1. Introduction
Animal venoms consist of a complex mixture of bioactives, including small molecules, peptides, and proteins [
1,
2,
3]. These natural libraries of compounds have evolved to target specific ion channels and receptors, and they are now actively being mined to discover new pharmacological probes but also potential drug and eco-friendly agrochemical candidates [
4]. Among the venomous arthropods, spiders represent one of the most speciose invertebrate group, with more than 48,000 species described to date [
1]. Spiders can be found in very diverse environments, having adapted to nearly all ground level niches and up to high in the canopy as well as under water [
5]. One of the reasons for the evolutionary success of spiders comes from their ability to produce complex venom to protect themselves from predators (defense), and to facilitate prey capture (predation) [
6,
7]. Besides a sister clade known as the Mesothelae, spiders are broadly divided into the mygalomorphs (“ancient spiders”), the araneomorphs (“modern spiders”), the latter containing the vast majority of described species, (with approximately 39,000 species or >90% of all spiders).
Only a few spider species can be lethal to a fully grown adult human being, including the “terrible trio” made of the black widows (
Latrodectus sp.), the wandering banana spider of South Americas (
Phoneutria nigriventer), and the infamous Australian funnel-web spiders (
Atrax robustus and
Hadronyche sp.) [
8]. Yet, any “black hairy spider” will almost instantaneously trigger uncontrollable fear in many people. As a result of their deadly potential, the venom of spiders has been the focus of many studies early on, notably to investigate the mode of action of the medically important venom components [
8]. Remarkably, in the case of the Australian funnel-web spiders, it is the male that is particularly dangerous [
9]. One explanation for this sexual dimorphism is that males need to leave the safety of their burrow to go find a female for mating, exposing themselves to predators and requiring “defensive toxins” rather than predatory toxins. These sex-driven venom intraspecific variations have since been demonstrated for other spider groups, including the ctenids and tetragnathids [
10,
11]. Other venom variations can be attributed to the method of collection [
