Thrombolome and Its Emerging Role in Chronic Kidney Diseases



Abstract
:1. Introduction
2. Hemostatic Disorders in CKD
3. Dysbiosis in CKD
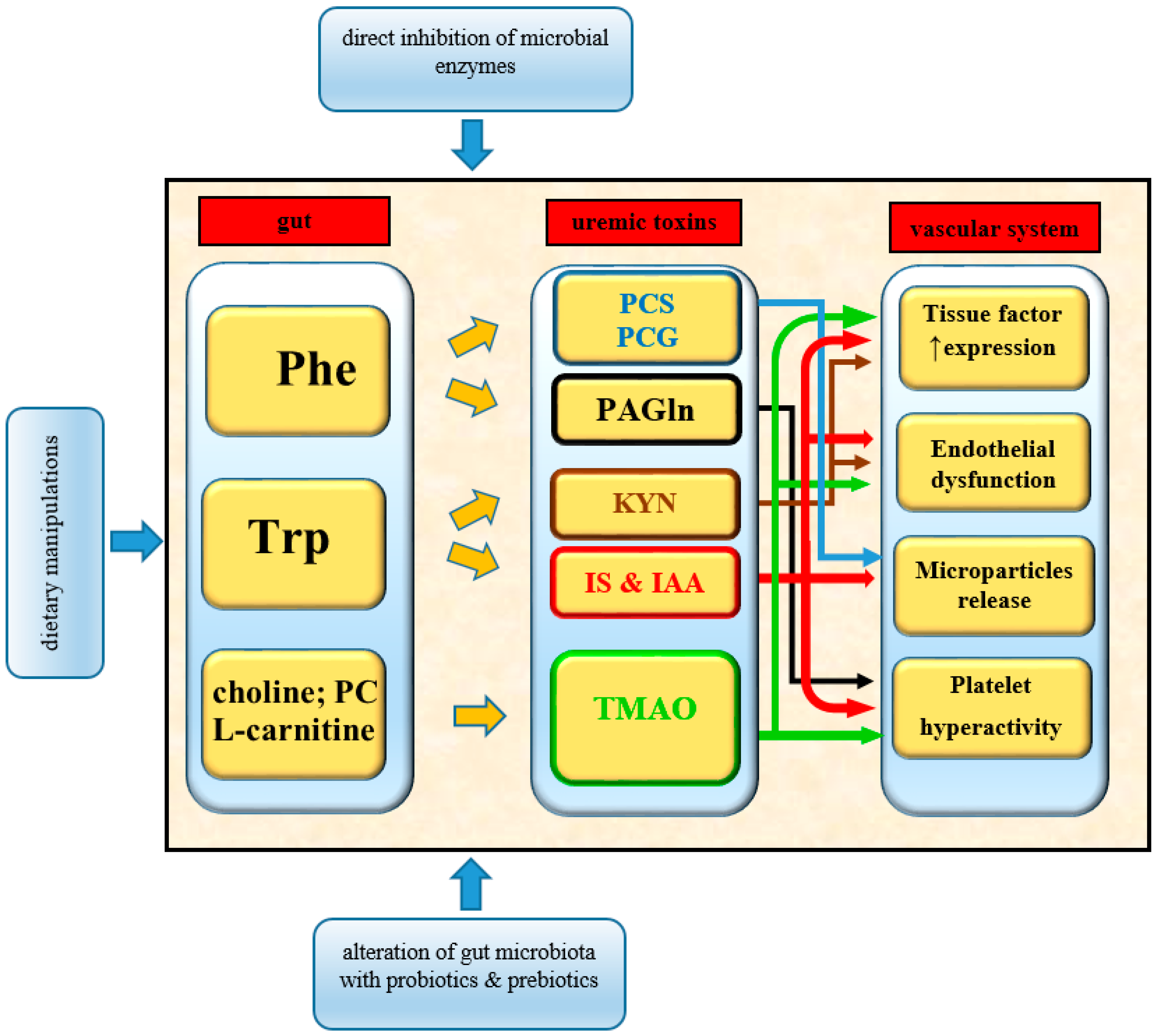
4. Gut Microbiota-Derived Uremic Toxins
5. Tryptophan-Derived Uremic Toxins
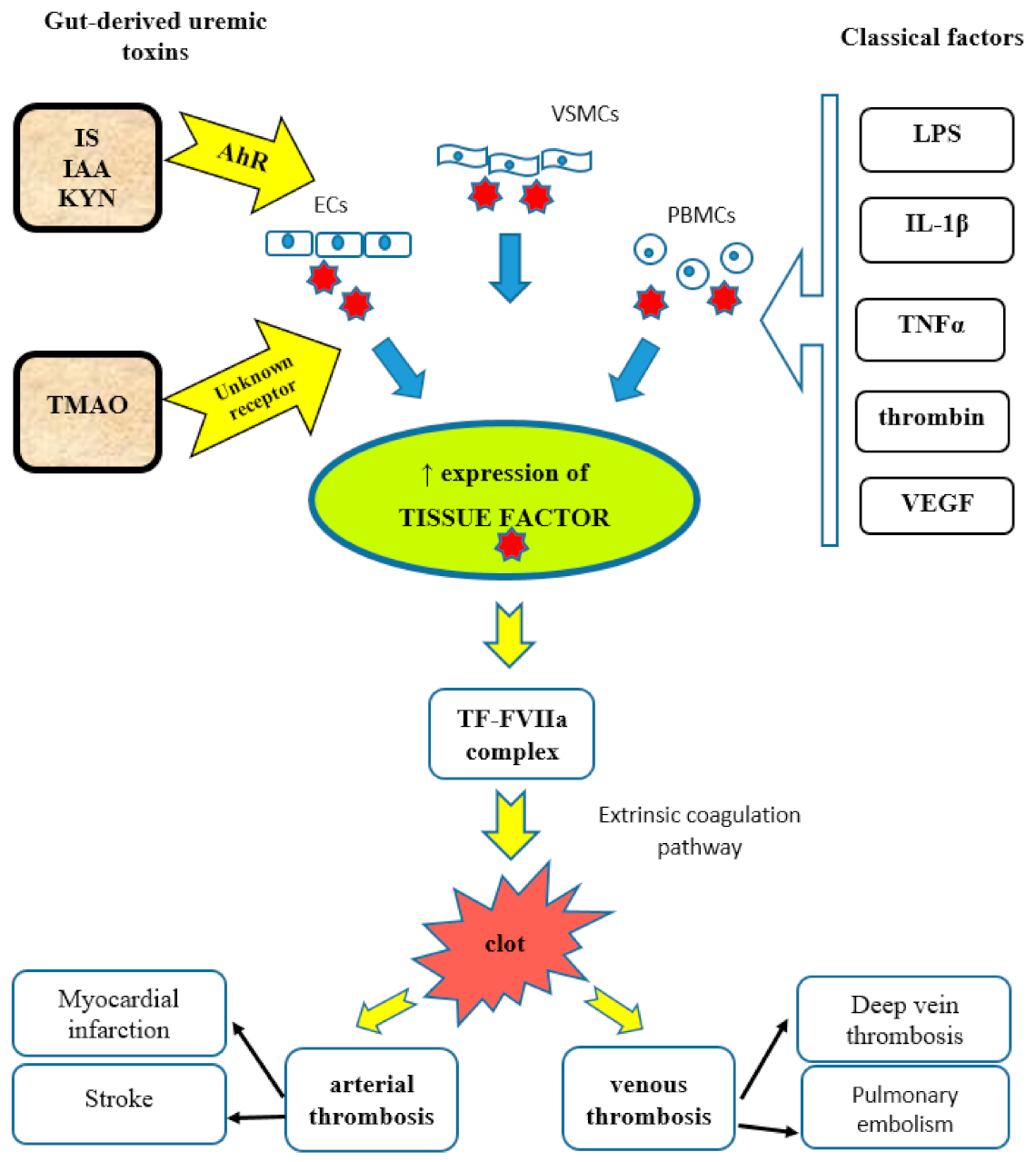
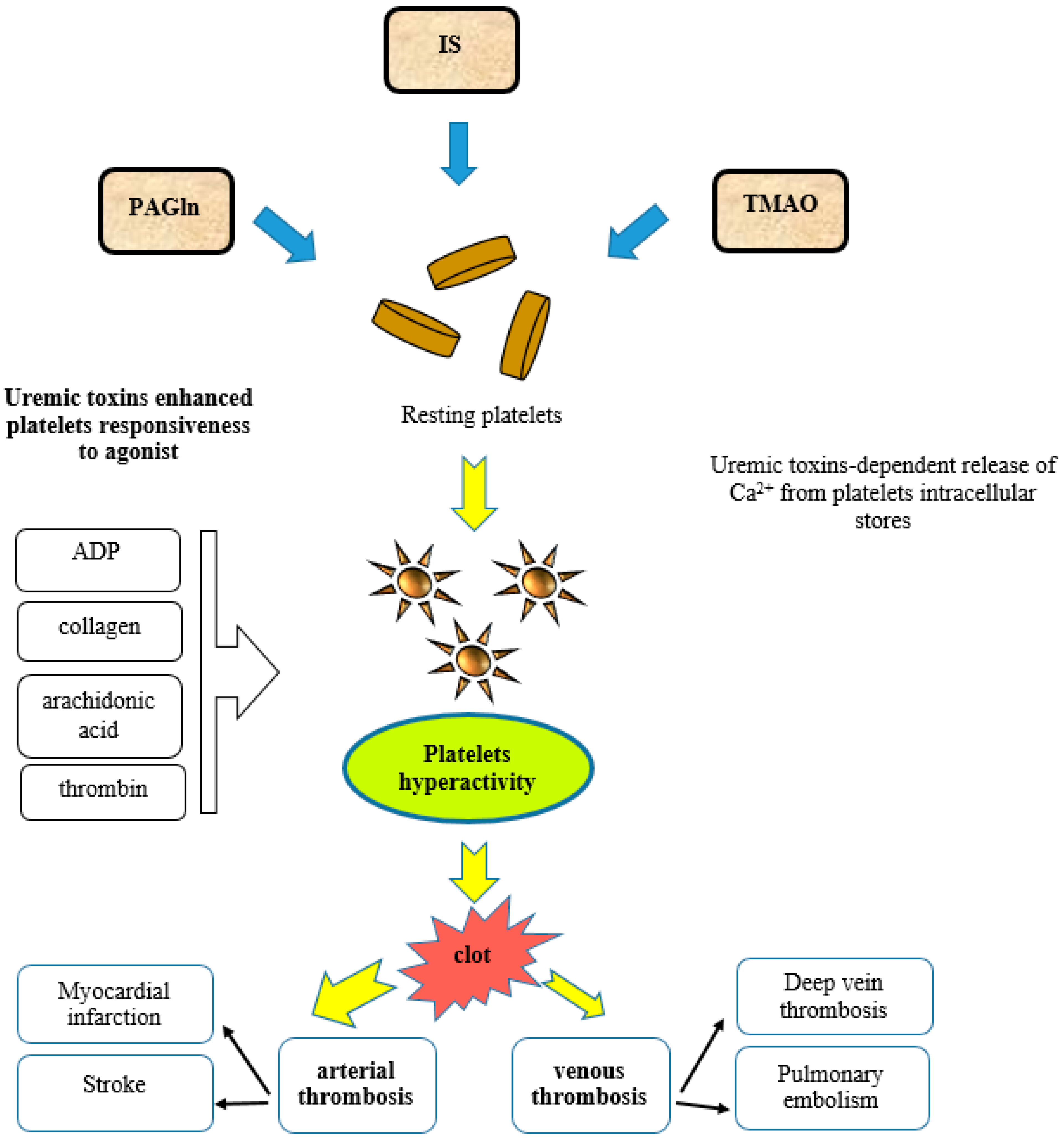
5.1. Indoxyl Sulfate and Indole-3-Acetic Acid
5.1.1. Platelet Hyperactivity
5.1.2. Endothelial Cells
5.1.3. Tissue Factor
5.2. Kynureine Metabolites
6. Phenylalanine/Tyrosine-Derived Uremic Toxins
6.1. P-Cresol Sulfate and P-Cresol Glucuronide
Endothelial Microparticles
6.2. Phenylacetylglutamine
Platelet Thrombosis via Adrenergic Receptor Signaling
7. Choline/Phosphatidylcholine-Derived Uremic Toxins
7.1. Trimethylamine N-Oxide
7.1.1. Platelet Hyperactivity
7.1.2. Endothelial Cells
7.1.3. Tissue Factor
8. Thrombosis Prevention
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jalal, D.I.; Chonchol, M.; Targher, G. Disorders of hemostasis associated with chronic kidney disease. Semin. Thromb. Hemost. 2010, 36, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.; Menke, J.; Sollinger, D.; Schinzel, H.; Thürmel, K. Haemostasis in chronic kidney disease. Nephrol. Dial. Transpl. 2014, 29, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Lutz, P.; Jurk, P. Platelets in Advanced Chronic Kidney Disease: Two Sides of the Coin. Semin. Thromb. Hemost. 2020, 46, 342–356. [Google Scholar] [CrossRef]
- Jeyaruban, A.; Hoy, W.; Cameron, A.; Healy, H.; Wang, Z.; Zhang, J.; Mallett, A. Impact of cardiovascular events on mortality and progression of renal dysfunction in a Queensland CKD cohort. Nephrology 2020, 25, 839–844. [Google Scholar] [CrossRef]
- Vanholder, R.; Massy, Z.; Argiles, A.; Spasovski, G.; Verbeke, F.; Lameire, N. Chronic kidney disease as cause of cardiovascular morbidity and mortality. Nephrol. Dial. Transpl. 2005, 20, 1048–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasquez, M.T.; Centron, P.; Barrows, I.; Dwivedi, R.; Raj, D.S. Gut Microbiota and Cardiovascular Uremic Toxicities. Toxins 2018, 10, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shashar, M.; Francis, J.; Chitalia, V. Thrombosis in the uremic milieu—Emerging role of “thrombolome”. Semin. Dial. 2015, 28, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Pavord, S.; Myers, B. Bleeding and thrombotic complications of kidney disease. Blood Rev. 2011, 25, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Rattazzi, M.; Villalta, S.; De Lucchi, L.; Sponchiado, A.; Galliazzo, S.; Faggin, E.; Pagliara, V.; Zilli, C.; Callegari, E.; Caberlotto, L.; et al. Chronic kidney disease is associated with increased risk of venous thromboembolism recurrence. Thromb. Res. 2017, 160, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Wattanakit, K.; Cushman, M.; Stehman-Breen, C.; Heckbert, S.R.; Folsom, A.R. Chronic kidney disease increases risk for venous thromboembolism. J. Am. Soc. Nephrol. 2008, 19, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Borawski, J.; Naumnik, B.; Myśliwiec, M. Circulating endothelial markers and probability of stroke in haemodialysis patients. Blood Coagul. Fibrinolys. 2003, 14, 315–317. [Google Scholar] [CrossRef]
- Ocak, G.; Lijfering, W.M.; Verduijn, M.; Dekker, F.W.; Rosendaal, F.R.; Cannegieter, S.C.; Vossen, C.Y. Risk of venous thrombosis in patients with chronic kidney disease: Identification of high-risk groups. J. Thromb. Haemost. 2013, 11, 627–633. [Google Scholar] [CrossRef]
