17, 430–431. [Google Scholar] [CrossRef] [PubMed]
- New, S.E.; Aikawa, E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ. Res. 2011, 108, 1381–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panuccio, V.; Enia, G.; Tripepi, R.; Aliotta, R.; Mallamaci, F.; Tripepi, G.; Zoccali, C. Pro-inflammatory cytokines and bone fractures in CKD patients. An exploratory single centre study. BMC. Nephrol. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.; Wunsch, R.; Turzer, M.; Bahner, M.; Raggi, P.; Querfeld, U.; Mehls, O.; Schaefer, F. Advanced coronary and carotid arteriopathy in young adults with childhood-onset chronic renal failure. Circulation 2002, 106, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Guerin, A.P.; London, G.M.; Marchais, S.J.; Metivier, F. Arterial stiffening and vascular calcifications in end-stage renal disease. Nephrol. Dial. Transplant. 2000, 15, 1014–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cauley, J.A.; Barbour, K.E.; Harrison, S.L.; Cloonan, Y.K.; Danielson, M.E.; Ensrud, K.E.; Fink, H.A.; Orwoll, E.S.; Boudreau, R. Inflammatory Markers and the Risk of Hip and Vertebral Fractures in Men: The Osteoporotic Fractures in Men (MrOS). J. Bone Miner. Res. 2016, 31, 2129–2138. [Google Scholar] [CrossRef] [Green Version]
- Al-Aly, Z.; Shao, J.S.; Lai, C.F.; Huang, E.; Cai, J.; Behrmann, A.; Cheng, S.L.; Towler, D.A. Aortic Msx2-Wnt Calcification Cascade Is Regulated by TNF-+¦GÇôDependent Signals in Diabetic LdlrGêÆ/GêÆ Mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2589–2596. [Google Scholar] [CrossRef] [Green Version]
- Henze, L.A.; Luong, T.T.D.; Boehme, B.; Masyout, J.; Schneider, M.P.; Brachs, S.; Lang, F.; Pieske, B.; Pasch, A.; Eckardt, K.U.; et al. Impact of C-reactive protein on osteo-/chondrogenic transdifferentiation and calcification of vascular smooth muscle cells. Aging (Albany. NY) 2019, 11, 5445–5462. [Google Scholar] [CrossRef]
- Ketteler, M.; Bongartz, P.; Westenfeld, R.; Wildberger, J.E.; Mahnken, A.H.; Böhm, R.; Metzger, T.; Wanner, C.; Jahnen-Dechent, W.; Floege, J. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: A cross-sectional study. Lancet 2003, 361, 827–833. [Google Scholar] [CrossRef]
- Feyen, J.H.; Elford, P.; Di Padova, F.E.; Trechsel, U. Interleukin-6 is produced by bone and modulated by parathyroid hormone. J. Bone Miner. Res. 1989, 4, 633–638. [Google Scholar] [CrossRef]
- Pfeilschifter, J.; Chenu, C.; Bird, A.; Mundy, G.R.; Roodman, G.D. Interleukin-1 and tumor necrosis factor stimulate the formation of human osteoclastlike cells in vitro. J. Bone Miner. Res. 1989, 4, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Saraiva, M.; Behets, G.; Macedo, A.; Galvao, M.; D’Haese, P.; Drueke, T.B. Evaluation of bone remodeling in hemodialysis patients: Serum biochemistry, circulating cytokines and bone histomorphometry. J. Nephrol. 2009, 22, 783–793. [Google Scholar] [PubMed]
- Cafiero, C.; Gigante, M.; Brunetti, G.; Simone, S.; Chaoul, N.; Oranger, A.; Ranieri, E.; Colucci, S.; Pertosa, G.B.; Grano, M.; et al. Inflammation induces osteoclast differentiation from peripheral mononuclear cells in chronic kidney disease patients: Crosstalk between the immune and bone systems. Nephrol. Dial. Transplant. 2018, 33, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofbauer, L.C.; Lacey, D.L.; Dunstan, C.R.; Spelsberg, T.C.; Riggs, B.L.; Khosla, S. Interleukin-1beta and tumor necrosis factor-alpha, but not interleukin-6, stimulate osteoprotegerin ligand gene expression in human osteoblastic cells. Bone 1999, 25, 255–259. [Google Scholar] [CrossRef]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 2000, 191, 275–286. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Moyses, R.M.; Neves, C.L.; Jorgetti, V.; Draibe, S.A.; Canziani, M.E.; Carvalho, A.B. Osteoporosis in hemodialysis patients revisited by bone histomorphometry: A new insight into an old problem. Kidney Int. 2006, 69, 1852–1857. [Google Scholar] [CrossRef] [Green Version]
- Tousen, Y.; Matsumoto, Y.; Nagahata, Y.; Kobayashi, I.; Inoue, M.; Ishimi, Y. Resistant Starch Attenuates Bone Loss in Ovariectomised Mice by Regulating the Intestinal Microbiota and Bone-Marrow Inflammation. Nutrients 2019, 11, 297. [Google Scholar] [CrossRef] [Green Version]
- McCabe, L.; Britton, R.A.; Parameswaran, N. Prebiotic and Probiotic Regulation of Bone Health: Role of the Intestine and its Microbiome. Curr. Osteoporos. Rep. 2015, 13, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, K.; Krautkramer, K.A.; Org, E.; Romano, K.A.; Kerby, R.L.; Vivas, E.I.; Mehrabian, M.; Denu, J.M.; Backhed, F.; Lusis, A.J.; et al. Interactions between Roseburia intestinalis and diet modulate atherogenesis in a murine model. Nat. Microbiol. 2018, 3, 1461–1471. [Google Scholar] [CrossRef]
- Lampe, J.W.; Navarro, S.L.; Hullar, M.A.; Shojaie, A. Inter-individual differences in response to dietary intervention: Integrating omics platforms towards personalised dietary recommendations. Proc. Nutr. Soc. 2013, 72, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Derrien, M.; Veiga, P. Rethinking Diet to Aid Human-Microbe Symbiosis. Trends Microbiol. 2017, 25, 100–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evenepoel, P.; Dejongh, S.; Verbeke, K.; Meijers, B. The Role of Gut Dysbiosis in the Bone–Vascular Axis in Chronic Kidney Disease. Toxins 2020, 12, 285. https://doi.org/10.3390/toxins12050285
Evenepoel P, Dejongh S, Verbeke K, Meijers B. The Role of Gut Dysbiosis in the Bone–Vascular Axis in Chronic Kidney Disease. Toxins. 2020; 12(5):285. https://doi.org/10.3390/toxins12050285
Chicago/Turabian StyleEvenepoel, Pieter, Sander Dejongh, Kristin Verbeke, and Bjorn Meijers. 2020. "The Role of Gut Dysbiosis in the Bone–Vascular Axis in Chronic Kidney Disease" Toxins 12, no. 5: 285. https://doi.org/10.3390/toxins12050285
APA StyleEvenepoel, P., Dejongh, S., Verbeke, K., & Meijers, B. (2020). The Role of Gut Dysbiosis in the Bone–Vascular Axis in Chronic Kidney Disease. Toxins, 12(5), 285. https://doi.org/10.3390/toxins12050285