12]. Largely employed, electric stimulation of the venom glands produces higher yields but may differ from naturally produced venom. Therefore, caution is required when interpreting the results of electrically stimulated venom, since proteins and other components released from damaged secretory cells can alter the composition.
Whereas many araneomorph spiders have evolved web-building skills to facilitate prey capture, some remain active hunters and rely on speed and fast acting venom to subdue their prey. From this latter group, the wolf spiders (Lycosidae) are widely distributed across the globe, with more than 2300 species described [
13].
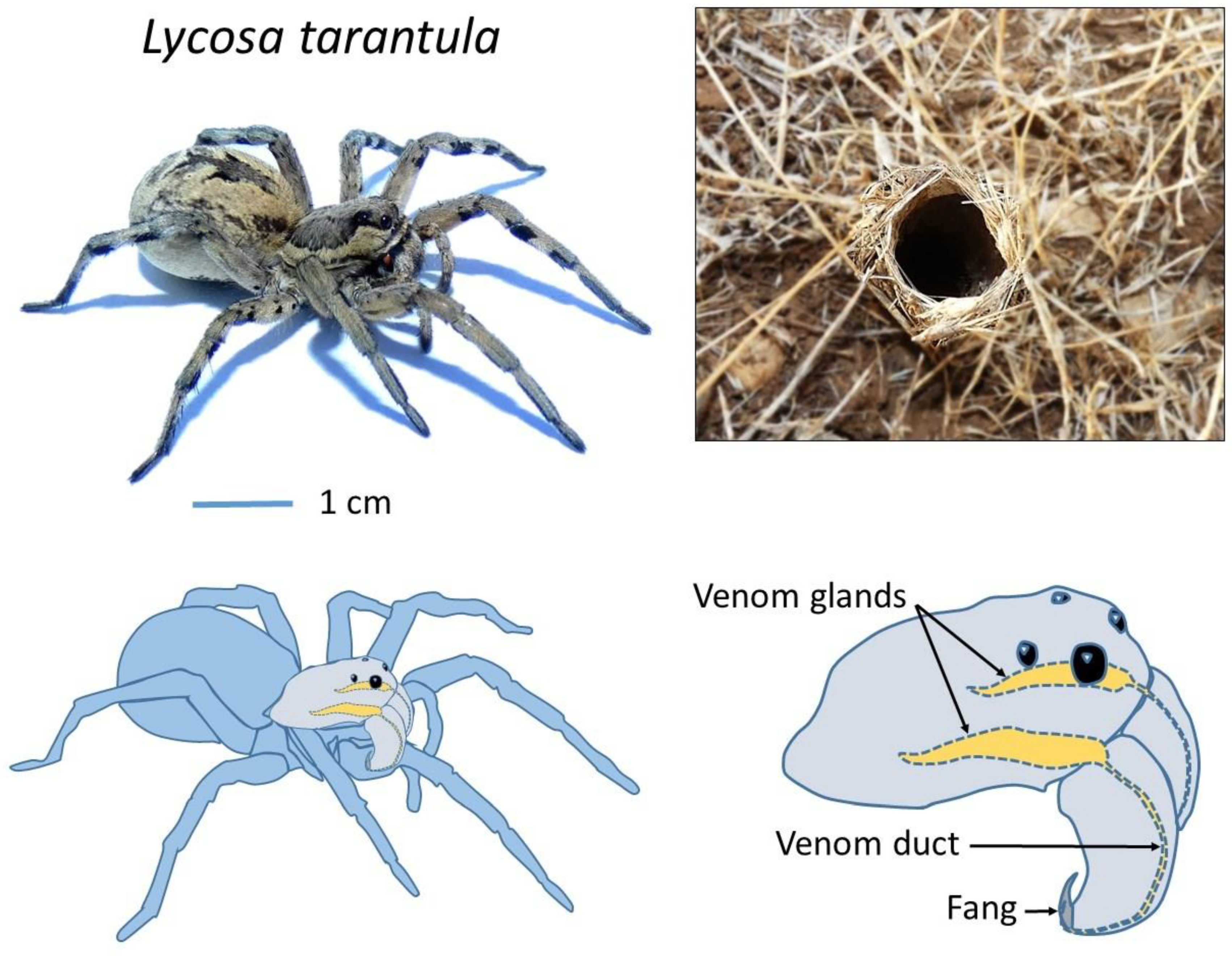
Lycosa tarantula is one of the largest representatives of the Lycosidae family in the Mediterranean basin [
14], reaching a body length of 30 mm. It lives in burrows up to 40 cm deep, which end with a “turret” made of twigs, plant debris and small pebbles agglomerated with silk (
Figure 1). From this burrow, the spider will ambush passing prey. It is found in southern Europe, including France, but also Italy, where this legendary spider was wrongly held responsible for the “tarantism”. A bite from this “tarantula” was supposed to cause a state of lethargy that may lead to death, and affected victims had to dance the “tarantella” if they were to survive [
15]. It was later suggested that the true culprit was likely the local black widow (
Latrodectus tredecimguttatus). Remarkably, although locally common in Mediterranean regions and relatively large, the venom of
L. tarantula is still mostly unknown. Despite being considered harmless to humans, early experiments from naturalist Jean-Henri Fabre demonstrated that its venom can have deleterious effects on small vertebrates [
16]. Indeed, a young sparrow and a mole bitten by an adult
L. tarantula would both succumb to the effect of its venom in less than 48 h, suggesting that this species should be handled carefully.
Accordingly, the venom of other large Lycosidae, such as
L. singoriensis, was shown to affect the physiology of vertebrates, including the contraction of a frog’s heart and the rat vas deferens [
17]. However, the class of toxins responsible for these biological effects are unknown, and the full toxin repertoire produced by a
Lycosa spider remains unclear. Although the venom gland transcriptomes of
L. singoriensis [
18] and
L. vittata [