- Mahmoodi, B.K.; Gansevoort, R.T.; Næss, I.A.; Lutsey, P.L.; Brækkan, S.K.; Veeger, N.J.; Brodin, E.E.; Meijer, K.; Sang, Y.; Matsushita, K.; et al. Association of mild to moderate chronic kidney disease with venous thromboembolism: Pooled analysis of five prospective general population cohorts. Circulation 2012, 126, 1964–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, G.; Sakhuja, A.; Taneja, A.; Majumdar, T.; Patel, J.; Whittle, J.; Nanchal, R. Pulmonary embolism in patients with CKD and ESRD. Clin. J. Am. Soc. Nephrol. 2012, 7, 1584–1590. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, K.; Pawlak, D.; Mysliwiec, M. Tissue factor and urokinase-type plasminogen activator system are related to the presence of cardiovascular disease in hemodialysis patients. Thromb. Res. 2007, 120, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Malyszko, J.; Malyszko, J.S.; Mysliwiec, M. Comparison of hemostatic disturbances between patients on CAPD and patients on hemodialysis. Perit. Dial Int. 2001, 21, 158–165. [Google Scholar] [CrossRef]
- Huang, M.J.; Wei, R.B.; Wang, Y.; Su, T.Y.; Di, P.; Li, Q.P.; Yang, X.; Li, P.; Chen, X.M. Blood coagulation system in patients with chronic kidney disease: A prospective observational study. BMJ Open 2017, 7, e014294. [Google Scholar] [CrossRef] [Green Version]
- Dumaine, R.L.; Montalescot, G.; Steg, P.G.; Ohman, E.M.; Eagle, K.; Bhatt, D.L. Renal function, atherothrombosis extent, and outcomes in high-risk patients. Am. Heart J. 2009, 158, 141–148.e141. [Google Scholar] [CrossRef] [PubMed]
- Reiner, M.F.; Müller, D.; Gobbato, S.; Stalder, O.; Limacher, A.; Bonetti, N.R.; Pasterk, L.; Méan, M.; Rodondi, N.; Aujesky, D.; et al. Gut microbiota-dependent trimethylamine-N-oxide (TMAO) shows a U-shaped association with mortality but not with recurrent venous thromboembolism. Thromb. Res. 2019, 174, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.L.; Bouchard, B.A.; Cushman, M. Venous thromboembolism, factor VIII and chronic kidney disease. Thromb. Res. 2018, 170, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolski, C.; Clark, E.G.; Sood, M.M. Venous thromboembolism in chronic kidney disease: Epidemiology, the role of proteinuria, CKD severity and therapeutics. J. Thromb Thrombolys. 2017, 43, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.L.; Zakai, N.A.; Folsom, A.R.; Kurella Tamura, M.; Peralta, C.A.; Judd, S.E.; Callas, P.W.; Cushman, M. Measures of Kidney Disease and the Risk of Venous Thromboembolism in the REGARDS (Reasons for Geographic and Racial Differences in Stroke) Study. Am. J. Kidney Dis. 2017, 70, 182–190. [Google Scholar] [CrossRef]
- Christiansen, C.F.; Schmidt, M.; Lamberg, A.L.; Horváth-Puhó, E.; Baron, J.A.; Jespersen, B.; Sørensen, H.T. Kidney disease and risk of venous thromboembolism: A nationwide population-based case-control study. J. Thromb. Haemost. 2014, 12, 1449–1454. [Google Scholar] [CrossRef]
- Wattanakit, K.; Cushman, M. Chronic kidney disease and venous thromboembolism: Epidemiology and mechanisms. Curr. Opin. Pulm. Med. 2009, 15, 408–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, C.; Katz, R.; Cushman, M.; Fried, L.F.; Shlipak, M. Association of kidney function with inflammatory and procoagulant markers in a diverse cohort: A cross-sectional analysis from the Multi-Ethnic Study of Atherosclerosis (MESA). BMC Nephrol. 2008, 9, 9. [Google Scholar] [CrossRef]
