Human Proximal Tubule Epithelial Cells (HK-2) as a Sensitive In Vitro System for Ochratoxin A Induced Oxidative Stress


Abstract
:1. Introduction
2. Results
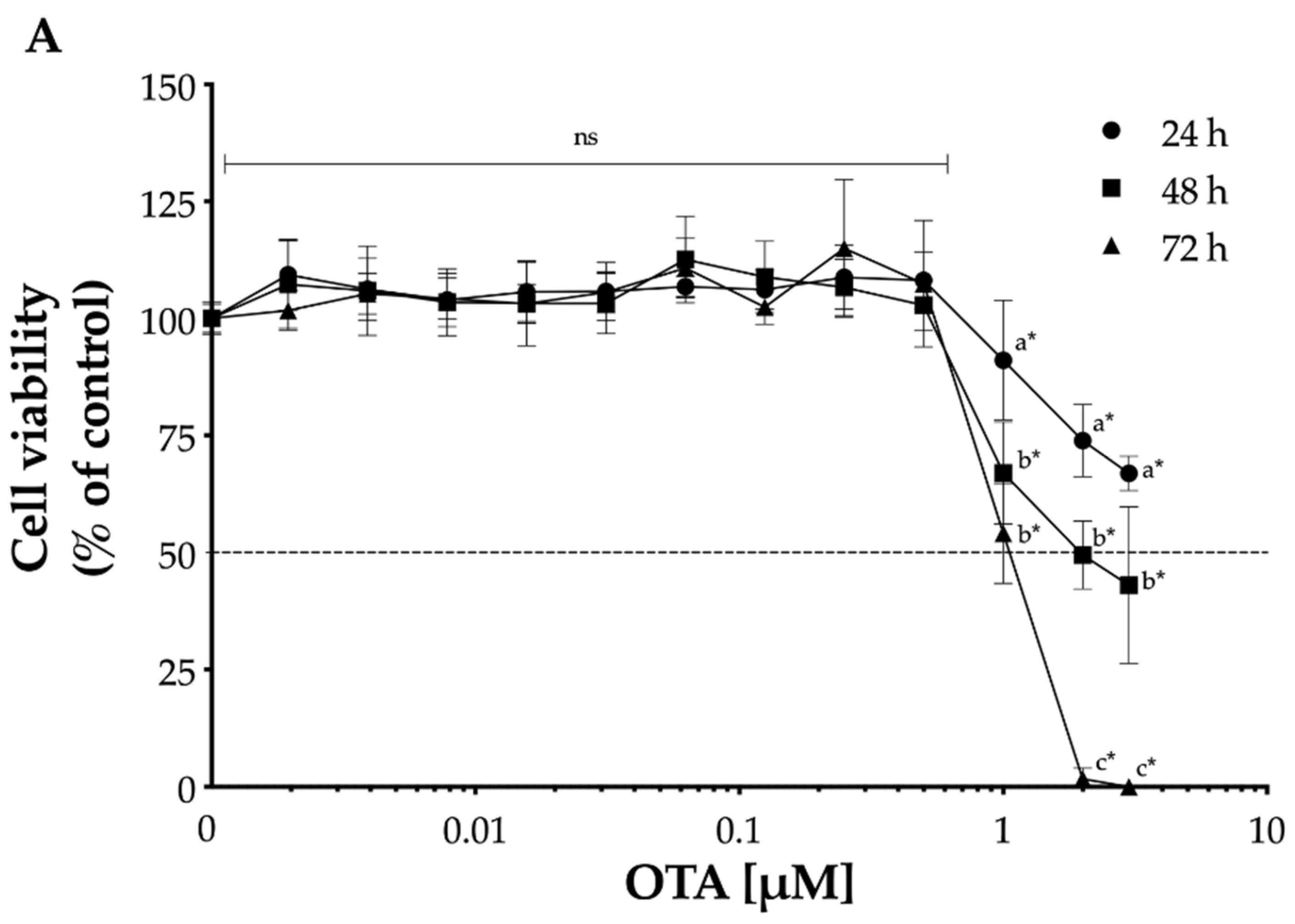
2.1. Effect of OTA on Cell Viability
2.2. Effect of OTA on Cellular Redox State
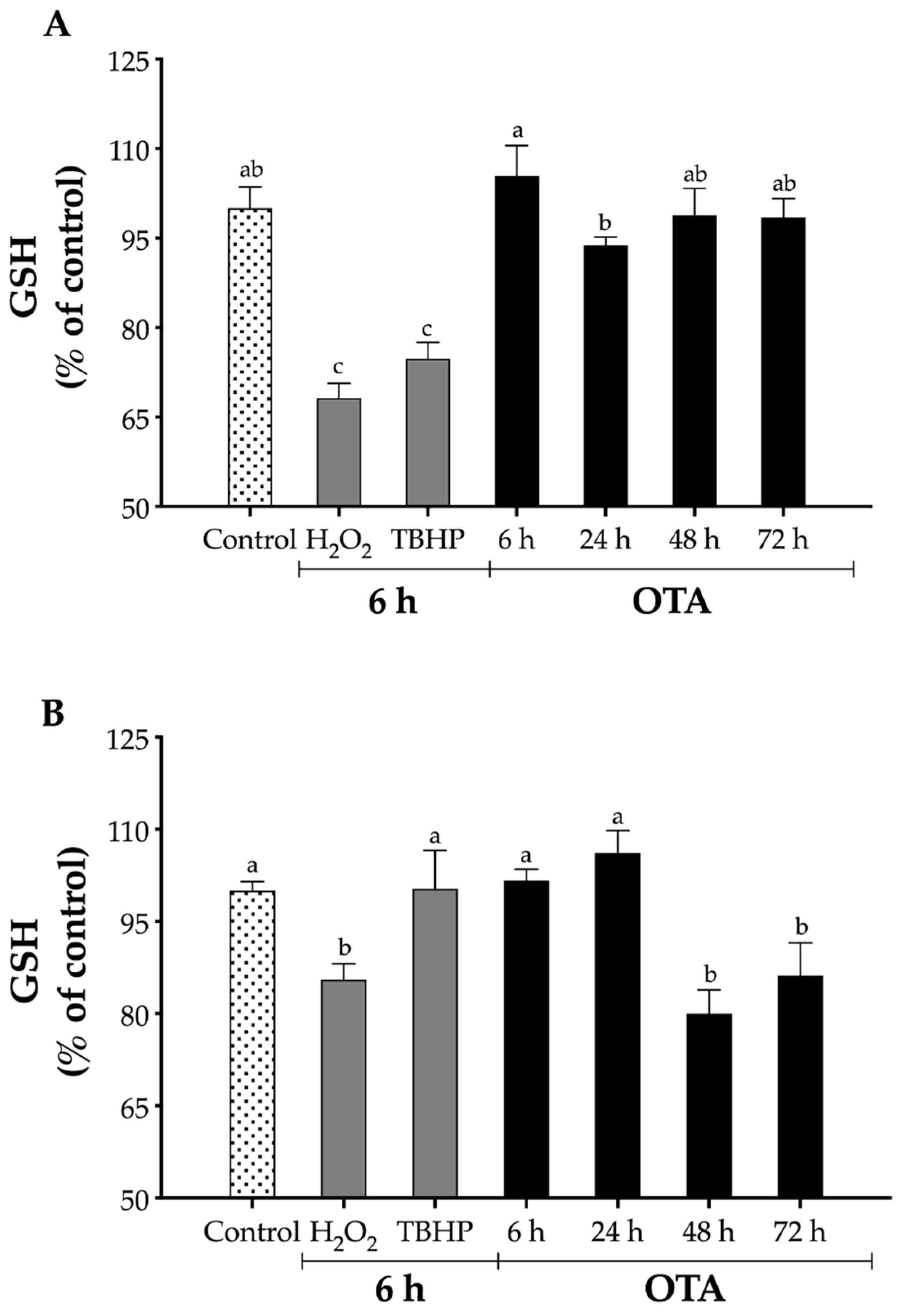
2.3. Effect of OTA on Intracellular GSH Levels
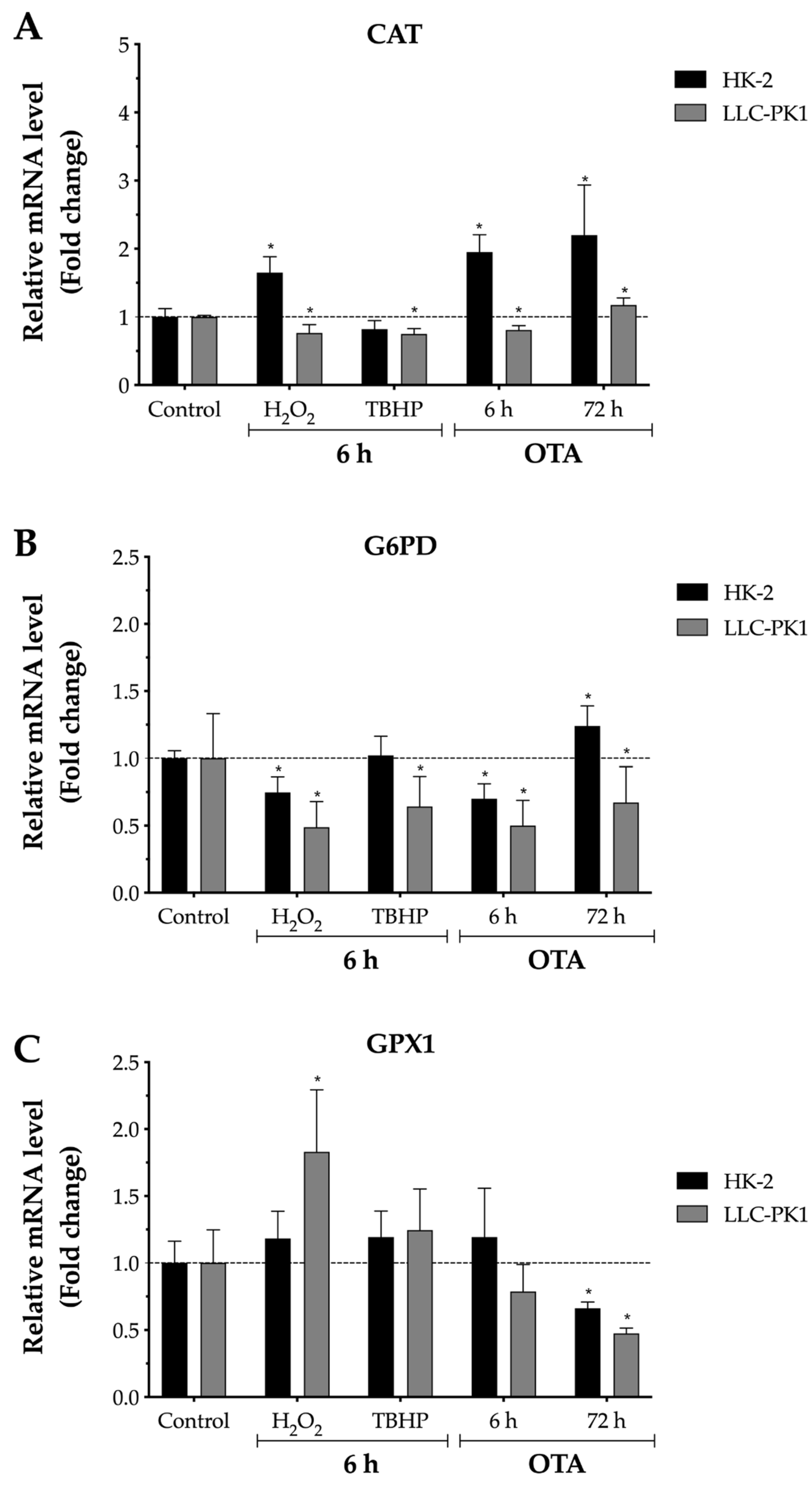
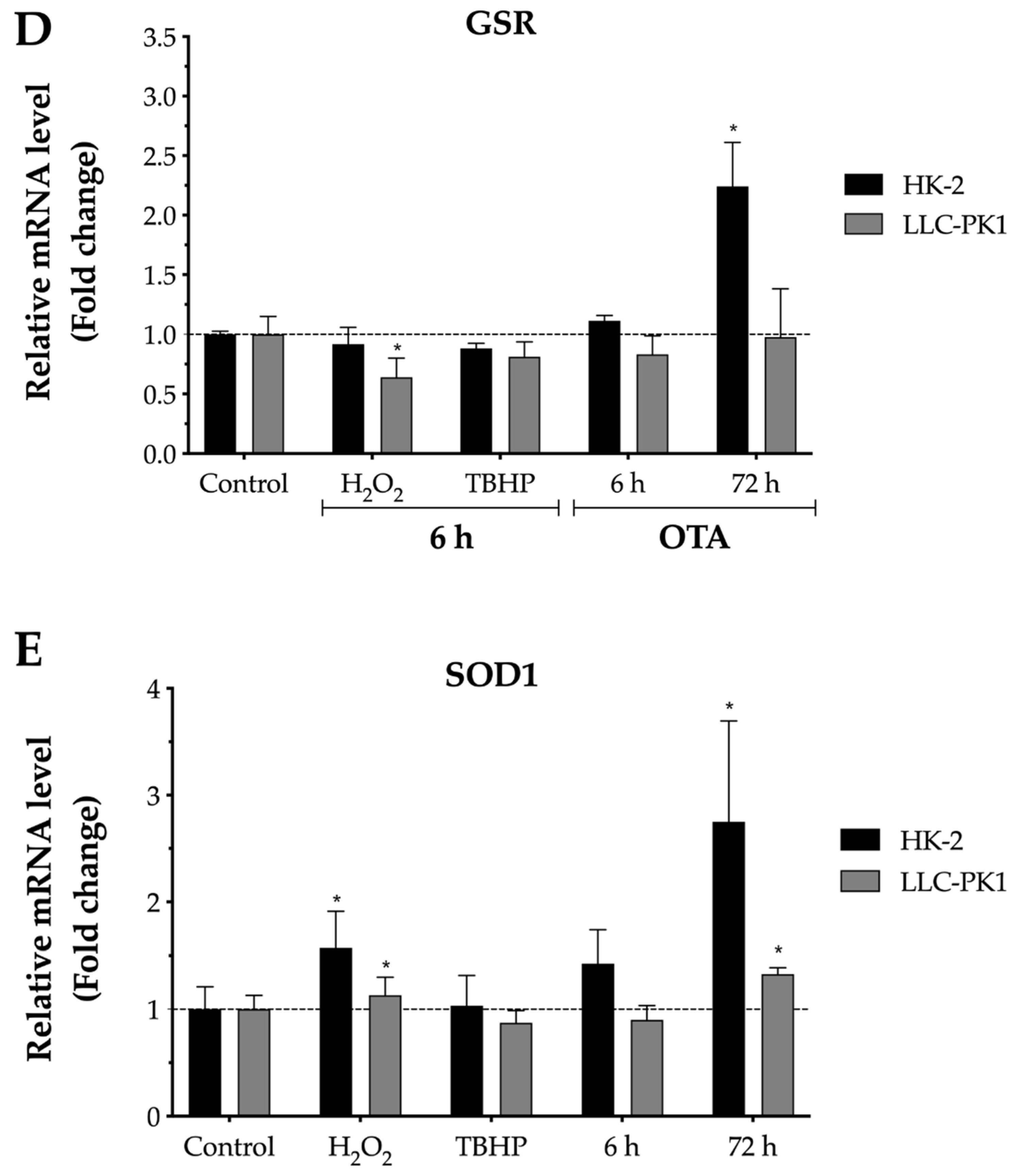
2.4. Effect of OTA on Antioxidant Enzymes Gene Expression
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemicals
5.2. Cell Culture
5.3. Cell Viability Assay
5.4. Intracellular ROS Analysis
5.5. Glutathione (GSH) Measurement
5.6. Real-Time Quantitative Reverse Transcription PCR (RT-qPCR)
5.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Agency for Research on Cancer (IARC). Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins; IARC: Lyon, France, 1993. [Google Scholar]
- Lee, H.J.; Ryu, D. Significance of ochratoxin A in breakfast cereals from the United States. J. Agric. Food Chem. 2015, 63, 9404–9409. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, J.D.; O’Keeffe, T.L.; Ho, Y.S.; Santillan, C.J. Occurrence of ochratoxin A contamination and detection of ochratoxigenic Aspergillus species in retail samples of dried fruits and nuts. J. Food Prot. 2015, 78, 836–842. [Google Scholar] [CrossRef]
- Solfrizzo, M.; Panzarini, G.; Visconti, A. Determination of ochratoxin A in grapes, dried vine fruits, and winery byproducts by high-performance liquid chromatography with fluorometric detection (HPLC−FLD) and immunoaffinity cleanup. J. Agric. Food Chem. 2008, 56, 11081–11086. [Google Scholar] [CrossRef] [PubMed]
- Pfohl-Leszkowicz, A.; Manderville, R.A. Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar] [CrossRef] [PubMed]
- National Toxicology Program (NTP). Toxicology and carcinogenesis studies of ochratoxin A (CAS No. 303-47-9) in F344/N Rats (Gavage studies). Natl. Toxicol. Program Tech. Rep. Ser. 1989, 358, 1–146. [Google Scholar]