19] have been obtained via traditional Sanger sequencing, the high throughput next generation sequencing technologies have only been applied to a single species to date, namely
Pardosa pseudoannulata [
20]. Yet, combination of venom gland transcriptomics and venom proteomic analysis has not been reported for any Lycosidae. Thus, we provide here our in-depth analysis of the venom composition of
L. tarantula using an integrated proteotranscriptomic approach. Bioinformatic-based identification of putative toxin-like and protein sequences coupled to LC-MS/MS proteomic analysis of the electrically stimulated venom provide an important resource for a better understanding of the biology of
L. tarantula and for the mining of novel pharmacological compounds of interest.
3. Discussion
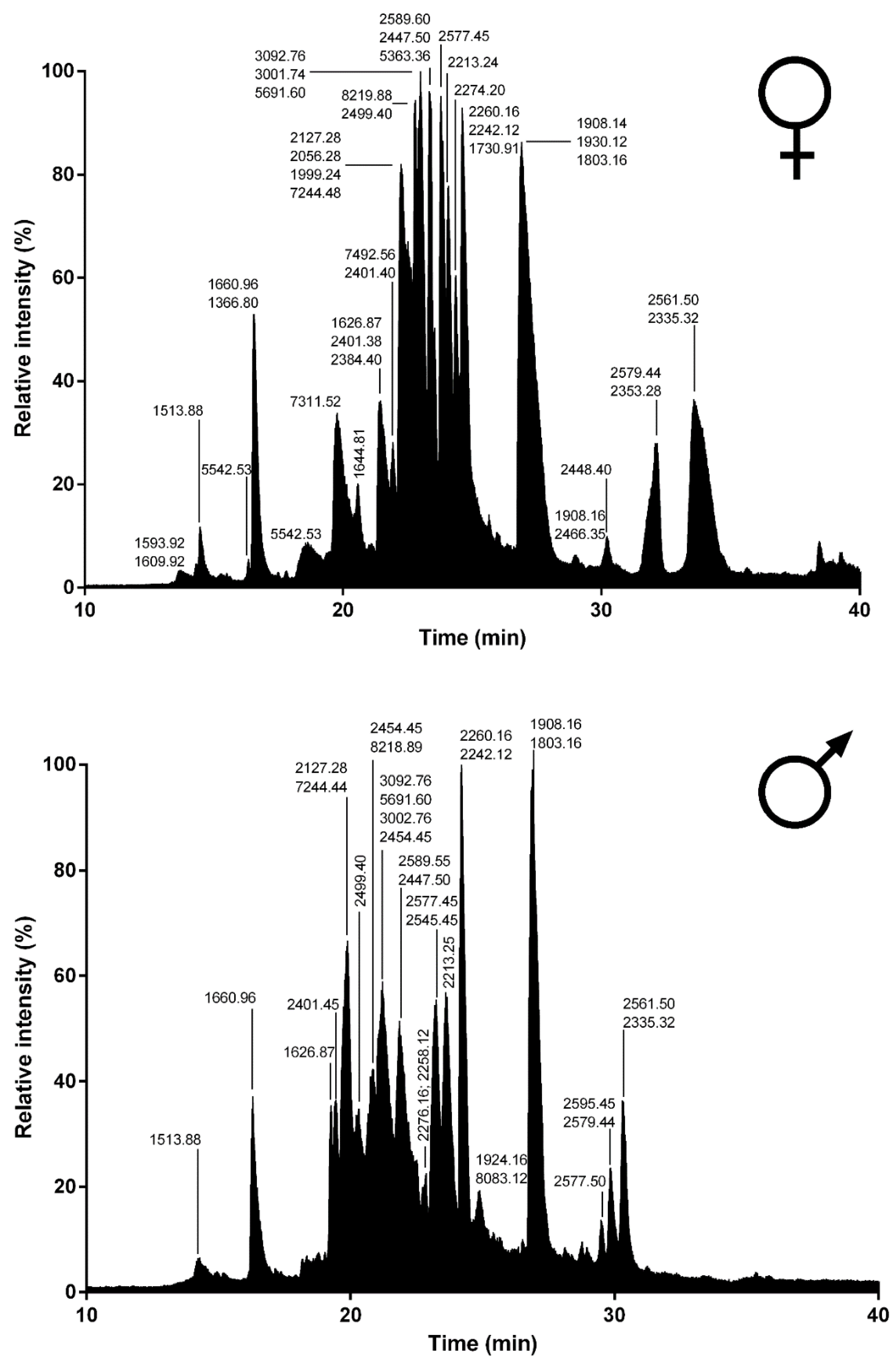
Spider venoms consist of complex mixtures of biologically active compounds that are for the most part gene encoded polypeptides and proteins. Therefore, combining venom gland transcriptomics with venom proteomics is a powerful method to accelerate the identification of full precursors and mature toxins for a better understanding of spider biology, venom-ecology relationships, and for the mining of useful pharmaceutical and agrochemical molecules. In this work, we used such proteotranscriptomics strategy to provide the first insights into the venom of one of the largest Lycosid spiders found in the Mediterranean region,
Lycosa tarantula. Automated bioinformatics analyses followed by manual validation of the venom gland transcriptome revealed 18 distinct venom-related sequences classified into 10 structural families. The disulfide rich neurotoxin-like peptides comprised 10 sequences from six families, whereas the venom proteins were grouped into four distinct classes. Besides these sequences, proteomics investigations also revealed the presence of common cellular proteins, confirming that electrically stimulated venom includes contaminants. Indeed, the manually stimulated venom from both male and female specimens showed a less complex LC-MS profile and a different mass distribution compared to electrically stimulated venom. Interestingly, more than 50% of the masses detected in female venom were unique and not found in the male’s venom, suggesting that some intraspecific variations may be due to sex. Such intraspecific variations between male and female has already been reported in several species of spiders [
27,
28,
29].