- Ocak, G.; Vossen, C.Y.; Lijfering, W.M.; Verduijn, M.; Dekker, F.W.; Rosendaal, F.R.; Cannegieter, S.C. Role of hemostatic factors on the risk of venous thrombosis in people with impaired kidney function. Circulation 2014, 129, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.L.; Zakai, N.A.; Callas, P.W.; Howard, G.; Mahmoodi, B.K.; Peralta, C.A.; Judd, S.E.; Kurella Tamura, M.; Cushman, M. Mechanisms and mitigating factors for venous thromboembolism in chronic kidney disease: The REGARDS study. J. Thromb. Haemost. 2018, 16, 1743–1752. [Google Scholar] [CrossRef]
- Boccardo, P.; Remuzzi, G.; Galbusera, M. Platelet dysfunction in renal failure. Semin. Thromb. Hemost. 2004, 30, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Kaw, D.; Malhotra, D. Platelet dysfunction and end-stage renal disease. Semin. Dial. 2006, 19, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Borawski, J.; Naumnik, B.; Mysliwiec, M. Tissue factor and thrombomodulin in hemodialysis patients: Associations with endothelial injury, liver disease, and erythropoietin therapy. Clin. Appl. Thromb Hemost. 2002, 8, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Borawski, J.; Naumnik, B.; Mysliwiec, M. Relationship between oxidative stress and extrinsic coagulation pathway in haemodialyzed patients. Thromb. Res. 2003, 109, 247–251. [Google Scholar] [CrossRef]
- Chitalia, V.C.; Shivanna, S.; Martorell, J.; Balcells, M.; Bosch, I.; Kolandaivelu, K.; Edelman, E.R. Uremic serum and solutes increase post-vascular interventional thrombotic risk through altered stability of smooth muscle cell tissue factor. Circulation 2013, 127, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [Green Version]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [Green Version]
- Simenhoff, M.L.; Saukkonen, J.J.; Burke, J.F.; Wesson, L.G., Jr.; Schaedler, R.W.; Gordon, S.J. Bacterial populations of the small intestine in uremia. Nephron 1978, 22, 63–68. [Google Scholar] [CrossRef]
- Tang, W.H.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.Y.; Tang, W.H.W. Contributory Role of Gut Microbiota and Their Metabolites Toward Cardiovascular Complications in Chronic Kidney Disease. Semin. Nephrol. 2018, 38, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, M.; Gallieni, M.; Rizzo, M.A.; Stucchi, A.; Delanaye, P.; Cavalier, E.; Moysés, R.M.A.; Jorgetti, V.; Iervasi, G.; Giannini, S.; et al. Vitamin K plasma levels determination in human health. Clin. Chem. Lab. Med. 2017, 55, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäckel, S.; Kiouptsi, K.; Lillich, M.; Hendrikx, T.; Khandagale, A.; Kollar, B.; Hörmann, N.; Reiss, C.; Subramaniam, S.; Wilms, E.; et al. Gut microbiota regulate hepatic von Willebrand factor synthesis and arterial thrombus formation via Toll-like receptor-2. Blood 2017, 130, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Bryniarski, M.A.; Hamarneh, F.; Yacoub, R. The role of chronic kidney disease-associated dysbiosis in cardiovascular disease. Exp. Biol. Med. 2019, 244, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Gryp, T.; Huys, G.R.B.; Joossens, M.; Van Biesen, W.; Glorieux, G.; Vaneechoutte, M. Isolation and Quantification of Uremic Toxin Precursor-Generating Gut Bacteria in Chronic Kidney Disease Patients. Int. J. Mol. Sci. 2020, 21, 1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mafra, D.; Borges, N.; Alvarenga, L.; Esgalhado, M.; Cardozo, L.; Lindholm, B.; Stenvinkel, P. Dietary Components That May Influence the Disturbed Gut Microbiota in Chronic Kidney Disease. Nutrients 2019, 11, 496. [Google Scholar] [CrossRef] [Green Version]