- Schrenk, D.; Bodin, L.; Chipman, J.K.; del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.; Leblanc, J.C.; Nebbia, C.S. Risk assessment of ochratoxin A in food. EFSA Panel Contam. Food Chain. EFSA J. 2020, 18, e06113. [Google Scholar]
- Lea, T.; Steien, K.; Størmer, F.C. Mechanism of ochratoxin A-induced immunosuppression. Mycopathologia 1989, 107, 153–159. [Google Scholar] [CrossRef]
- Mayura, K.; Parker, R.; Berndt, W.; Phillips, T. Ochratoxin A-induced teratogenesis in rats: Partial protection by phenylalanine. Appl. Environ. Microbiol. 1984, 48, 1186–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meisner, H.; Chan, S. Ochratoxin A, an inhibitor of mitochondrial transport systems. Biochemistry 1974, 13, 2795–2800. [Google Scholar] [CrossRef]
- Wei, Y.; Lu, C.; Lin, T.; Wei, R. Effect of ochratoxin A on rat liver mitochondrial respiration and oxidative phosphorylation. Toxicology 1985, 36, 119–130. [Google Scholar] [CrossRef]
- Creppy, E.; Kern, D.; Steyn, P.; Vleggaar, R.; Röschenthaler, R.; Dirheimer, G. Comparative study of the effect of ochratoxin A analogues on yeast aminoacyl-tRNA synthetases and on the growth and protein synthesis of hepatoma cells. Toxicol. Lett. 1983, 19, 217–224. [Google Scholar] [CrossRef]
- Creppy, E.; Størmer, F.; Kern, D.; Röschenthaler, R.; Dirheimer, G. Effects of ochratoxin A metabolites on yeast phenylalanyl-tRNA synthetase and on the growth and in vivo protein synthesis of hepatoma cells. Chem. Biol. Interact. 1983, 47, 239–247. [Google Scholar] [CrossRef]
- Kamp, H.G.; Eisenbrand, G.; Janzowski, C.; Kiossev, J.; Latendresse, J.R.; Schlatter, J.; Turesky, R.J. Ochratoxin A induces oxidative DNA damage in liver and kidney after oral dosing to rats. Mol. Nutr. Food Res. 2005, 49, 1160–1167. [Google Scholar] [CrossRef]