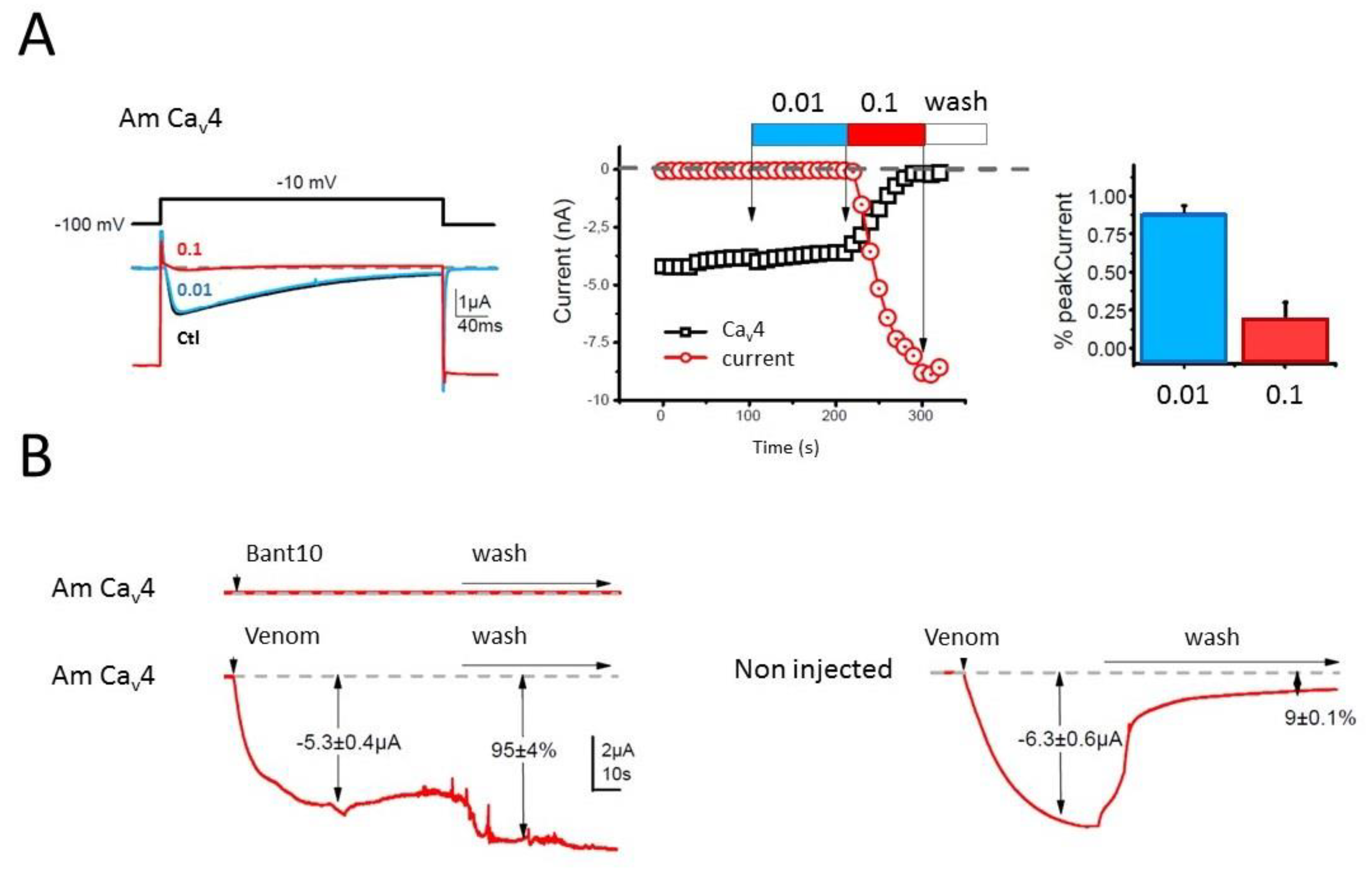
Whereas the biological activity of the neurotoxin-like peptides remains to be elucidated, our preliminary investigation of the crude venom on honeybee Ca
V4 ion channel indicated the possible presence of selective blockers. However, further deconvolution of the crude venom will be necessary to uncover the peptides responsible for this activity, since the cytolytic activity present in the venom prevented accurate electrical measurement. Indeed, application of the crude venom to injected and non-injected oocytes induced a strong leak current, consistent with the cytolytic activity described for several other Lycosidae venoms. The molecular entities responsible for this cytolytic activity are known as antimicrobial peptides (AMPs), which are usually small, highly positively charged linear peptides adopting an amphipathic secondary structure in lipid membrane. Several such AMPs have been isolated and sequenced from Lycosidae venom [
25]. Often, only the mature peptide sequences are available, not the full precursors, raising the question about the molecular origin of these AMPs. Interestingly, in the recently published high throughput sequencing of the venom gland of the Lycosidae
Pardosa pseudoannulata, there is no mention of AMPs. However, a closer inspection of the reported sequences reveals that family A resembles the “inhibitory cysteine knot (ICK) + α-helix” modular toxin described from a Zodariidae spider,
Lachesana tarabaevi. In these modular toxins, the C-terminal fragment synthesized separately was shown to possess membrane-binding activity consistent with a cytolytic effect [
30]. These AMPs are often major components of the venom in Lycosidae, as seen with LyeTx I, a peptide isolated from
Lycosa erythrognatha [
31]. In our transcriptome, family SN_19 also displays the same architecture, with a N-terminal ICK motif and a C-terminal AMP-like sequence, and LC-MS of the venom shows a major contribution of peptides in the 1500–2500 Da range. Interestingly, the C-terminal peptide (QQPKSHKIAEKIVDKAKTVI) of U2-lycotoxin Lt19a has a mass (2260.32 Da) that corresponds to the major peak present in the venom (see
Figure 9). The C-terminal peptides of the other SN_19 family sequences are also in the same mass range of 2000–2500 Da. Further work, including HPLC fractionation and purification steps, will be necessary to confirm this hypothesis.
