Immunotoxins Immunotherapy against Hepatocellular Carcinoma: A Promising Prospect


Abstract
:1. Introduction
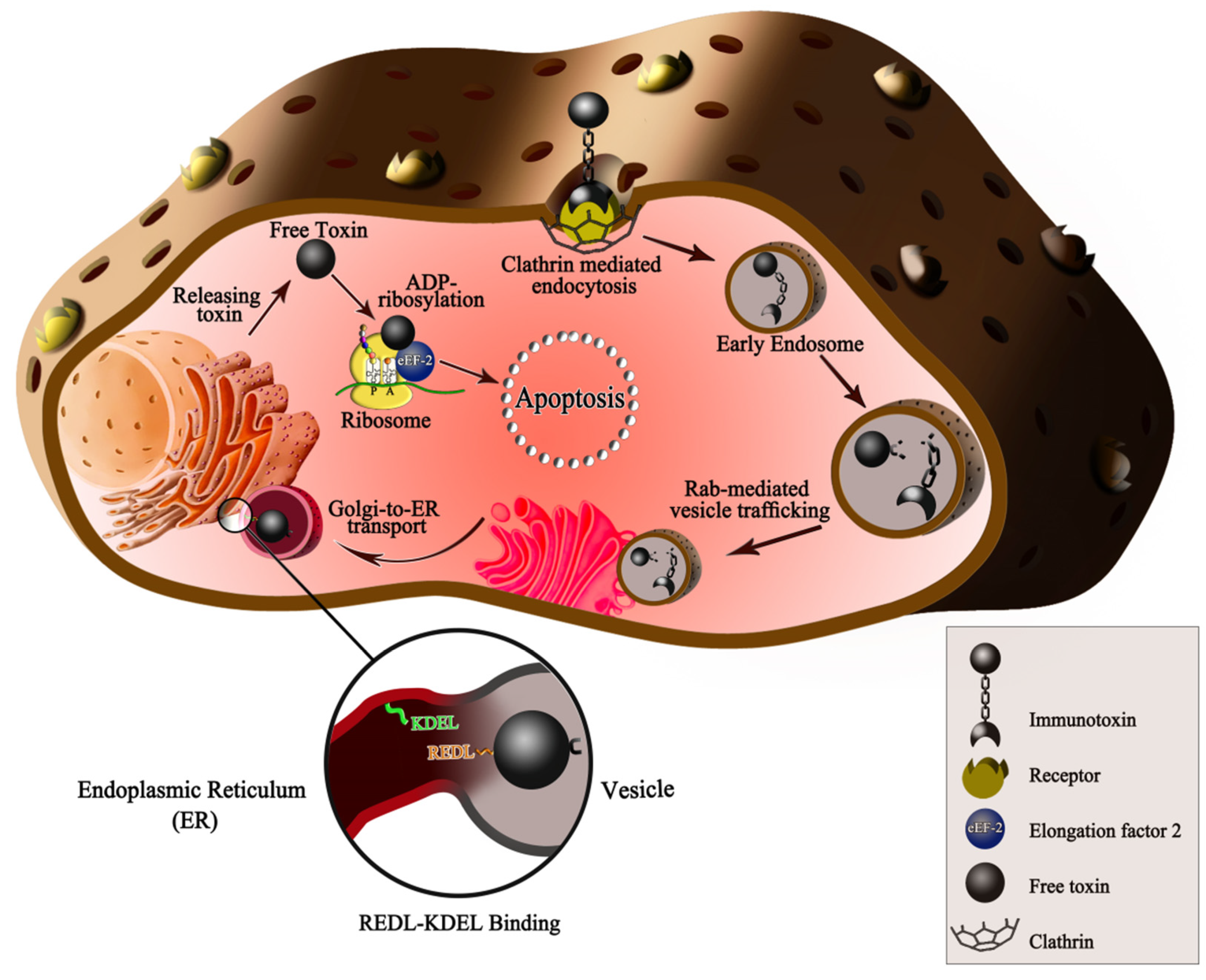
2. Immunotoxins
3. Factors Influencing Immunotoxins Efficiency in HCC
4. HCC Tumor Markers
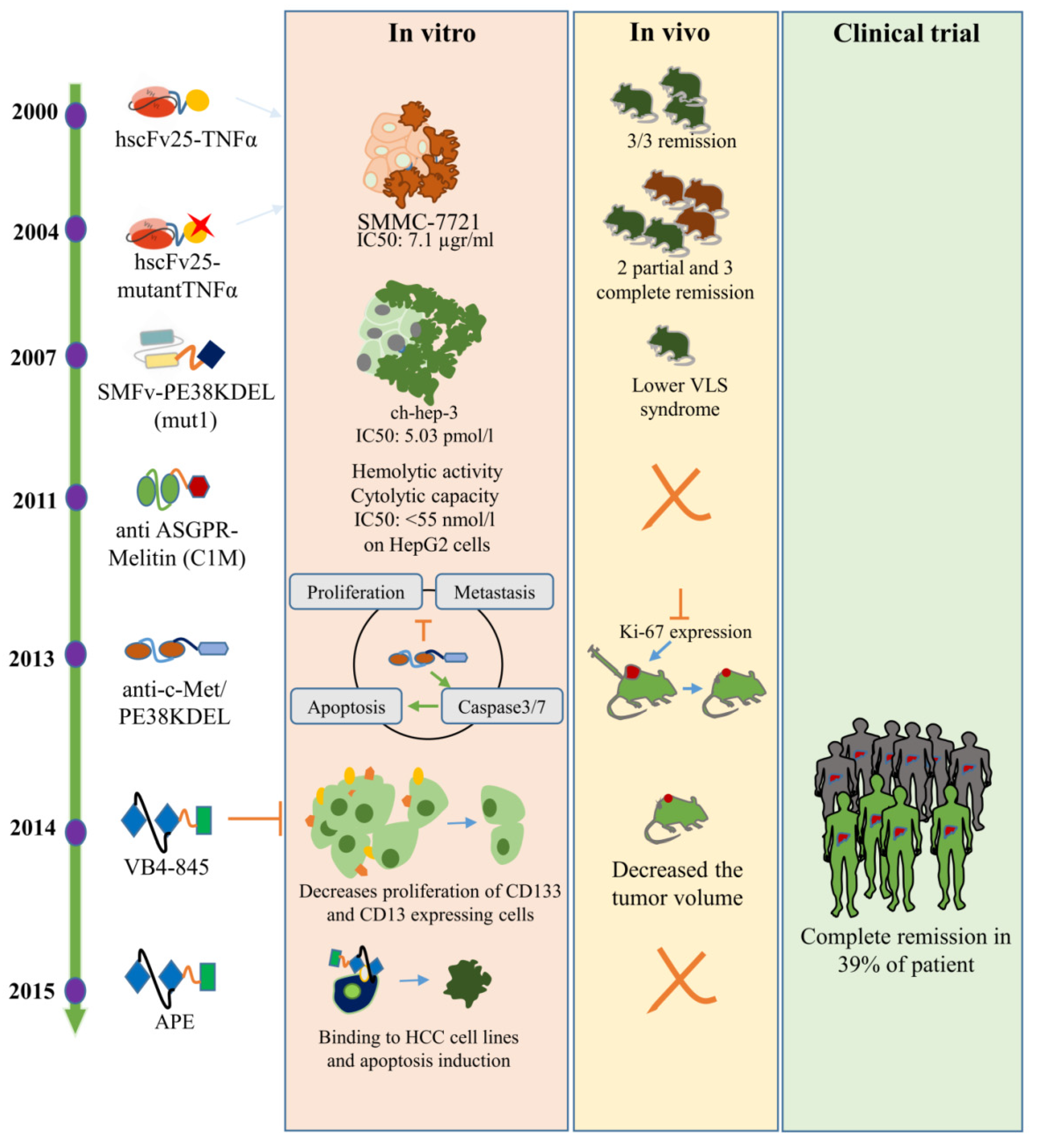
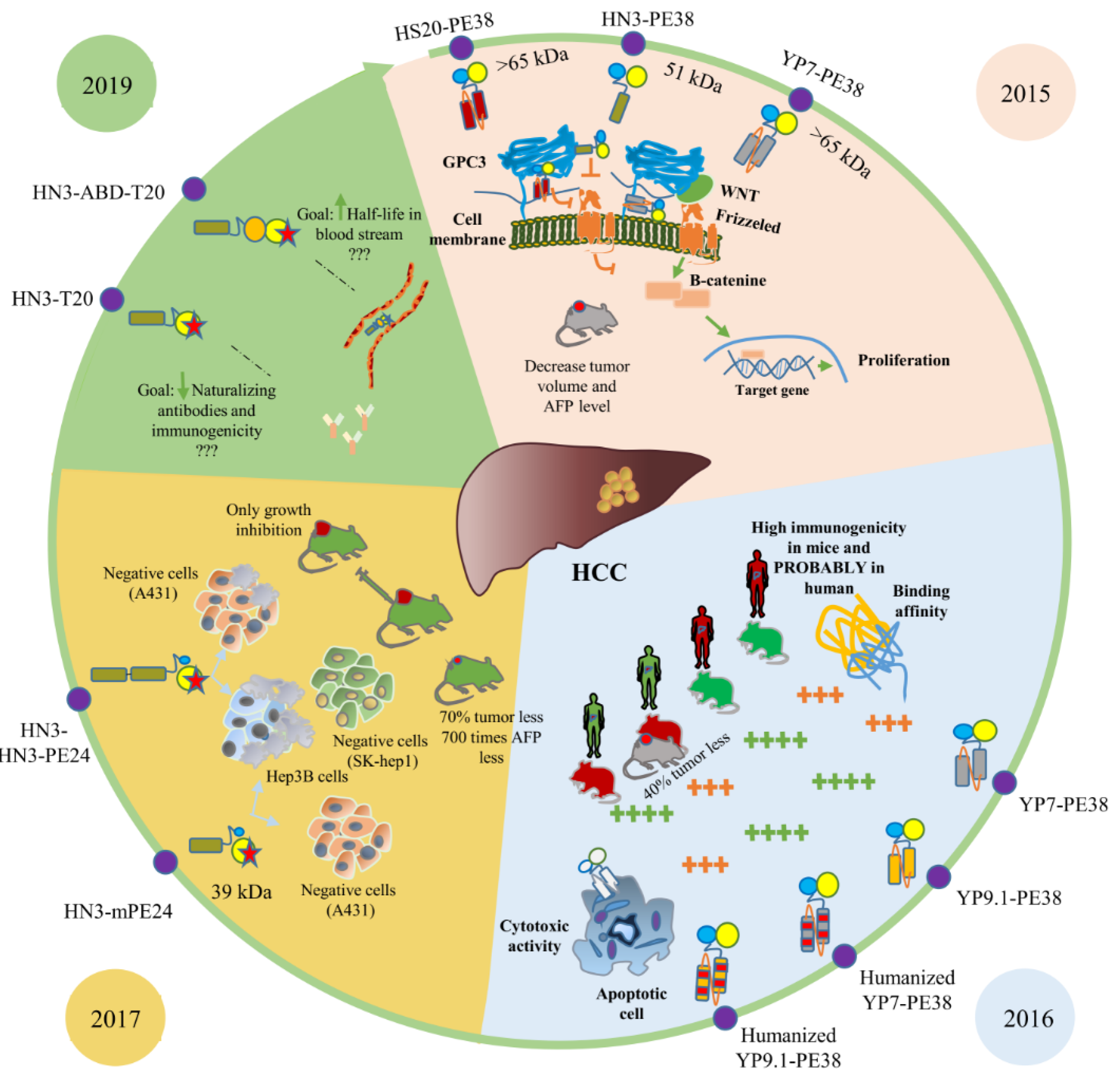
5. Anti-HCC Immunotoxins
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fleming, B.; Ho, M. Glypican-3 Targeting Immunotoxins for the Treatment of Liver Cancer. Toxins 2016, 8, 274. [Google Scholar] [CrossRef] [PubMed]
- Vasanthakumar, S.; Sasikala, P.; Padma, M.; Balachandar, V.; Venkatesh, B.; Ganesan, S. EpCAM as a Novel Therapeutic Target for Hepatocellular Carcinoma. J. Oncol. Sci. 2017, 3, 71–76. [Google Scholar]
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of Hepatocellular Carcinoma: Epidemiology, Etiology, and Carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of Worldwide Burden of Cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Gao, W.; Tang, Z.; Zhang, Y.F.; Feng, M.; Qian, M.; Dimitrov, D.S.; Ho, M. Immunotoxin Targeting Glypican-3 Regresses Liver Cancer via Dual Inhibition of Wnt Signalling and Protein Synthesis. Nat. Commun. 2015, 6, 6536. [Google Scholar] [CrossRef] [Green Version]
- Ozakyol, A. Global Epidemiology of Hepatocellular Carcinoma (HCC Epidemiology). J. Gastrointest. Cancer 2017, 48, 238–240. [Google Scholar] [CrossRef]
- Kawakami, K.; Aggarwal, B.B.; Puri, R.K. Cytotoxins and Immunotoxins for Cancer Therapy; CRC Press: Boca Raton, FL, USA, 2004; ISBN 0415263654. [Google Scholar]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Khafaei, M.; Rezaie, E.; Mohammadi, A.; Shahnazi Gerdehsang, P.; Ghavidel, S.; Kadkhoda, S.; Zorrieh Zahra, A.; Forouzanfar, N.; Arabameri, H.; Tavallaie, M. MiR-9: From Function to Therapeutic Potential in Cancer. J. Cell. Physiol. 2019, 9, 14651–14665. [Google Scholar] [CrossRef]
- Amani, N.; Dorkoosh, F.A.; Mobedi, H. ADCs, as Novel Revolutionary Weapons for Providing a Step Forward in Targeted Therapy of Malignancies. Curr. Drug Deliv. 2020, 17, 23–51. [Google Scholar] [CrossRef]
- Knödler, M.; Buyel, J.F. Plant-Made Immunotoxin Building Blocks: A Roadmap for Producing Therapeutic Antibody-Toxin Fusions. Biotechnol. Adv. 2021, 47, 107683. [Google Scholar] [CrossRef]
- Foss, F. Clinical Experience with Denileukin Diftitox (ONTAK). Proc. Semin. Oncol. 2006, 33, 11–16. [Google Scholar] [CrossRef]
- Vallera, D.A.; Kreitman, R.J. Immunotoxins Targeting B Cell Malignancy—Progress and Problems with Immunogenicity. Biomedicines 2019, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezaie, E.; Nekoie, H.; Miri, A.; Oulad, G.; Ahmadi, A.; Saadati, M.; Bozorgmehr, M.; Ebrahimi, M.; Salimian, J. Different Frequencies of Memory B-Cells Induced by Tetanus, Botulinum, and Heat-Labile Toxin Binding Domains. Microb. Pathog. 2019, 127, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Marvig, R.L.; Sommer, L.M.; Molin, S.; Johansen, H.K. Convergent Evolution and Adaptation of Pseudomonas Aeruginosa within Patients with Cystic Fibrosis. Nat. Genet. 2015, 47, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Michalska, M.; Wolf, P. Pseudomonas Exotoxin A: Optimized by Evolution for Effective Killing. Front. Microbiol. 2015, 6, 963. [Google Scholar] [CrossRef] [Green Version]
- Mazor, R.; Pastan, I. Immunogenicity of Immunotoxins Containing Pseudomonas Exotoxin A: Causes, Consequences, and Mitigation. Front. Immunol. 2020, 11, 1261. [Google Scholar] [CrossRef]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R. The Immunogenicity of Humanized and Fully Human Antibodies. MAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [Green Version]
- FitzGerald, D.J.; Kreitman, R.; Wilson, W.; Squires, D.; Pastan, I. Recombinant Immunotoxins for Treating Cancer. Int. J. Med. Microbiol. 2004, 293, 577–582. [Google Scholar] [CrossRef]