- Mally, A.; Pepe, G.; Ravoori, S.; Fiore, M.; Gupta, R.C.; Dekant, W.; Mosesso, P. Ochratoxin A causes DNA damage and cytogenetic effects but no DNA adducts in rats. Chem. Res. Toxicol. 2005, 18, 1253–1261. [Google Scholar] [CrossRef] [Green Version]
- Omar, R.F.; Hasinoff, B.B.; Mejilla, F.; Rahimtula, A.D. Mechanism of ochratoxin A stimulated lipid peroxidation. Biochem. Pharmacol. 1990, 40, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Omar, R.F.; Rahimtula, A.D.; Bartsch, H. Role of cytochrome P-450 in ochratoxin a-stimulated lipid peroxidation. J. Biochem. Toxicol. 1991, 6, 203–209. [Google Scholar] [CrossRef]
- Rahimtula, A.; Bereziat, J.; Bussacchini-Griot, V.; Bartsch, H. Lipid peroxidation as a possible cause of ochratoxin A toxicity. Biochem. Pharmacol. 1988, 37, 4469–4477. [Google Scholar] [CrossRef]
- Cavin, C.; Delatour, T.; Marin-Kuan, M.; Holzhäuser, D.; Higgins, L.; Bezencon, C.; Guignard, G.; Junod, S.; Richoz-Payot, J.; Gremaud, E. Reduction in antioxidant defenses may contribute to ochratoxin A toxicity and carcinogenicity. Toxicol. Sci. 2007, 96, 30–39. [Google Scholar] [CrossRef]
- Petrik, J.; Žanić-Grubišić, T.; Barišić, K.; Pepeljnjak, S.; Radić, B.; Ferenčić, Ž.; Čepelak, I. Apoptosis and oxidative stress induced by ochratoxin A in rat kidney. Arch. Toxicol. 2003, 77, 685–693. [Google Scholar] [CrossRef]
- Schaaf, G.; Nijmeijer, S.; Maas, R.; Roestenberg, P.; De Groene, E.; Fink-Gremmels, J. The role of oxidative stress in the ochratoxin A-mediated toxicity in proximal tubular cells. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2002, 1588, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Klaunig, J.E.; Kamendulis, L.M. The role of oxidative stress in carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 239–267. [Google Scholar] [CrossRef]
- Marin-Kuan, M.; Cavin, C.; Delatour, T.; Schilter, B. Ochratoxin A carcinogenicity involves a complex network of epigenetic mechanisms. Toxicon 2008, 52, 195–202. [Google Scholar] [CrossRef]