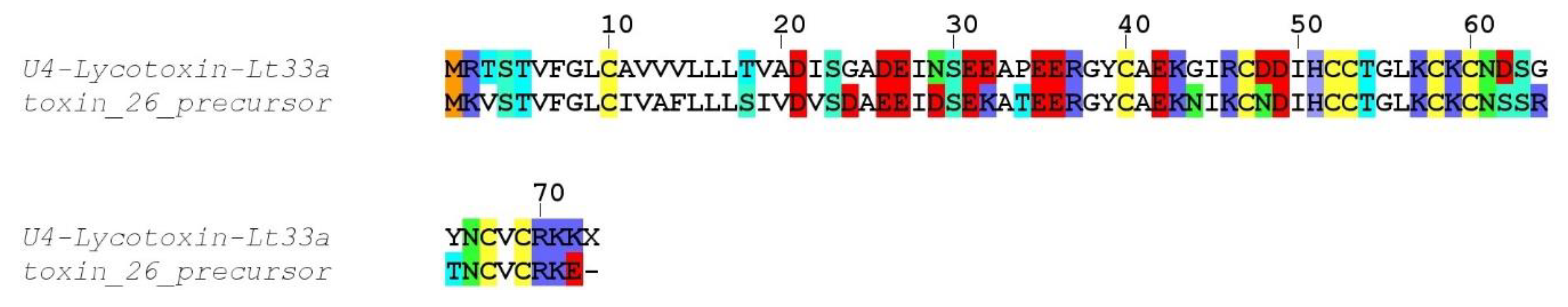
Compared to the transcriptomes of other Lycosidae, such as
Lycosa singoriensis or
Lycosa vittata, our
Lycosa tarantula transcriptome revealed a similar number of structural families, but fewer paralogs for each family [
18,
19]. However, it should be noted that in these studies, many of the reported paralogs were actually often single substitution sequence variants, and several of these substitutions were located in the propeptide, therefore producing identical mature toxin. We suspect that the assembly step of our Illumina reads eliminated the majority of these minor substitution variants that were otherwise picked up by the traditional Sanger sequencing technology used in these studies. However, we cannot exclude that additional neurotoxin-like sequences were missed. For instance, interrogation of the PEAKS “de novo” peptides that did not match any sequence from our transcriptome revealed a number of fragments that show high similarity to known neurotoxins, such as YPESGEGELCTCQQPK (75% U3-lycotoxin-Ls1h,
Lycosa singoriensis), CTPLLHDCSHDR (92% U4-lycotoxin-Ls1b,
Lycosa singoriensis), GCGFLDFNYPGDGR (93% Venom allergen 5,
Lycosa singoriensis), and CCWPWSCVCWSQTLS (87% Omega-lycotoxin-Gsp2671e,
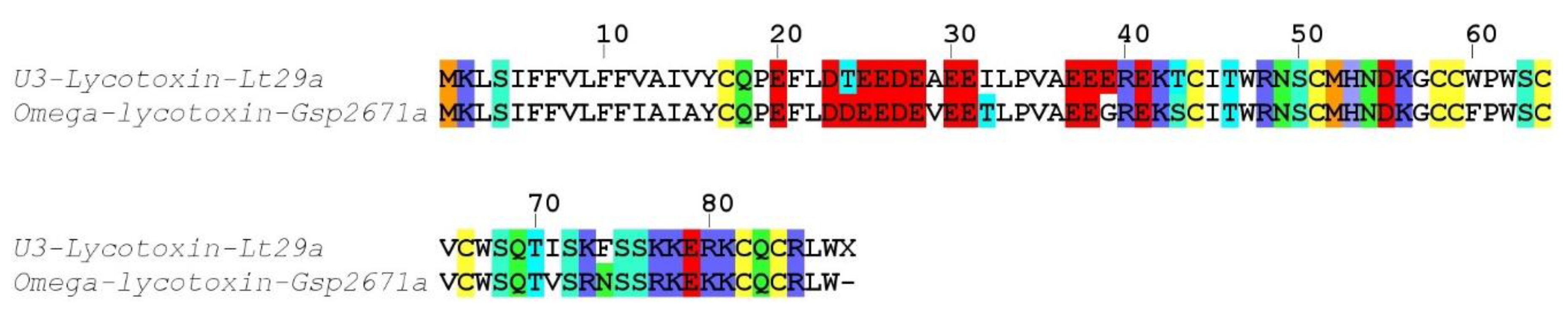
Alopecosa marikovskyi). These unmatched yet high quality proteomic sequences may arise from the different specimens used for venom gland transcriptomics and venom proteomics.
In summary, we have reported here the first proteotranscriptomics analysis of Lycosa tarantula venom, including 18 distinct sequences of short neurotoxin-like peptides and venom proteins from 10 structural families. Future works should focus on the synthesis and pharmacological characterization of some of the neurotoxin-like peptides, as well as the cytolytic activity of some C-terminal fragments. Our data contribute to a treasure trove for the mining of useful pharmacological compounds.
4. Materials and Methods
4.1. Spiders, Venom Collection, and Venom Gland Dissection
Twelve specimens, including two mature males and 10 adult females of Lycosa tarantula were collected in the scrublands around Montpellier, France. These spiders (except males, caught wandering in open areas) were lured out of their burrow using a small stick wiggling around the entrance and caught into plastic jars. Specimens were then individually isolated in small boxes and maintained in the laboratory at room temperature. They were watered twice a week and fed once a week with commercially available mealworms.
To collect the venom from these specimens, two methods were used. First, a “manual stimulation” was applied similar to that described by Liu et al. [
17], where each spider was presented with a piece of soft tubing (0.5 cm in diameter) and aggravated with tweezers to trigger a bite. Venom drops deposited on the tube were recovered using a pipette and diluted in distilled water. Secondly, electrostimulation was carried out on several specimens (
n > 7) using an electric venom extractor based on the Arduino
® Mega 2560 board, specifically designed for the extraction of venom from arthropods and other small size animals [
32].
Spiders were not fed for at least a week prior to the milking session. Specimens were anesthetized before milking (with 5% CO2). Chelicerae were stimulated by electrical impulses (3 to 7 V and approximately 0.5 to 2 A) discharged in a 2 s “working time” and 2 s of “rest time” steps. Released venom was collected from the tip of the fangs using a pipette and transferred to a 1.5 mL microcentrifuge tube containing approximately 20 μL of distilled water. Protein concentration of the venom samples were assessed using a nanophotometer N60 (Implen GmbH, München, Germany). Venom collected from individual spiders was pooled, freeze-dried and stored at −20 °C for subsequent use (proteomic characterization and electrophysiology).