- Lv, M.H.; Qiu, F.; Li, T.T.; Sun, Y.J.; Zhang, C.M.; Zhu, P.; Qi, X.; Wan, J.; Yang, K.; Zhang, K. Construction, Expression, and Characterization of a Recombinant Immunotoxin Targeting EpCAM. Mediat. Inflamm. 2015. [Google Scholar] [CrossRef]
- Hu, W.; Li, F.; Yang, X.; Li, Z.; Xia, H.; Li, G.; Wang, Y.; Zhang, Z. A Flexible Peptide Linker Enhances the Immunoreactivity of Two Copies HBsAg PreS1 (21–47) Fusion Protein. J. Biotechnol. 2004, 107, 83–90. [Google Scholar] [CrossRef]
- Rezaie, E.; Mohammadi, M.; Sakhteman, A.; Bemani, P.; Ahrari, S. Application of Molecular Dynamics Simulations to Design a Dual-Purpose Oligopeptide Linker Sequence for Fusion Proteins. J. Mol. Model. 2018, 24, 313. [Google Scholar] [CrossRef]
- Masuda, K.; Takahashi, K.; Hirano, K.; Takagishi, Y. Selective Antitumor Effect of Thioether-Linked Immunotoxins Composed of Gelonin and Monoclonal Antibody to Alpha-Fetoprotein or Its F(Ab’)2 Fragment. Tumor Biol. 1994, 15, 175–183. [Google Scholar] [CrossRef]
- Wang, Q.C.; Ying, W.B.; Xie, H.; Zhang, Z.C.; Yang, Z.H.; Ling, L.Q. Trichosanthin-Monoclonal Antibody Conjugate Specifically Cytotoxic to Human Hepatoma Cells in Vitro. Cancer Res. 1991, 51, 3353–3355. [Google Scholar] [PubMed]
- Khan, D.A. Hypersensitivity and Immunologic Reactions to Biologics: Opportunities for the Allergist. Ann. Allergy Asthma Immunol. 2016, 117, 115–120. [Google Scholar] [CrossRef]
- Baluna, R.; Vitetta, E.S. Vascular Leak Syndrome: A Side Effect of Immunotherapy. Immunopharmacology 1997, 37, 117–132. [Google Scholar] [CrossRef]
- Schroeder, B.; McNiven, M.A. Importance of Endocytic Pathways in Liver Function and Disease. Compr. Physiol. 2014, 4, 1403. [Google Scholar] [PubMed] [Green Version]
- El Hage, T.; Lorin, S.; Decottignies, P.; Djavaheri-Mergny, M.; Authier, F. Proteolysis of Pseudomonas Exotoxin A within Hepatic Endosomes by Cathepsins B and D Produces Fragments Displaying in Vitro ADP-ribosylating and Apoptotic Effects. FEBS J. 2010, 277, 3735–3749. [Google Scholar] [CrossRef]
- Weldon, J.E.; Pastan, I. A Guide to Taming a Toxin–Recombinant Immunotoxins Constructed from Pseudomonas Exotoxin A for the Treatment of Cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alewine, C.; Hassan, R.; Pastan, I. Advances in Anticancer Immunotoxin Therapy. Oncologist 2015. [Google Scholar] [CrossRef] [Green Version]
- Wayne, A.S.; FitzGerald, D.J.; Kreitman, R.J.; Pastan, I. Immunotoxins for Leukemia. Blood 2014, 123, 2470–2477. [Google Scholar] [CrossRef] [Green Version]
- Pai-Scherf, L.H.; Villa, J.; Pearson, D.; Watson, T.; Liu, E.; Willingham, M.C.; Pastan, I. Hepatotoxicity in Cancer Patients Receiving Erb-38, a Recombinant Immunotoxin That Targets the ErbB2 Receptor. Clin. Cancer Res. 1999, 5, 2311–2315. [Google Scholar]
- Coulie, P.G.; Van den Eynde, B.J.; Van Der Bruggen, P.; Boon, T. Tumour Antigens Recognized by T Lymphocytes: At the Core of Cancer Immunotherapy. Nat. Rev. Cancer 2014, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, N. Human Tumor Antigens and Cancer Immunotherapy. Biomed. Res. Int. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerutti, P.; Hussain, P.; Pourzand, C.; Aguilar, F. Mutagenesis of the H-Ras Protooncogene and the P53 Tumor Suppressor Gene. Cancer Res. 1994, 54, 1934–1938. [Google Scholar]
- Wargalla, U.C.; Reisfeld, R.A. Rate of Internalization of an Immunotoxin Correlates with Cytotoxic Activity against Human Tumor Cells. Proc. Natl. Acad. Sci. USA 1989, 86, 5146–5150. [Google Scholar] [CrossRef] [Green Version]
- Cohen, K.A.; Liu, T.F.; Cline, J.M.; Wagner, J.D.; Hall, P.D.; Frankel, A.E. Safety Evaluation of DT 388 IL3, a Diphtheria Toxin/Interleukin 3 Fusion Protein, in the Cynomolgus Monkey. Cancer Immunol. Immunother. 2005, 54, 799–806. [Google Scholar] [CrossRef]
- Du, X.; Ho, M.; Pastan, I. New Immunotoxins Targeting CD123, a Stem Cell Antigen on Acute Myeloid Leukemia Cells. J. Immunother. 2007, 30, 607–613. [Google Scholar] [CrossRef]