- Czakai, K.; Müller, K.; Mosesso, P.; Pepe, G.; Schulze, M.; Gohla, A.; Patnaik, D.; Dekant, W.; Higgins, J.M.; Mally, A. Perturbation of mitosis through inhibition of histone acetyltransferases: The key to ochratoxin a toxicity and carcinogenicity? Toxicol. Sci. 2011, 122, 317–329. [Google Scholar] [CrossRef] [Green Version]
- Hibi, D.; Kijima, A.; Kuroda, K.; Suzuki, Y.; Ishii, Y.; Jin, M.; Nakajima, M.; Sugita-Konishi, Y.; Yanai, T.; Nohmi, T. Molecular mechanisms underlying ochratoxin A-induced genotoxicity: Global gene expression analysis suggests induction of DNA double-strand breaks and cell cycle progression. J. Toxicol. Sci. 2013, 38, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfohl-Leszkowicz, A.; Manderville, R.A. An update on direct genotoxicity as a molecular mechanism of ochratoxin a carcinogenicity. Chem. Res. Toxicol. 2012, 25, 252–262. [Google Scholar] [CrossRef]
- Lee, H.J.; Pyo, M.C.; Shin, H.S.; Ryu, D.; Lee, K.-W. Renal toxicity through AhR, PXR, and Nrf2 signaling pathway activation of ochratoxin A-induced oxidative stress in kidney cells. Food Chem. Toxicol. 2018, 122, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Pyo, M.C.; Choi, I.-G.; Lee, K.-W. Transcriptome Analysis Reveals the AhR, Smad2/3, and HIF-1α Pathways as the Mechanism of Ochratoxin A Toxicity in Kidney Cells. Toxins 2021, 13, 190. [Google Scholar] [CrossRef]
- Erikstein, B.S.; Hagland, H.R.; Nikolaisen, J.; Kulawiec, M.; Singh, K.K.; Gjertsen, B.T.; Tronstad, K.J. Cellular stress induced by resazurin leads to autophagy and cell death via production of reactive oxygen species and mitochondrial impairment. J. Cell. Biochem. 2010, 111, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Sreemannarayana, O.; Frohlich, A.; Vitti, T.; Marquardt, R.; Abramson, D. Studies of the tolerance and disposition of ochratoxin A in young calves. J. Anim. Sci. 1988, 66, 1703–1711. [Google Scholar] [CrossRef]