To obtain the amount of mRNA required for the transcriptome sequencing, venom glands of six anesthetized adult female spiders were dissected on ice and placed in a 1.5 mL microcentrifuge tube containing 500 µL of lysis buffer. Next, the mRNA was extracted using a commercial kit (Magnetic mRNA isolation kit, Biolabs) following the manufacturer’s instructions. After extraction, the mRNA concentration was measured using a nanophotometer N60 (Implen GmbH, München, Germany).
4.2. Library prepaRation and Illumina Sequencing
RNA-Seq libraries were constructed with the Truseq stranded mRNA sample preparation (low throughput protocol) kit from Illumina (San Diego, CA, USA). Depending on the samples, 100 or 200 ng of mRNA was used for the construction of the libraries. Next, the mRNA was fragmented into small pieces using divalent cations under elevated temperature. The cleaved RNA fragments were copied into first strand cDNA using SuperScript II reverse transcriptase, Actinomycin D and random hexamer primers. The second strand cDNA was synthesized by replacing deoxythymidine triphosphate (dTTP) with deoxyuridine triphosphate (dUTP). These cDNA fragments have the addition of a single ‘A’ base and subsequent ligation of the adapter. The products are then purified and enriched with 15 cycles of PCR. The final cDNA libraries were validated with a Fragment Analyzer (Agilent Santa Clara, CA, USA) and quantified with a KAPA qPCR kit (Kapa Biosystems, Wilmington, MA, USA).
The transcriptome of L. tarantula was sequenced as part of a larger project comprising 15 other venom gland transcriptomes. On three sequencing lanes of V2 flowcells, the 16 libraries were pooled in equal proportions, denatured with NaOH and diluted to 18 pM before clustering. Cluster formation, primer hybridization and single-end read, 125 cycles sequencing were performed on cBot and HiSeq2500 (Illumina, San Diego, CA, USA) respectively.
Image analysis and base calling were performed using the HiSeq Control Software v.2.2.68 (Illumina, San Diego, CA, USA) and Real-Time Analysis component v.1.18.66.3 (Illumina, San Diego, CA, USA). Demultiplexing was performed using Illumina’s conversion software (bcl2fastq 2.18). The quality of the data was assessed using FastQC from the Babraham Institute v.0.11.5 and the Illumina software SAV (Sequencing Analysis Viewer) v. 2.1.8 (Illumina, San Diego, CA, USA). Potential contaminants were investigated with the FastQ Screen software from the Babraham Institute v.0.9.5.
4.3. Bioinformatics Sequence Analysis
Data issued from the sequencing platform were trimmed using the Trinity trimmomatic tool with default parameters. Reads were assembled using the Trinity software (version 2.1.1) [
33]. Obtained contigs were translated in-silico into their six reading frames and annotated using the following procedure. An in-house database composed of all spider toxins from Arachnoserver, UniprotKB/SwissProt and Venomzone were created using makeblastdb of BLAST+ package after redundancy removal using CD-HIT [
34,
35] at the threshold of 1.00.
All Contigs were searched using an improved version of the previously published Ekenda Hidden Markov Models (HMM) library and the hmmcompete program [
21].
All Contigs were submitted to a first BLAST step against this database to provide an annotated subset of the transcriptome. Annotated contigs were again BLASTed against the whole UniprotKB/SwissProt database to confirm the exactitude of obtained hits and remove false positive hits (BlastP against UniProtKB with e-threshold = 0.0001; matrix BLOSUM-62, non-filtering and gapped; UniProtKB/SwissProt 2019_03).
Spider toxin-related sequences were identified and classified into toxins family using Ekenda Hidden Markov Models (HMM) and the hmmcompete program. Signal Peptide and propeptide were predicted using respectively SignalP version 5.0 [
22] and SpiderP [
23] directly from the Arachnoserver web server at
http://www.arachnoserver.org/spiderP.html. A final manual validation step was performed: multiple sequence alignments using MAFFT Version 7 [
36], variant identification, and cleavage site validation. All peptide hits as well as their corresponding contigs sequences were further analyzed at nucleotide level to detect eventual mutations. Nucleotide sequence variants that obviously resulted from sequencing errors, assembly errors or frame shifts were excluded.