- Liu, T.F.; Urieto, J.O.; Moore, J.E.; Miller, M.S.; Lowe, A.C.; Thorburn, A.; Frankel, A.E. Diphtheria Toxin Fused to Variant Interleukin-3 Provides Enhanced Binding to the Interleukin-3 Receptor and More Potent Leukemia Cell Cytotoxicity. Exp. Hematol. 2004, 32, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Hexham, J.M.; Dudas, D.; Hugo, R.; Thompson, J.; King, V.; Dowling, C.; Neville, D.M., Jr.; Digan, M.E.; Lake, P. Influence of Relative Binding Affinity on Efficacy in a Panel of Anti-CD3 ScFv Immunotoxins. Mol. Immunol. 2001, 38, 397–408. [Google Scholar] [CrossRef]
- Hou, S.-C.; Chen, H.-S.; Lin, H.-W.; Chao, W.-T.; Chen, Y.-S.; Fu, C.-Y.; Yu, C.-M.; Huang, K.-F.; Wang, A.H.-J.; Yang, A.-S. High Throughput Cytotoxicity Screening of Anti-HER2 Immunotoxins Conjugated with Antibody Fragments from Phage-Displayed Synthetic Antibody Libraries. Sci. Rep. 2016, 6, 31878. [Google Scholar] [CrossRef] [Green Version]
- Sohrabi, E.; Moslemi, M.; Rezaie, E.; Nafissi, N.; Khaledi, M.; Afkhami, H.; Fathi, J.; Zekri, A. The Tissue Expression of MCT3, MCT8, and MCT9 Genes in Women with Breast Cancer. Genes Genom. 2021, 43, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, E.; Rezaie, E.; Heiat, M.; Sefidi-Heris, Y. An Integrated Data Analysis of MRNA, MiRNA and Signaling Pathways in Pancreatic Cancer. Biochem. Genet. 2021, 59, 1326–1358. [Google Scholar] [CrossRef] [PubMed]
- Gires, O.; Seliger, B. Tumor-Associated Antigens: Identification, Characterization, and Clinical Applications; John Wiley & Sons: Hoboken, NJ, USA, 2009; ISBN 3527625984. [Google Scholar]
- Lander, A.D.; Selleck, S.B. The Elusive Functions of Proteoglycans: In Vivo Veritas. J. Cell Biol. 2000, 148, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G. E-Cadherin-Integrin Crosstalk in Cancer Invasion and Metastasis. J. Cell Sci. 2013, 126, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.K.; Poon, R.T.P.; Yuen, A.P.; Ling, M.T.; Kwok, W.K.; Wang, X.H.; Wong, Y.C.; Guan, X.Y.; Man, K.; Chau, K.L.; et al. Twist Overexpression Correlates with Hepatocellular Carcinoma Metastasis through Induction of Epithelial-Mesenchymal Transition. Clin. Cancer Res. 2006, 12, 5369–5376. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Gao, W.; Feng, M.; Pastan, I.; Ho, M. Construction of an Immunotoxin, HN3-MPE24, Targeting Glypican-3 for Liver Cancer Therapy. Oncotarget 2017, 8, 32450–32460. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Takahashi, T.; Motoya, S.; Ishida, T.; Itoh, F.; Adachi, M.; Hinoda, Y.; Imai, K. MUC1 Mucin Core Protein Binds to the Domain 1 of ICAM-1. Digestion 2001, 63, 87–92. [Google Scholar] [CrossRef]
- Haugsten, E.M.; Malecki, J.; Bjørklund, S.M.S.; Olsnes, S.; Wesche, J. Ubiquitination of Fibroblast Growth Factor Receptor 1 Is Required for Its Intracellular Sorting but Not for Its Endocytosis. Mol. Biol. Cell 2008, 19, 3390–3403. [Google Scholar] [CrossRef] [Green Version]
- Kapeller, R.; Chakrabarti, R.; Cantley, L.; Fay, F.; Corvera, S. Internalization of Activated Platelet-Derived Growth Factor Receptor-Phosphatidylinositol-3′ Kinase Complexes: Potential Interactions with the Microtubule Cytoskeleton. Mol. Cell. Biol. 1993, 13, 6052–6063. [Google Scholar] [CrossRef] [Green Version]
- Bryant, D.M.; Wylie, F.G.; Stow, J.L. Regulation of Endocytosis, Nuclear Translocation, and Signaling of Fibroblast Growth Factor Receptor 1 by E-Cadherin. Mol. Biol. Cell 2005, 16, 14–23. [Google Scholar] [CrossRef]
- Hsu, T.; Adereth, Y.; Kose, N.; Dammai, V. Endocytic Function of von Hippel-Lindau Tumor Suppressor Protein Regulates Surface Localization of Fibroblast Growth Factor Receptor 1 and Cell Motility. J. Biol. Chem. 2006, 281, 12069–12080. [Google Scholar] [CrossRef] [Green Version]