- Brunet, P.; Gondouin, B.; Duval-Sabatier, A.; Dou, L.; Cerini, C.; Dignat-George, F.; Jourde-Chiche, N.; Argiles, A.; Burtey, S. Does uremia cause vascular dysfunction? Kidney Blood Press. Res. 2011, 34, 284–290. [Google Scholar] [CrossRef]
- Cunha, R.S.D.; Santos, A.F.; Barreto, F.C.; Stinghen, A.E.M. How do Uremic Toxins Affect the Endothelium? Toxins 2020, 12, 412. [Google Scholar] [CrossRef]
- Addi, T.; Dou, L.; Burtey, S. Tryptophan-Derived Uremic Toxins and Thrombosis in Chronic Kidney Disease. Toxins 2018, 10, 412. [Google Scholar] [CrossRef] [Green Version]
- Lano, G.; Burtey, S.; Sallée, M. Indoxyl Sulfate, a Uremic Endotheliotoxin. Toxins 2020, 12, 229. [Google Scholar] [CrossRef] [Green Version]
- Bush, K.T.; Singh, P.; Nigam, S.K. Gut-derived uremic toxin handling in vivo requires OAT-mediated tubular secretion in chronic kidney disease. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Holle, J.; Kirchner, M.; Okun, J.; Bayazit, A.K.; Obrycki, L.; Canpolat, N.; Bulut, I.K.; Azukaitis, K.; Duzova, A.; Ranchin, B.; et al. Serum indoxyl sulfate concentrations associate with progression of chronic kidney disease in children. PLoS ONE 2020, 15, e0240446. [Google Scholar] [CrossRef]
- Lin, C.J.; Chen, H.H.; Pan, C.F.; Chuang, C.K.; Wang, T.J.; Sun, F.J.; Wu, C.J. p-Cresylsulfate and indoxyl sulfate level at different stages of chronic kidney disease. J. Clin. Lab. Anal. 2011, 25, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Ezawa, A.; Kikuchi, K.; Tsuruta, Y.; Niwa, T. Protein-bound uremic toxins in hemodialysis patients measured by liquid chromatography/tandem mass spectrometry and their effects on endothelial ROS production. Anal. Bioanal. Chem. 2012, 403, 1841–1850. [Google Scholar] [CrossRef]
- Karbowska, M.; Kaminski, T.W.; Marcinczyk, N.; Misztal, T.; Rusak, T.; Smyk, L.; Pawlak, D. The Uremic Toxin Indoxyl Sulfate Accelerates Thrombotic Response after Vascular Injury in Animal Models. Toxins 2017, 9, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karbowska, M.; Kaminski, T.W.; Znorko, B.; Domaniewski, T.; Misztal, T.; Rusak, T.; Pryczynicz, A.; Guzinska-Ustymowicz, K.; Pawlak, K.; Pawlak, D. Indoxyl Sulfate Promotes Arterial Thrombosis in Rat Model via Increased Levels of Complex TF/VII, PAI-1, Platelet Activation as Well as Decreased Contents of SIRT1 and SIRT3. Front. Physiol. 2018, 9, 1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolachalama, V.B.; Shashar, M.; Alousi, F.; Shivanna, S.; Rijal, K.; Belghasem, M.E.; Walker, J.; Matsuura, S.; Chang, G.H.; Gibson, C.M.; et al. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. J. Am. Soc. Nephrol. 2018, 29, 1063–1072. [Google Scholar] [CrossRef]