4.4. Proteomics
4.4.1. Liquid Chromatography Coupled Mass Spectrometry (LC-MS)
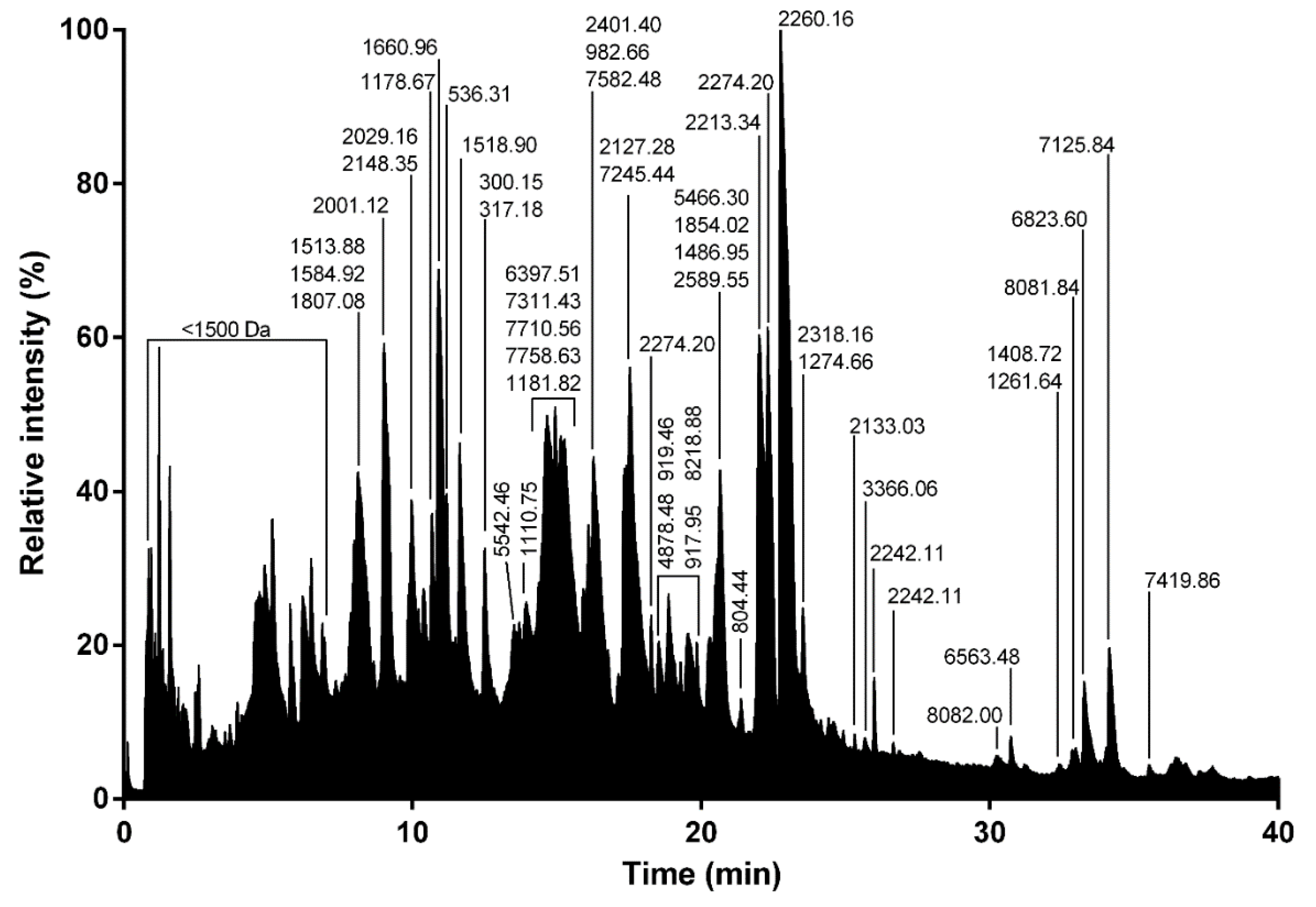
RP-UPLC was operated on an Acquity H-Class ultrahigh performance liquid chromatography (UPLC) system (Waters, Corp., Milford, MA, United States) fitted with a UV detector (diode array detector) under the control of Waters MassLynx software (version 4.1). Separation of the L. tarantula venom (~600 μg) was achieved using a Kinetex C18 100 Å column (2.1 × 150 mm, 3 µm) fitted with a pre-column. Elution was carried out using a gradient of 0–80% B (0.1% formic acid in acetonitrile) in 80 min. Samples eluting from the UPLC were introduced into the mass spectrometer at a flow rate of 500 µL/min. Acquisitions were carried out over the range 50 Da to 1800 Da m/z every 0.1 s on a Synapt-G2-S high-definition MS system (Waters, Corp., Milford, MA, United States). To obtain the molecular masses of the venom components eluting between 0 and 40 min, each peak from the total ion current (TIC) chromatogram was analyzed with Waters Mass Lynx software (version 4.1) (Waters, Milford, MA, USA).
4.4.2. Shotgun Proteomics (LC-MS/MS)
Prior to shotgun proteomics, venom protein extracts were denatured, reduced, and alkylated. Briefly, each sample (~50 μg) was dissolved in 89 μL of triethylammonium bicarbonate (TEABC) 100 mM. One microliter of dithiothreitol (DTT) 1 M was added and incubation was performed for 30 min at 60 °C. A volume of 10 μL of iodoacetamide (IAA) 0.5 M was added (incubation for 30 min in the dark). Enzymatic digestion was performed by addition of 2 μg trypsin (Gold, Promega, Madison, WI, USA) in TEABC 100 mM and incubation overnight at 30 °C. After completing the digestion step, peptides were purified and concentrated using OMIX Tips C18 reverse-phase resin (Agilent Technologies Inc., Santa Clara, CA, USA) according to the manufacturer’s specifications. Peptides were dehydrated in a vacuum centrifuge.