- Galtier, P.; Alvinerie, M.; Charpenteau, J. The pharmacokinetic profiles of ochratoxin A in pigs, rabbits and chickens. Food Cosmet. Toxicol. 1981, 19, 735–738. [Google Scholar] [CrossRef]
- Studer-Rohr, I.; Schlatter, J.; Dietrich, D.R. Kinetic parameters and intraindividual fluctuations of ochratoxin A plasma levels in humans. Arch. Toxicol. 2000, 74, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Authority, E.F.S. Opinion of the Scientific Panel on contaminants in the food chain [CONTAM] related to ochratoxin A in food. EFSA J. 2006, 4, 365. [Google Scholar] [CrossRef]
- Chu, F.S. Interaction of ochratoxin A with bovine serum albumin. Arch. Biochem. Biophys. 1971, 147, 359–366. [Google Scholar] [CrossRef]
- Bondy, G.; Armstrong, C. Cytotoxicity of nephrotoxic fungal toxins to kidney-derived LLC-PK 1 and OK cell lines. Cell Biol. Toxicol. 1998, 14, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Özcan, Z.; Gül, G.; Yaman, I. Ochratoxin A activates opposing c-MET/PI3K/Akt and MAPK/ERK 1-2 pathways in human proximal tubule HK-2 cells. Arch. Toxicol. 2015, 89, 1313–1327. [Google Scholar] [CrossRef] [PubMed]
- Boesch-Saadatmandi, C.; Loboda, A.; Józkowicz, A.; Huebbe, P.; Blank, R.; Wolffram, S.; Dulak, J.; Rimbach, G. Effect of ochratoxin A on redox-regulated transcription factors, antioxidant enzymes and glutathione-S-transferase in cultured kidney tubulus cells. Food Chem. Toxicol. 2008, 46, 2665–2671. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, D.R.; O’Brien, E.; Stack, M.E.; Heussner, A.H. Species-and sex-specific renal cytotoxicity of ochratoxin A and B in vitro. Exp. Toxicol. Pathol. 2001, 53, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Arbillaga, L.; Azqueta, A.; van Delft, J.H.; de Cerain, A.L. In vitro gene expression data supporting a DNA non-reactive genotoxic mechanism for ochratoxin A. Toxicol. Appl. Pharmacol. 2007, 220, 216–224. [Google Scholar] [CrossRef]