- Wu, C.C.; Hsieh, M.Y.; Hung, S.C.; Kuo, K.L.; Tsai, T.H.; Lai, C.L.; Chen, J.W.; Lin, S.J.; Huang, P.H.; Tarng, D.C. Serum Indoxyl Sulfate Associates with Postangioplasty Thrombosis of Dialysis Grafts. J. Am. Soc. Nephrol. 2016, 27, 1254–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.J.; Pan, C.F.; Liu, H.L.; Chuang, C.K.; Jayakumar, T.; Wang, T.J.; Chen, H.H.; Wu, C.J. The role of protein-bound uremic toxins on peripheral artery disease and vascular access failure in patients on hemodialysis. Atherosclerosis 2012, 225, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Ji, S.; Dong, W.; Qi, Y.; Song, W.; Cui, D.; Shi, J. Indolic uremic solutes enhance procoagulant activity of red blood cells through phosphatidylserine exposure and microparticle release. Toxins 2015, 7, 4390–4403. [Google Scholar] [CrossRef] [Green Version]
- Hamza, E.; Metzinger, L.; Metzinger-Le Meuth, V. Uremic Toxins Affect Erythropoiesis during the Course of Chronic Kidney Disease: A Review. Cells 2020, 9, 2039. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Abed, M.; Voelkl, J.; Lang, F. Triggering of suicidal erythrocyte death by uremic toxin indoxyl sulfate. BMC Nephrol. 2013, 14, 244. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Du, C.; Wang, X.; Li, F.; Xu, Y.; Wang, S.; Chen, S.; Chen, F.; Shen, M.; Chen, M.; et al. Indoxyl sulfate induces platelet hyperactivity and contributes to chronic kidney disease-associated thrombosis in mice. Blood 2017, 129, 2667–2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krötz, F.; Sohn, H.Y.; Pohl, U. Reactive oxygen species: Players in the platelet game. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1988–1996. [Google Scholar] [CrossRef] [PubMed]
- Dias, G.F.; Bonan, N.B.; Steiner, T.M.; Tozoni, S.S.; Rodrigues, S.; Nakao, L.S.; Kuntsevich, V.; Pecoits Filho, R.; Kotanko, P.; Moreno-Amaral, A.N. Indoxyl Sulfate, a Uremic Toxin, Stimulates Reactive Oxygen Species Production and Erythrocyte Cell Death Supposedly by an Organic Anion Transporter 2 (OAT2) and NADPH Oxidase Activity-Dependent Pathways. Toxins 2018, 10, 280. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.H.; Lai, Y.H.; Kuo, C.H.; Lin, Y.L.; Tsai, J.P.; Hsu, B.G. Association between Serum Indoxyl Sulfate Levels and Endothelial Function in Non-Dialysis Chronic Kidney Disease. Toxins 2019, 11, 589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumur, Z.; Niwa, T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am. J. Nephrol. 2009, 29, 551–557. [Google Scholar] [CrossRef]
- Lin, C.J.; Wu, C.J.; Wu, P.C.; Pan, C.F.; Wang, T.J.; Sun, F.J.; Liu, H.L.; Chen, H.H.; Yeh, H.I. Indoxyl Sulfate Impairs Endothelial Progenitor Cells and Might Contribute to Vascular Dysfunction in Patients with Chronic Kidney Disease. Kidney Blood Press. Res. 2016, 41, 1025–1036. [Google Scholar] [CrossRef]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Yoo, T.H.; Hwang, Y.; Lee, G.H.; Kim, B.; Jang, J.; Yu, H.T.; Kim, M.C.; Cho, J.Y.; Lee, C.J.; et al. Indoxyl sulfate (IS)-mediated immune dysfunction provokes endothelial damage in patients with end-stage renal disease (ESRD). Sci. Rep. 2017, 7, 3057. [Google Scholar] [CrossRef] [Green Version]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-kappaB activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [Green Version]
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef] [Green Version]