Samples were then subjected to nano-flow liquid chromatography coupled to tandem mass spectrometry (NanoLC-MS/MS). Samples were resuspended in 20 μL formic acid (0.1%, buffer A) and 1 µL was loaded onto an analytical 25 cm reversed-phase column (75 mm inner diameter, Acclaim Pepmap 100® C18, Thermo Fisher Scientific) and separated with an Ultimate 3000 RSLC system (Thermo Fisher Scientific, Waltham, MA, USA) coupled to a Q Exactive HF-X (Thermo Fisher Scientific, Waltham, MA, USA) via a nano-electrospray source, using a 123 min gradient of 6% to 40% of buffer B (80% ACN, 0.1% formic acid) and a flow rate of 300 nL/min. MS/MS analyses were performed in a data-dependent mode. Full scans (375–1500 m/z) were acquired in the Orbitrap mass analyzer (Thermo Fisher Scientific, Waltham, MA, USA) with a 60,000 resolution at 200 m/z. For the full scans, 3 × 106 ions were accumulated within a maximum injection time of 60 ms and detected in the Orbitrap mass analyzer. The twelve most intense ions with charge states ≥2 were sequentially isolated to a target value of 1 × 105 with a maximum injection time of 45 ms and fragmented by higher-energy collisional dissociation (HCD) in the collision cell (normalized collision energy of 28%) and detected in the Orbitrap mass analyzer at 30,000 resolution.
4.4.3. Bioinformatic Integration of Proteomic and Transcriptomic Data
PEAKS Studio 8.5 software (Bioinformatics solutions, Waterloo, ON, Canada) was used to match MS/MS spectra obtained from proteomic analysis of L. tarantula venom. MS spectra were elucidated based on a personalized database resulting from assembled contigs translated into their six reading frames. Carbamidomethylation was set as fixed modification, while oxidation (M) was set as variable modifications, with maximum missed cleavages at 3 for trypsin digestion. Parent mass and fragment mass error tolerance were set at 5 ppm and 0.015 Da respectively. False discovery rate (FDR) of 1% and unique peptide ≥2 were used for filtering out inaccurate proteins. A −10lgP > 120 was used to estimate whether the detected proteins was identified by enough reliable peptides MS/MS spectra. In order to identify more relevant sequences, the Spider algorithm from PEAKS Studio software was used to find additional mutations or to correct the sequences. This algorithm corrects the sequences stored in transcriptomic database with de novo sequences based on MS/MS spectra, which allowed to identify post-translational modifications (PTMs) and mutations. Minimum ion intensity for mutation and PTMs was set to 5%, and ALC score ≥ 90 for de novo sequences leading to low precursor mass error in order to identify reliable PTM’s and potential mutations.
4.5. Electrophysiology
Ovaries were surgically removed from Xenopus laevis female, anesthetized using a 0.2% MS222 solution (Sigma Saint-Louis, MO, USA). After a first mechanical dissociation and extensive washing using the OR-2 solution (containing in mM: NaCl, 100; MgCl2, 2; KCl, 2; HEPES, 10), oocytes were isolated by approximately 2 h enzymatic dissociation using 1 mg/mL collagenase IA (Sigma Saint-Louis, MO, USA) dissolved in OR-2. Oocytes were then washed several times with OR-2 and selected in the survival medium (containing in mM: NaCl, 96; MgCl2, 2; KCl, 2; CaCl2, 1.8; HEPES, 10; pyruvic acid, 2.5; gentamycin, 50 ~μg/mL; neutralized at pH 7.2 using NaOH).
Oocytes injection was performed in the equatorial region by employing a home-made pneumatic injectory. Xenopus oocytes were microinjected with RNA corresponding to the AmCaV4 channel (1 µg/µL) of domestic honeybee, Apis mellifera. About 40 oocytes were injected with 1 µL of solution, and these injected oocytes were incubated at 18 °C in OR-2 solution for at least 24 h for 2–7 days at 19 °C under gentle agitation before recording. The survival medium was renewed daily.
Whole cell Ba2+ currents were recorded under two electrode voltage-clamp by employing the GeneClamp 500 amplifier (Axon Inst., Burlingame, CA, USA). Current and voltage electrodes were filled with a solution containing: KCl 3M; KOH. The bath-clamp head stage was connected to the bath using two agar bridges filled with 2% agar in 3M KCl, and the extracellular solution (physiological solution) was BANT10 (BaOH: 10 mM, TEAOH 20%: 12 mL, NMDG: 30 mM, CsOH: 2 mM, HEPES: 10 mM, pH = 7.2 with methane sulfonate). Injection of BAPTA (in mM: BAPTA-free acid (Sigma Saint-Louis, MO, USA), 100; CsOH, 10; HEPES, 10; pH 7.2) into oocytes was performed using a third microelectrode (in order to eliminate any Ca2+-activated Cl current). Under these conditions uncontaminated Ba2+ currents can be recorded. Ba2+ currents were elicited by series of depolarizing steps of 400 ms duration from a holding potential of −100 mV, to 10 mV every 10 s. Voltage-protocol and ionic currents were generated and recorded using the Clampex software (pClamp, ver 7.0, Axon Inst) (Molecular Devices, San Jose, CA, USA). Venom solution to be tested were prepared just prior to the experiment by adding the desired concentration in the BANT10 physiological solution. The different concentrations of venom (0.01, 0.1, and 1 µM) were then applied manually in a static bath using a pipette delivering a dose from about 20 μL.
The effect of administered venom was measured when steady state was reached (after about 1 to 2 min, i.e., 6–12 depolarizations) as a percentage of inhibition of the peak Ba2+ current amplitude recorded during a depolarizing pulse ranging of −100 to 10 mV. Data are presented as means ± S.E.M. from at least three oocytes.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}