- Kawada, K.; Upadhyay, G.; Ferandon, S.; Janarthanan, S.; Hall, M.; Vilardaga, J.-P.; Yajnik, V. Cell Migration Is Regulated by Platelet-Derived Growth Factor Receptor Endocytosis. Mol. Cell. Biol. 2009, 29, 4508–4518. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H.; Glennie, M.J. Agonistic CD40 Antibodies and Cancer Therapy. Clin. Cancer Res. 2013, 19, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonderheide, R.H.; Flaherty, K.T.; Khalil, M.; Stumacher, M.S.; Bajor, D.L.; Hutnick, N.A.; Sullivan, P.; Mahany, J.J.; Gallagher, M.; Kramer, A.; et al. Clinical Activity and Immune Modulation in Cancer Patients Treated with CP-870,893, a Novel CD40 Agonist Monoclonal Antibody. J. Clin. Oncol. 2007, 25, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Maksym, R.B.; Tarnowski, M.; Grymula, K.; Tarnowska, J.; Wysoczynski, M.; Liu, R.; Czerny, B.; Ratajczak, J.; Kucia, M.; Ratajczak, M.Z. The Role of Stromal-Derived Factor-1--CXCR7 Axis in Development and Cancer. Eur. J. Pharmacol. 2009, 625, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, M.; Zheng, J.; Hou, K.; Wang, J.; Chen, X.; Lu, X.; Bo, J.; Xu, C.; Shen, K.; Wang, J. Role of Chemokine Receptor CXCR7 in Bladder Cancer Progression. Biochem. Pharmacol. 2012, 84, 204–214. [Google Scholar] [CrossRef]
- Luker, K.E.; Steele, J.M.; Mihalko, L.A.; Ray, P.; Luker, G.D. Constitutive and Chemokine-Dependent Internalization and Recycling of CXCR7 in Breast Cancer Cells to Degrade Chemokine Ligands. Oncogene 2010, 29, 4599–4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, R.; Kreitman, R.J.; Pastan, I.; Willingham, M.C. Localization of Mesothelin in Epithelial Ovarian Cancer. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Bharadwaj, U.; Zhang, R.; Zhang, S.; Mu, H.; Fisher, W.E.; Brunicardi, F.C.; Chen, C.; Yao, Q. Mesothelin Is a Malignant Factor and Therapeutic Vaccine Target for Pancreatic Cancer. Mol. Cancer Ther. 2008, 7, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I Study of SS1P, a Recombinant Anti-Mesothelin Immunotoxin given as a Bolus IV Infusion to Patients with Mesothelin-Expressing Mesothelioma, Ovarian, and Pancreatic Cancers. Clin. Cancer Res. 2007, 13, 5144–5149. [Google Scholar] [CrossRef] [Green Version]
- Kruyt, F.A.E. TRAIL and Cancer Therapy. Cancer Lett. 2008, 263, 14–25. [Google Scholar] [CrossRef]
- Moran, A.E.; Kovacsovics-Bankowski, M.; Weinberg, A.D. The TNFRs OX40, 4-1BB, and CD40 as Targets for Cancer Immunotherapy. Curr. Opin. Immunol. 2013, 25, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Sethi, G.; Sung, B.; Aggarwal, B.B. TNF: A Master Switch for Inflammation to Cancer. Front. Biosci. 2008, 13, 5094–5107. [Google Scholar] [CrossRef] [Green Version]
- Pranatharthiharan, S.; Patel, M.D.; Malshe, V.C.; Pujari, V.; Gorakshakar, A.; Madkaikar, M.; Ghosh, K.; Devarajan, P.V. Asialoglycoprotein Receptor Targeted Delivery of Doxorubicin Nanoparticles for Hepatocellular Carcinoma. Drug Deliv. 2017, 24, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Hui, H.; Jin, Y.; Dong, D.; Liang, X.; Yang, X.; Tan, K.; Dai, Z.; Cheng, Z.; Tian, J. Enhanced Immunotherapy of SM5-1 in Hepatocellular Carcinoma by Conjugating with Gold Nanoparticles and Its in Vivo Bioluminescence Tomographic Evaluation. Biomaterials 2016, 87, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pompella, A.; De Tata, V.; Paolicchi, A.; Zunino, F. Expression of γ-Glutamyltransferase in Cancer Cells and Its Significance in Drug Resistance. Biochem. Pharmacol. 2006, 71, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Franzini, M.; Paolicchi, A.; Pompella, A. Gamma-Glutamyltransferase of Cancer Cells at the Crossroads of Tumor Progression, Drug Resistance and Drug Targeting. Anticancer Res. 2010, 30, 1169–1181. [Google Scholar] [PubMed]
- Tsutsumi, M.; Sakamuro, D.; Takada, A.; Zang, S.; Furukawa, T.; Taniguchi, N. Detection of a Unique Γ-glutamyl Transpeptidase Messenger RNA Species Closely Related to the Development of Hepatocellular Carcinoma in Humans: A New Candidate for Early Diagnosis of Hepatocellular Carcinoma. Hepatology 1996, 23, 1093–1097. [Google Scholar] [PubMed]