- Owens, A.P., 3rd; Mackman, N. Tissue factor and thrombosis: The clot starts here. Thromb Haemost. 2010, 104, 432–439. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.P.; et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J. Am. Soc. Nephrol. 2016, 27, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Shashar, M.; Belghasem, M.E.; Matsuura, S.; Walker, J.; Richards, S.; Alousi, F.; Rijal, K.; Kolachalama, V.B.; Balcells, M.; Odagi, M.; et al. Targeting STUB1-tissue factor axis normalizes hyperthrombotic uremic phenotype without increasing bleeding risk. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addi, T.; Poitevin, S.; McKay, N.; El Mecherfi, K.E.; Kheroua, O.; Jourde-Chiche, N.; de Macedo, A.; Gondouin, B.; Cerini, C.; Brunet, P.; et al. Mechanisms of tissue factor induction by the uremic toxin indole-3 acetic acid through aryl hydrocarbon receptor/nuclear factor-kappa B signaling pathway in human endothelial cells. Arch. Toxicol. 2019, 93, 121–136. [Google Scholar] [CrossRef]
- Wirleitner, B.; Rudzite, V.; Neurauter, G.; Murr, C.; Kalnins, U.; Erglis, A.; Trusinskis, K.; Fuchs, D. Immune activation and degradation of tryptophan in coronary heart disease. Eur. J. Clin. Investig. 2003, 33, 550–554. [Google Scholar] [CrossRef]
- Mangge, H.; Stelzer, I.; Reininghaus, E.Z.; Weghuber, D.; Postolache, T.T.; Fuchs, D. Disturbed tryptophan metabolism in cardiovascular disease. Curr. Med. Chem. 2014, 21, 1931–1937. [Google Scholar] [CrossRef] [PubMed]
- Sulo, G.; Vollset, S.E.; Nygård, O.; Midttun, Ø.; Ueland, P.M.; Eussen, S.J.; Pedersen, E.R.; Tell, G.S. Neopterin and kynurenine-tryptophan ratio as predictors of coronary events in older adults, the Hordaland Health Study. Int. J. Cardiol. 2013, 168, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Darlington, L.G.; Mackay, G.M.; Forrest, C.M.; Stoy, N.; George, C.; Stone, T.W. Altered kynurenine metabolism correlates with infarct volume in stroke. Eur. J. Neurosci. 2007, 26, 2211–2221. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Fujigaki, S.; Heyes, M.P.; Shibata, K.; Takemura, M.; Fujii, H.; Wada, H.; Noma, A.; Seishima, M. Mechanism of increases in L-kynurenine and quinolinic acid in renal insufficiency. Am. J. Physiol. Renal Physiol. 2000, 279, F565–F572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, T.; Yoshizumi, H.; Emoto, Y.; Miyazaki, T.; Hashimoto, N.; Takeda, N.; Tatematsu, A.; Maeda, K. Accumulation of quinolinic acid in uremic serum and its removal by hemodialysis. Clin. Chem. 1991, 37, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Mysliwiec, M.; Pawlak, D. Haemostatic system, biochemical profiles, kynurenines and the prevalence of cardiovascular disease in peritoneally dialyzed patients. Thromb. Res. 2010, 125, e40–e45. [Google Scholar] [CrossRef]
- Pawlak, K.; Mysliwiec, M.; Pawlak, D. Hypercoagulability is independently associated with kynurenine pathway activation in dialysed uraemic patients. Thromb Haemost. 2009, 102, 49–55. [Google Scholar] [CrossRef]
- Pawlak, K.; Domaniewski, T.; Mysliwiec, M.; Pawlak, D. Kynurenines and oxidative status are independently associated with thrombomodulin and von Willebrand factor levels in patients with end-stage renal disease. Thromb. Res. 2009, 124, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Sallée, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef]
- Pawlak, K.; Myśliwiec, M.; Pawlak, D. Kynurenine pathway—A new link between endothelial dysfunction and carotid atherosclerosis in chronic kidney disease patients. Adv. Med. Sci. 2010, 55, 196–203. [Google Scholar] [CrossRef]