- Hadjeba-Medjdoub, K.; Tozlovanu, M.; Pfohl-Leszkowicz, A.; Frenette, C.; Paugh, R.J.; Manderville, R.A. Structure–activity relationships imply different mechanisms of action for Ochratoxin A-mediated cytotoxicity and genotoxicity. Chem. Res. Toxicol. 2012, 25, 181–190. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Tozlovanu, M.; Manderville, R.; Peraica, M.; Castegnaro, M.; Stefanovic, V. New molecular and field evidences for the implication of mycotoxins but not aristolochic acid in human nephropathy and urinary tract tumor. Mol. Nutr. Food Res. 2007, 51, 1131–1146. [Google Scholar] [CrossRef]
- Baldi, A.; Losio, M.; Cheli, F.; Rebucci, R.; Sangalli, L.; Fusi, E.; Bertasi, B.; Pavoni, E.; Carli, S.; Politis, I. Evaluation of the protective effects of α-tocopherol and retinol against ochratoxin A cytotoxicity. Br. J. Nutr. 2004, 91, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.; Utan, A.; Cervellati, R.; Speroni, E.; Guerra, M. Catechins: Natural free-radical scavengers against ochratoxin A-induced cell damage in a pig kidney cell line (LLC-PK1). Food Chem. Toxicol. 2007, 45, 1910–1917. [Google Scholar] [CrossRef]
- Arbillaga, L.; Azqueta, A.; Ezpeleta, O.; Cerain, A.L.d. Oxidative DNA damage induced by ochratoxin A in the HK-2 human kidney cell line: Evidence of the relationship with cytotoxicity. Mutagenesis 2007, 22, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, D.J.; Smyth, G.K. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 2009, 25, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- National Institutes of Health (NTP). National Toxicology Program Chemical Repository Database; NTP: Research Triangle Park, NC, USA, 1992. Available online: https://ntp.niehs.nih.gov/go/tr358 (accessed on 11 May 2021).
- United States Environmental Protection Agency (US EPA). Estimation Program Interface (EPI) Suite. Ver. 4.1. 2012. Available online: https://www.epa.gov/tsca-screening-tools/download-epi-suitetm-estimation-program-interface-v411 (accessed on 11 May 2021).