- Masoud, G.N.; Li, W. HIF-1α Pathway: Role, Regulation and Intervention for Cancer Therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Zeng, C.-M.; Chen, Z.; Fu, L. Frizzled Receptors as Potential Therapeutic Targets in Human Cancers. Int. J. Mol. Sci. 2018, 19, 1543. [Google Scholar] [CrossRef] [Green Version]
- Gujral, T.S.; Chan, M.; Peshkin, L.; Sorger, P.K.; Kirschner, M.W.; MacBeath, G. A Noncanonical Frizzled2 Pathway Regulates Epithelial-Mesenchymal Transition and Metastasis. Cell 2014, 159, 844–856. [Google Scholar] [CrossRef] [Green Version]
- Sideras, K.; Bots, S.J.; Biermann, K.; Sprengers, D.; Polak, W.G.; IJzermans, J.N.M.; De Man, R.A.; Pan, Q.; Sleijfer, S.; Bruno, M.J. Tumour Antigen Expression in Hepatocellular Carcinoma in a Low-Endemic Western Area. Br. J. Cancer 2015, 112, 1911. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.S.; Noh, S.J.; Jang, K.Y.; Park, H.S.; Chung, M.J.; Park, C.K.; Moon, W.S. Expression and Role of Epithelial Cell Adhesion Molecule in Dysplastic Nodule and Hepatocellular Carcinoma. Int. J. Oncol. 2012, 41, 2150–2158. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Yang, J.; Li, X.; Huang, H.; Guo, X.; Zhou, G.; Xu, X.; Cai, Y.; Zhu, M.; Wang, P. Hepatocellular Carcinoma Treated with Anti-Epidermal Growth Factor Receptor Antibody Nimotuzumab: A Case Report. Medicine 2017, 96. [Google Scholar] [CrossRef]
- Tsou, A.-P.; Wu, K.-M.; Tsen, T.-Y.; Chi, C.-W.; Chiu, J.-H.; Lui, W.-Y.; Hu, C.-P.; Chang, C.; Chou, C.-K.; Tsai, S.-F. Parallel Hybridization Analysis of Multiple Protein Kinase Genes: Identification of Gene Expression Patterns Characteristic of Human Hepatocellular Carcinoma. Genomics 1998, 50, 331–340. [Google Scholar] [CrossRef]
- Chen, L.; Shi, Y.; Jiang, C.; Wei, L.; Lv, Y.; Wang, Y.; Dai, G. Coexpression of PDGFR-Alpha, PDGFR-Beta and VEGF as a Prognostic Factor in Patients with Hepatocellular Carcinoma. Int. J. Biol. Markers 2011, 26, 108–116. [Google Scholar] [CrossRef]
- Greten, T.F.; Ott, M. CD40 in Hepatocellular Carcinoma: Relevant or Not? Eur. J. Gastroenterol. Hepatol. 2003, 15, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.-F.; Li, K.-Z.; Wang, L.; Dou, K.-F.; Yan, Z.; Han, W.; Zhang, Y.-Q. Expression of MUC1 and Its Significance in Hepatocellular and Cholangiocarcinoma Tissue. World J. Gastroenterol. WJG 2005, 11, 4661. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Feng, M.; Kim, H.; Phung, Y.; Kleiner, D.E.; Gores, G.J.; Qian, M.; Wang, X.W.; Ho, M. Mesothelin as a Potential Therapeutic Target in Human Cholangiocarcinoma. J. Cancer 2010, 1, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraki, K.; Yamanaka, T.; Inoue, H.; Kawakita, T.; Enokimura, N.; Okano, H.; Sugimoto, K.; Murata, K.; Nakano, T. Expression of TNF-Related Apoptosis-Inducing Ligand in Human Hepatocellular Carcinoma. Int. J. Oncol. 2005, 26, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ma, S.; Qi, X.; Tang, X.; Cui, D.; Wang, Z.; Chi, J.; Li, P.; Zhai, B. Knockdown of the Differentially Expressed Gene TNFRSF12A Inhibits Hepatocellular Carcinoma Cell Proliferation and Migration in Vitro. Mol. Med. Rep. 2017, 15, 1172–1178. [Google Scholar] [CrossRef]
- Wei, Y.; Van Nhieu, J.T.; Prigent, S.; Srivatanakul, P.; Tiollais, P.; Buendia, M. Altered Expression of E-cadherin in Hepatocellular Carcinoma: Correlations with Genetic Alterations, Β-catenin Expression, and Clinical Features. Hepatology 2002, 36, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; He, J.; Zhang, F.; Wang, B.; Ding, H.; Shen, H.; Li, Y.; Chen, X. Diagnostic Value of Protein Induced by Vitamin K Absence (PIVKAII) and Hepatoma-Specific Band of Serum Gamma-Glutamyl Transferase (GGTII) as Hepatocellular Carcinoma Markers Complementary to α-Fetoprotein. Br. J. Cancer 2003, 88, 1878. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.-L.; Zeng, Z.-C.; Fan, J.; Tang, Z.-Y.; Zeng, H.-Y.; Gao, D.-M. Gene Expression Profiling of Fixed Tissues Identified Hypoxia-Inducible Factor-1α, VEGF, and Matrix Metalloproteinase-2 as Biomarkers of Lymph Node Metastasis in Hepatocellular Carcinoma. Clin. Cancer Res. 2011, 17, 5463–5472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tien, L.T.; Ito, M.; Nakao, M.; Niino, D.; Serik, M.; Nakashima, M.; Wen, C.-Y.; Yatsuhashi, H.; Ishibashi, H. Expression of β-Catenin in Hepatocellular Carcinoma. World J. Gastroenterol. WJG 2005, 11, 2398. [Google Scholar] [CrossRef]