- Pawlak, K.; Brzosko, S.; Mysliwiec, M.; Pawlak, D. Kynurenine, quinolinic acid--the new factors linked to carotid atherosclerosis in patients with end-stage renal disease. Atherosclerosis 2009, 204, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Tankiewicz, J.; Mysliwiec, M.; Pawlak, D. Tissue factor/its pathway inhibitor system and kynurenines in chronic kidney disease patients on conservative treatment. Blood Coagul. Fibrinolys. 2009, 20, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Kowalewska, A.; Mysliwiec, M.; Pawlak, D. Kynurenine and its metabolites—Kynurenic acid and anthranilic acid are associated with soluble endothelial adhesion molecules and oxidative status in patients with chronic kidney disease. Am. J. Med. Sci. 2009, 338, 293–300. [Google Scholar] [CrossRef]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef]
- Mutsaers, H.A.; Caetano-Pinto, P.; Seegers, A.E.; Dankers, A.C.; van den Broek, P.H.; Wetzels, J.F.; van den Brand, J.A.; van den Heuvel, L.P.; Hoenderop, J.G.; Wilmer, M.J.; et al. Proximal tubular efflux transporters involved in renal excretion of p-cresyl sulfate and p-cresyl glucuronide: Implications for chronic kidney disease pathophysiology. Toxicol. In Vitro 2015, 29, 1868–1877. [Google Scholar] [CrossRef]
- Poesen, R.; Evenepoel, P.; de Loor, H.; Kuypers, D.; Augustijns, P.; Meijers, B. Metabolism, Protein Binding, and Renal Clearance of Microbiota-Derived p-Cresol in Patients with CKD. Clin J. Am. Soc. Nephrol. 2016, 11, 1136–1144. [Google Scholar] [CrossRef] [Green Version]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transpl. 2010, 25, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Wu, I.W.; Hsu, K.H.; Hsu, H.J.; Lee, C.C.; Sun, C.Y.; Tsai, C.J.; Wu, M.S. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients—A prospective cohort study. Nephrol. Dial. Transpl. 2012, 27, 1169–1175. [Google Scholar] [CrossRef] [Green Version]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Xie, R.; Yu, C.; Ma, R.; Dong, W.; Meng, H.; Zhang, Y.; Si, Y.; Zhang, Z.; Novakovic, V.; et al. Thrombotic Role of Blood and Endothelial Cells in Uremia through Phosphatidylserine Exposure and Microparticle Release. PLoS ONE 2015, 10, e0142835. [Google Scholar] [CrossRef]
- Mörtberg, J.; Lundwall, K.; Mobarrez, F.; Wallén, H.; Jacobson, S.H.; Spaak, J. Increased concentrations of platelet- and endothelial-derived microparticles in patients with myocardial infarction and reduced renal function—A descriptive study. BMC Nephrol. 2019, 20, 71. [Google Scholar] [CrossRef]
- Favretto, G.; Cunha, R.S.D.; Dalboni, M.A.; Oliveira, R.B.; Barreto, F.C.; Massy, Z.A.; Stinghen, A.E.M. Endothelial Microparticles in Uremia: Biomarkers and Potential Therapeutic Targets. Toxins 2019, 11, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faure, V.; Dou, L.; Sabatier, F.; Cerini, C.; Sampol, J.; Berland, Y.; Brunet, P.; Dignat-George, F. Elevation of circulating endothelial microparticles in patients with chronic renal failure. J. Thromb. Haemost. 2006, 4, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; Van Kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef]
- Ryu, J.H.; Park, H.; Kim, S.J. The effects of indoxyl sulfate-induced endothelial microparticles on neointimal hyperplasia formation in an ex vivo model. Ann. Surg. Treat. Res. 2017, 93, 11–17. [Google Scholar] [CrossRef]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef]