- O’Brien, E.; Heussner, A.H.; Dietrich, D.R. Species-, sex-, and cell type-specific effects of ochratoxin A and B. Toxicol. Sci. 2001, 63, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennings, P.; Weiland, C.; Limonciel, A.; Bloch, K.M.; Radford, R.; Aschauer, L.; McMorrow, T.; Wilmes, A.; Pfaller, W.; Ahr, H.J. Transcriptomic alterations induced by Ochratoxin A in rat and human renal proximal tubular in vitro models and comparison to a rat in vivo model. Arch. Toxicol. 2012, 86, 571–589. [Google Scholar] [CrossRef]
- Brandsch, M.; Ganapathy, V.; Leibach, F.H. H (+)-peptide cotransport in Madin-Darby canine kidney cells: Expression and calmodulin-dependent regulation. Am. J. Physiol. Ren. Physiol. 1995, 268, F391–F397. [Google Scholar] [CrossRef]
- Schwerdt, G.; Freudinger, R.; Silbernagl, S.; Gekle, M. Apical uptake of radiolabelled ochratoxin A into Madin–Darby canine kidney cells. Toxicology 1998, 131, 193–202. [Google Scholar] [CrossRef]
- Schwerdt, G.; Gekle, M.; Freudinger, R.; Mildenberger, S.; Silbernagl, S. Apical-to-basolateral transepithelial transport of ochratoxin A by two subtypes of Madin-Darby canine kidney cells. Biochim. Biophys. Acta (BBA) Biomembr. 1997, 1324, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Dürst, M.; Dzarlieva-Petrusevska, R.; Boukamp, P.; Fusenig, N.; Gissmann, L. Molecular and cytogenetic analysis of immortalized human primary keratinocytes obtained after transfection with human papillomavirus type 16 DNA. Oncogene 1987, 1, 251–256. [Google Scholar]
- Halbert, C.; Demers, G.; Galloway, D. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J. Virol. 1991, 65, 473–478. [Google Scholar] [CrossRef] [Green Version]
- Hawley-Nelson, P.; Vousden, K.H.; Hubbert, N.L.; Lowy, D.R.; Schiller, J.T. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 1989, 8, 3905–3910. [Google Scholar] [CrossRef]
- Howley, P.M. Role of the human papillomaviruses in human cancer. Cancer Res. 1991, 51, 5019s–5022s. [Google Scholar] [PubMed]
- Pirisi, L.; Creek, K.E.; Doniger, J.; Dipaolo, J.A. Continuous cell lines with altered growth and differentiation properties originate after transfection of human keratinocytes with human papillomavirus type 16 DNA. Carcinogenesis 1988, 9, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Pirisi, L.; Yasumoto, S.; Feller, M.; Doniger, J.; DiPAOLO, J.A. Transformation of human fibroblasts and keratinocytes with human papillomavirus type 16 DNA. J. Virol. 1987, 61, 1061–1066. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.E. In vitro transformation of human epithelial cells. Biochim. Biophys. Acta (BBA) Rev. Cancer 1986, 823, 161–194. [Google Scholar] [CrossRef]
- Graham, F.L.; Smiley, J.; Russell, W.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Nanus, D.; Lynch, S.; Rao, P.; Anderson, S.; Jhanwar, S.; Albino, A. Transformation of human kidney proximal tubule cells by a src-containing retrovirus. Oncogene 1991, 6, 2105–2111. [Google Scholar]
- Robinson, P.S.; Goochee, C.F. Kidney-specific enzyme expression by human kidney cell lines generated through oncogene transfection. J. Cell. Physiol. 1991, 148, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Schwerdt, G.; Bauer, K.; Gekle, M.; Silbernagl, S. Accumulation of ochratoxin A in rat kidney in vivo and in cultivated renal epithelial cells in vitro. Toxicology 1996, 114, 177–185. [Google Scholar] [CrossRef]
- Schwerdt, G.; Freudinger, R.; Silbernagl, S.; Gekle, M. Ochratoxin A-binding proteins in rat organs and plasma and in different cell lines of the kidney. Toxicology 1999, 135, 1–10. [Google Scholar] [CrossRef]
- Heussner, A.H.; O’Brien, E.; Dietrich, D.R. Species-and sex-specific variations in binding of ochratoxin A by renal proteins in vitro. Exp. Toxicol. Pathol. 2002, 54, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Galtier, P.; Charpenteau, J.-L.; Alvinerie, M.; Labouche, C. The pharmacokinetic profile of ochratoxin A in the rat after oral and intravenous administration. Drug Metab. Dispos. 1979, 7, 429–434. [Google Scholar] [PubMed]
- Hagelberg, S.; Hult, K.; Fuchs, R. Toxicokinetics of ochratoxin A in several species and its plasma-binding properties. J. Appl. Toxicol. 1989, 9, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, M.; Kurz, T.; Brunk, U.T.; Nilsson, S.E.; Frennesson, C.I. What does the commonly used DCF test for oxidative stress really show? Biochem. J. 2010, 428, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Schwerdt, G.; Freudinger, R.; Mildenberger, S.; Silbernagl, S.; Gekle, M. The nephrotoxin ochratoxin A induces apoptosis in cultured human proximal tubule cells. Cell Biol. Toxicol. 1999, 15, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Klarić, M.Š.; Rumora, L.; Ljubanović, D.; Pepeljnjak, S. Cytotoxicity and apoptosis induced by fumonisin B 1, beauvericin and ochratoxin A in porcine kidney PK15 cells: Effects of individual and combined treatment. Arch. Toxicol. 2008, 82, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Barišić, K.; Rumora, L.; Petrik, J.; Čepelak, I.; Žanić-Grubišić, T. Ochratoxin A induces apoptosis in LLC-PK1 cells via JNK and p38 MAPK activation. Croat. Chem. Acta 2005, 78, 385–392. [Google Scholar]