- Bengochea, A.; De Souza, M.M.; Lefrancois, L.; Le Roux, E.; Galy, O.; Chemin, I.; Kim, M.; Wands, J.R.; Trepo, C.; Hainaut, P. Common Dysregulation of Wnt/Frizzled Receptor Elements in Human Hepatocellular Carcinoma. Br. J. Cancer 2008, 99, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Liu, Y.; Yuan, Q.; Yu, W.; Huang, C.; Ma, L.; Yu, J. Targeting studies of humanized scFv25 fusing to TNFalpha against hepatocellular carcinoma. Chin. J. Hepatol. 2000, 8, 352–354. [Google Scholar]
- Zhang, J.; Liu, Y.F.; Yang, S.J.; Qiao, Q.; Cheng, H.; Zhang, C.S.; Ma, F.C.; Guo, H.Z. Primary Targeting of Recombinant Fv-Immunotoxin HscFv(25)-MTNF Alpha against Hepatocellular Carcinoma. World J. Gastroenterol. 2004, 10, 1872–1875. [Google Scholar] [CrossRef]
- Gao, J.; Kou, G.; Chen, H.; Wang, H.; Li, B.; Lu, Y.; Zhang, D.; Wang, S.; Hou, S.; Qian, W.; et al. Treatment of Hepatocellular Carcinoma in Mice with PE38KDEL Type I Mutant-Loaded Poly(Lactic-Co-Glycolic Acid) Nanoparticles Conjugated with Humanized SM5-1 F(Ab’) Fragments. Mol. Cancer Ther. 2008, 7, 3399–3407. [Google Scholar] [CrossRef] [Green Version]
- Rezaie, E.; Pour, A.B.; Amani, J.; Hosseini, H.M. Bioinformatics Predictions, Expression, Purification and Structural Analysis of the PE38KDEL-Scfv Immunotoxin Against EPHA2 Receptor. Int. J. Pept. Res. Ther. 2020, 26, 979–996. [Google Scholar] [CrossRef]
- Rezaie, E.; Amani, J.; Bidmeshki Pour, A.; Mahmoodzadeh Hosseini, H. A New Scfv-Based Recombinant Immunotoxin against EPHA2-Overexpressing Breast Cancer Cells; High in Vitro Anti-Cancer Potency. Eur. J. Pharmacol. 2020, 870, 172912. [Google Scholar] [CrossRef] [PubMed]
- Keshtvarz, M.; Salimian, J.; Yaseri, M.; Bathaie, S.Z.; Rezaie, E.; Aliramezani, A.; Norouzbabaei, Z.; Amani, J.; Douraghi, M. Bioinformatic Prediction and Experimental Validation of a PE38-Based Recombinant Immunotoxin Targeting the Fn14 Receptor in Cancer Cells. Immunotherapy 2017, 9, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, S.C.; Kou, G.; Li, B.H.; Zhang, D.P.; Hou, S.; Qian, W.Z.; Dai, J.X.; Tian, L.; Zhao, J.; et al. Treatment of Hepatocellular Carcinoma in a Mouse Xenograft Model with an Immunotoxin Which Is Engineered to Eliminate Vascular Leak Syndrome. Cancer Immunol. Immunother. 2007, 56, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Feng, Z.Q.; Zhu, X.J.; Xu, W.; Zhu, J.; Zhang, X.H.; Fan, Z.N.; Ji, G.Z. Construction, Expression, and Characterization of an Anti-Tumor Immunotoxin Containing the Human Anti-c-Met Single-Chain Antibody and PE38KDEL. Immunol. Lett. 2013, 149, 30–40. [Google Scholar] [CrossRef]
- Ogawa, K.; Tanaka, S.; Matsumura, S.; Murakata, A.; Ban, D.; Ochiai, T.; Irie, T.; Kudo, A.; Nakamura, N.; Tanabe, M.; et al. EpCAM-Targeted Therapy for Human Hepatocellular Carcinoma. Ann. Surg. Oncol. 2014, 21, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-F.; Ho, M. Humanization of High-Affinity Antibodies Targeting Glypican-3 in Hepatocellular Carcinoma. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Fleming, B.D.; Urban, D.J.; Hall, M.D.; Longerich, T.; Greten, T.F.; Pastan, I.; Ho, M. Engineered Anti-GPC3 Immunotoxin, HN3-ABD-T20, Produces Regression in Mouse Liver Cancer Xenografts Through Prolonged Serum Retention. Hepatology 2020, 71, 1696–1711. [Google Scholar] [CrossRef]
- Zhao, X.; Yu, Z.; Dai, W.; Yao, Z.; Zhou, W.; Zhou, J.; Yang, Y.; Zhu, Y.; Chen, S.; Cao, L. Construction and Characterization of an Anti-Asialoglycoprotein Receptor Single-Chain Variable-Fragment-Targeted Melittin. Biotechnol. Appl. Biochem. 2011, 58, 405–411. [Google Scholar] [CrossRef]