- Liang, R.; Shen, X.L.; Zhang, B.; Li, Y.; Xu, W.; Zhao, C.; Luo, Y.; Huang, K. Apoptosis signal-regulating kinase 1 promotes Ochratoxin A-induced renal cytotoxicity. Sci. Rep. 2015, 5, 8078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Shi, L.; Huang, K.; Xu, W. Protective effect of N-acetylcysteine against DNA damage and S-phase arrest induced by ochratoxin A in human embryonic kidney cells (HEK-293). Food Chem. Toxicol. 2014, 70, 40–47. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Gachhui, R.; Sil, P.C. Hepatoprotective properties of kombucha tea against TBHP-induced oxidative stress via suppression of mitochondria dependent apoptosis. Pathophysiology 2011, 18, 221–234. [Google Scholar] [CrossRef]
- Sarkar, M.K.; Sil, P.C. Prevention of tertiary butyl hydroperoxide induced oxidative impairment and cell death by a novel antioxidant protein molecule isolated from the herb, Phyllanthus niruri. Toxicol. In Vitro 2010, 24, 1711–1719. [Google Scholar] [CrossRef]
- Zavodnik, L.B.; Zavodnik, I.B.; Niekurzak, A.; Szosland, K.; Bryszewska, M. Activation of red blood cell glutathione peroxidase and morphological transformation of erythrocytes under the action of tert-butyl hydroperoxide. IUBMB Life 1998, 44, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Benatti, U.; Morelli, A.; Damiani, G.; De Flora, A. A methemoglobin-dependent and plasma-stimulated experimental model of oxidative hemolysis. Biochem. Biophys. Res. Commun. 1982, 106, 1183–1190. [Google Scholar] [CrossRef]
- Deuticke, B.; Heller, K.B.; Haest, C.W. Progressive oxidative membrane damage in erythrocytes after pulse treatment with t-butylhydroperoxide. Biochim. Biophys. Acta (BBA) Biomembr. 1987, 889, 113–124. [Google Scholar] [CrossRef]
- Yu, S.-W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly (ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Klarić, M.Š.; Medić, N.; Hulina, A.; Grubišić, T.Ž.; Rumora, L. Disturbed Hsp70 and Hsp27 expression and thiol redox status in porcine kidney PK15 cells provoked by individual and combined ochratoxin A and citrinin treatments. Food Chem. Toxicol. 2014, 71, 97–105. [Google Scholar] [CrossRef]
- Raghubeer, S.; Nagiah, S.; Phulukdaree, A.; Chuturgoon, A. The phytoalexin resveratrol ameliorates ochratoxin A toxicity in human embryonic kidney (HEK293) cells. J. Cell. Biochem. 2015, 116, 2947–2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Cao, M.; Yang, W.; Sun, F.; Xu, C.; Yin, L.; Pu, Y. Inhibition of glucose-6-phosphate dehydrogenase could enhance 1, 4-benzoquinone-induced oxidative damage in K562 cells. Oxid. Med. Cell. Longev. 2016, 2016, 3912515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012, 64, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yin, S.; Dong, Y.; Fan, L.; Hu, H. p53 activation inhibits ochratoxin A-induced apoptosis in monkey and human kidney epithelial cells via suppression of JNK activation. Biochem. Biophys. Res. Commun. 2011, 411, 458–463. [Google Scholar] [CrossRef]
- Gan, F.; Xue, H.; Huang, Y.; Pan, C.; Huang, K. Selenium alleviates porcine nephrotoxicity of ochratoxin A by improving selenoenzyme expression in vitro. PLoS ONE 2015, 10, e0119808. [Google Scholar] [CrossRef]
- Roussel, A.M.; Favier, A.; Anderson, R. Trace Elements in Man and Animals 10; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Riss, T.; Moravec, R.; Niles, A.; Duellman, S.; Benink, H.; Worzella, T.; Minor, L. Cell Viability Assays. In Assay Guidance Manual [Internet]; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Wang, H.; Joseph, J.A. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic. Biol. Med. 1999, 27, 612–616. [Google Scholar] [CrossRef]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell line | IC50 (μM OTA) | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| LLC-PK1 | >3 | 2 | 1 |
| HK-2 | 2 | 0.382 | 0.125 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Pérez, E.; Ryu, D.; Kim, H.-Y.; Kim, H.D.; Lee, H.J. Human Proximal Tubule Epithelial Cells (HK-2) as a Sensitive In Vitro System for Ochratoxin A Induced Oxidative Stress. Toxins 2021, 13, 787. https://doi.org/10.3390/toxins13110787
García-Pérez E, Ryu D, Kim H-Y, Kim HD, Lee HJ. Human Proximal Tubule Epithelial Cells (HK-2) as a Sensitive In Vitro System for Ochratoxin A Induced Oxidative Stress. Toxins. 2021; 13(11):787. https://doi.org/10.3390/toxins13110787
Chicago/Turabian StyleGarcía-Pérez, Enrique, Dojin Ryu, Hwa-Young Kim, Hae Dun Kim, and Hyun Jung Lee. 2021. "Human Proximal Tubule Epithelial Cells (HK-2) as a Sensitive In Vitro System for Ochratoxin A Induced Oxidative Stress" Toxins 13, no. 11: 787. https://doi.org/10.3390/toxins13110787
APA StyleGarcía-Pérez, E., Ryu, D., Kim, H. -Y., Kim, H. D., & Lee, H. J. (2021). Human Proximal Tubule Epithelial Cells (HK-2) as a Sensitive In Vitro System for Ochratoxin A Induced Oxidative Stress. Toxins, 13(11), 787. https://doi.org/10.3390/toxins13110787