- Wei, X.; Juan, Z.X.; Min, F.X.; Nan, C.; Hua, Z.X.; Qing, F.Z.; Zheng, L. Recombinant Immunotoxin Anti-c-Met/PE38KDEL Inhibits Proliferation and Promotes Apoptosis of Gastric Cancer Cells. J. Exp. Clin. Cancer Res. 2011, 30, 67. [Google Scholar] [CrossRef] [Green Version]
- Biggers, K.; Scheinfeld, N. VB4-845, a Conjugated Recombinant Antibody and Immunotoxin for Head and Neck Cancer and Bladder Cancer. Curr. Opin. Mol. Ther. 2008, 10, 176–186. [Google Scholar]
- Li, M.; Liu, Z.-S.; Liu, X.-L.; Hui, Q.; Lu, S.-Y.; Qu, L.-L.; Li, Y.-S.; Zhou, Y.; Ren, H.-L.; Hu, P. Clinical Targeting Recombinant Immunotoxins for Cancer Therapy. OncoTargets Ther. 2017, 10, 3645. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.-L.; Cheng, Y.-L.; Qiu, Y.; Shen, C.-H.; Yi, B.; Peng, C. Purification and Characterization of a Novel Type i Ribosome Inactivating Protein, Pachyerosin, from Pachyrhizus Erosus Seeds, and Preparation of Its Immunotoxin against Human Hepatoma Cells. Planta Med. 2014, 80, 896–901. [Google Scholar] [CrossRef]
- Fleming, B.D.; Ho, M. Development of Glypican-3 Targeting Immunotoxins for the Treatment of Liver Cancer: An Update. Biomolecules 2020, 10, 934. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gao, F.; Jiang, L.; Jia, M.; Ao, L.; Lu, M.; Gou, L.; Ho, M.; Jia, S.; Chen, F.; et al. 32A9, a Novel Human Antibody for Designing an Immunotoxin and CAR-T Cells against Glypican-3 in Hepatocellular Carcinoma. J. Transl. Med. 2020, 18, 295. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Miller, A.C.; Sharon, E.; Thomas, A.; Reynolds, J.C.; Ling, A.; Kreitman, R.J.; Miettinen, M.M.; Steinberg, S.M.; Fowler, D.H. Major Cancer Regressions in Mesothelioma after Treatment with an Anti-Mesothelin Immunotoxin and Immune Suppression. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Tumor Marker | Type | Location | The Expression Pattern in Liver Cancer Tissues | Samples | Role | History of Targeting by Immunotoxin | Ref. |
|---|---|---|---|---|---|---|---|
| Glypican 3 (GPC3) | Onco-fetal | Cell surface | 90% | 133 | Cell growth, development, differentiation, and migration | Yes (Y) | [74] |
| Epithelial cell adhesion molecule (EPCAM) | Overexpressed antigen | Cell surface | 34.1% | 132 | Cell–cell adhesion, cell proliferation, tumorogenisity, metastasis | Y | [75] |
| Epidermal growth factor receptor (EGFR) | Overexpressed antigen | Cell surface | 68% | Tumor cell proliferation, apoptosis, invasion, metastasis, and angiogenesis | Y | [76] | |
| Fibroblast growth factor receptor (FGFR) | Overexpressed antigen | Cell surface | Nearly 50% | Five HCC cell lines § | Regulation of the tumor microenvironment and angiogenesis, morphology changes from epithelial to mesenchymal (EMT) | Y | [77] |
| Platelet-derived growth factor receptor (PDGFR-α and β) | Overexpressed antigen | Intracellular and Cell surface | 68–73% | 63 | Blood vessel formation, regulation of cell growth and division, wound healing, and metastasis | Y | [78] |
| CD40 | Overexpressed antigen | Cell surface | 60% | 45 | Survival, proliferation, differentiation in B cells, chemotherapy resistance, and angiogenesis | Y | [79] |
| Mucin core protein 1 (MUC1) | Overexpressed antigen | Intracellular and Cell surface | 70.8% | 59 HCC and 37 CC | Preventing the pathogen from reaching the cell surface | Y | [80] |
| The C-X-C chemokine receptor type 7 (CXCR7) | Overexpressed antigen | Cell surface | - | Three cell lines ¶¶ | Pro-angiogenic role in HCC | Y | [80] |
| Mesothelin | Tumor differentiation antigen, Overexpressed antigen | Cell surface | 33% CCA and no expression in HCC | 87 | Cell adhesion | Y | [81] |
| TNF-related apoptosis-inducing ligand (TRAIL) | Overexpressed antigen | Cell surface | - | 10 | Lymphocyte cytotoxicity and the maintenance of immunological homeostasis in various tissues | Y | [82] |
| Tumor necrosis factor receptor superfamily member 12A (TNFRSF12A) | Overexpressed antigen | Cell surface | - | 20 | Tumor growth and metastasis | Y | [83] |
| Asialoglycoprotein receptor (ASGPR) | Overexpressed antigen | Cell surface | 500,000 ASGPR/hepatocyte | - | Internalization of galactose (Gal) | Y | [66] |
| Receptor protein p230 | Overexpressed antigen | Cell surface | - | - | Early tissues development | Y | [67] |
| E-cadherin | Overexpressed antigen | Cell surface | 40% | 37 | Cell adhesion protein | Y | [84] |
| Axl receptor tyrosine kinase | Overexpressed antigen | Cell surface | - | Five HCC cell lines § | Tumor development and progression, differentiation, invasion, chemotherapy resistance | No (N) | [76,77] |
| γ-glutamyl transferase (GGT) | Overexpressed antigen | Cell surface | 43.8% | 120 | Embryonic enzyme | N | [85] |
| Hypoxia-inducible factor (HIF)-1α | Overexpressed in HCC | Cell surface: Intracellular, Nucleoplasm, and Nuclear bodies | - | 309 | Tumor growth and metastasis | N | [86] |
| Ig gamma-1 chain C region | Overexpressed antigen | Cell surface and Secreted | - | 25 | - | N | [74] |
| β-catenin | Overexpressed antigen | Cell surface | 78% | 32 | Generation/differentiation of many tissues | N | [87] |
| Frizzled receptors-2/7 | Overexpressed antigen | Intracellular and Cell surface | 95% | 62 | Mammalian hair follicle development | N | [88] |
| Label | Year | Targeting Moiety | Toxin Moiety | Receptor Type | Receptor Expression | Minimum Cell IC50 | Ref. |
|---|---|---|---|---|---|---|---|
| hscFv25-TNFα | 2000 | scFv | TNFα | Unknown | --- | --- | [89] |
| hscFv25-mTNFα | 2004 | scFv | Mutant TNFα | Unknown | --- | --- | [90] |
| mut1 | 2007 | SM5-1 single chain antibody (SMFv) | PE38KDEL | p230 | Overexpressed | 5.03 pmol/L | [95] |
| C1M | 2011 | scFv of anti- ASGPR | Melittin | ASGPR ¶ | Overexpressed | <55 nmol/L | [91] |
| anti-c-Met/PE38KDEL | 2013 | scFv | PE38KDEL | c-Met | Overexpressed | 150 pmol/L | [96] |
| VB4-845 | 2014 | scFv | PE | EpCAM | Onco-fetal | <1 pmol/L | [97] |
| APE | 2015 | scFv | PE38KDEL | EpCAM | Onco-fetal | 50 pmol/L | [20] |
| HN3-PE38 | 2015 | VH domain | PE38 | GPC3 | Onco-fetal | 0.28 nmol/L | [5] |
| YP7-PE38 | 2015 | scFv | PE38 | GPC3 | Onco-fetal | 1.59 nmol/L | [5] |
| HS20-PE38 | 2015 | scFv | PE38 | GPC3 | Onco-fetal | --- | [5] |
| YP7-PE38 | 2016 | scFv | PE38 | GPC3 | Onco-fetal | 7.8 ng/mL | [98] |
| Humanized YP7-PE38 | 2016 | scFv | PE38 | GPC3 | Onco-fetal | 28 ng/mL | [98] |
| YP9.1-PE38 | 2016 | scFv | PE38 | GPC3 | Onco-fetal | 2.9 ng/mL | [98] |
| Humanized YP9.1-PE38 | 2016 | scFv | PE38 | GPC3 | Onco-fetal | 77 ng/mL | [98] |
| HN3-mPE24 | 2017 | VH domain | mPE24 | GPC3 | Onco-fetal | 0.2 nM | [48] |
| HN3- HN3-mPE24 | 2017 | VH domain | mPE24 | GPC3 | Onco-fetal | 0.4 nM | [48] |
| HN3-T20 | 2019 | VH domain | mPE24 | GPC3 | Onco-fetal | 1.6 nM | [99] |
| HN3-ABD-T20 | 2019 | VH domain | mPE24 | GPC3 | Onco-fetal | --- | [99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heiat, M.; Hashemi Yeganeh, H.; Alavian, S.M.; Rezaie, E. Immunotoxins Immunotherapy against Hepatocellular Carcinoma: A Promising Prospect. Toxins 2021, 13, 719. https://doi.org/10.3390/toxins13100719
Heiat M, Hashemi Yeganeh H, Alavian SM, Rezaie E. Immunotoxins Immunotherapy against Hepatocellular Carcinoma: A Promising Prospect. Toxins. 2021; 13(10):719. https://doi.org/10.3390/toxins13100719
Chicago/Turabian StyleHeiat, Mohammad, Hamid Hashemi Yeganeh, Seyed Moayed Alavian, and Ehsan Rezaie. 2021. "Immunotoxins Immunotherapy against Hepatocellular Carcinoma: A Promising Prospect" Toxins 13, no. 10: 719. https://doi.org/10.3390/toxins13100719
APA StyleHeiat, M., Hashemi Yeganeh, H., Alavian, S. M., & Rezaie, E. (2021). Immunotoxins Immunotherapy against Hepatocellular Carcinoma: A Promising Prospect. Toxins, 13(10), 719. https://doi.org/10.3390/toxins13100719