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y.; et al. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 2020, 180, 862–877.e822. [Google Scholar] [CrossRef] [PubMed]
- Poesen, R.; Claes, K.; Evenepoel, P.; de Loor, H.; Augustijns, P.; Kuypers, D.; Meijers, B. Microbiota-Derived Phenylacetylglutamine Associates with Overall Mortality and Cardiovascular Disease in Patients with CKD. J. Am. Soc. Nephrol. 2016, 27, 3479–3487. [Google Scholar] [CrossRef] [Green Version]
- Saeed, S.A.; Rasheed, H.; Fecto, F.A.; Achakzai, M.I.; Ali, R.; Connor, J.D.; Gilani, A.U. Signaling mechanisms mediated by G-protein coupled receptors in human platelets. Acta Pharmacol. Sin. 2004, 25, 887–892. [Google Scholar]
- Stalker, T.J.; Newman, D.K.; Ma, P.; Wannemacher, K.M.; Brass, L.F. Platelet signaling. Handb. Exp. Pharmacol. 2012. [Google Scholar] [CrossRef] [Green Version]
- Ziff, O.J.; Samra, M.; Howard, J.P.; Bromage, D.I.; Ruschitzka, F.; Francis, D.P.; Kotecha, D. Beta-blocker efficacy across different cardiovascular indications: An umbrella review and meta-analytic assessment. BMC Med. 2020, 18, 103. [Google Scholar] [CrossRef] [PubMed]
- Petrikova, M.; Jancinova, V.; Nosal, R.; Majekova, M.; Fabryova, V. Carvedilol—A beta-blocker with considerable antiaggregatory effect on human blood platelets. Bratisl. Lek Listy 2005, 106, 20–25. [Google Scholar]
- Ilardi, F.; Gargiulo, G.; Schiattarella, G.G.; Giugliano, G.; Paolillo, R.; Menafra, G.; De Angelis, E.; Scudiero, L.; Franzone, A.; Stabile, E.; et al. Effects of Carvedilol Versus Metoprolol on Platelet Aggregation in Patients with Acute Coronary Syndrome: The PLATE-BLOCK Study. Am. J. Cardiol. 2018, 122, 6–11. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Bergeron, N.; Levison, B.S.; Li, X.S.; Chiu, S.; Jia, X.; Koeth, R.A.; Li, L.; Wu, Y.; Tang, W.H.W.; et al. Impact of chronic dietary red meat, white meat, or non-meat protein on trimethylamine N-oxide metabolism and renal excretion in healthy men and women. Eur. Heart J. 2019, 40, 583–594. [Google Scholar] [CrossRef]
- Rhee, E.P.; Clish, C.B.; Ghorbani, A.; Larson, M.G.; Elmariah, S.; McCabe, E.; Yang, Q.; Cheng, S.; Pierce, K.; Deik, A.; et al. A combined epidemiologic and metabolomic approach improves CKD prediction. J. Am. Soc. Nephrol. 2013, 24, 1330–1338. [Google Scholar] [CrossRef] [Green Version]
- Miyake, T.; Mizuno, T.; Mochizuki, T.; Kimura, M.; Matsuki, S.; Irie, S.; Ieiri, I.; Maeda, K.; Kusuhara, H. Involvement of Organic Cation Transporters in the Kinetics of Trimethylamine N-oxide. J. Pharm. Sci. 2017, 106, 2542–2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, C.C.; Croyal, M.; Ene, L.; Aguesse, A.; Billon-Crossouard, S.; Krempf, M.; Lemoine, S.; Guebre-Egziabher, F.; Juillard, L.; Soulage, C.O. Elevation of Trimethylamine-N-Oxide in Chronic Kidney Disease: Contribution of Decreased Glomerular Filtration Rate. Toxins 2019, 11, 635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liabeuf, S.; Cheddani, L.; Massy, Z.A. Uremic Toxins and Clinical Outcomes: The Impact of Kidney Transplantation. Toxins 2018, 10, 229. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.D.; Lee, J.A.; Lee, H.A.; Sadler, P.J.; Wilkie, D.R.; Woodham, R.H. Nuclear magnetic resonance studies of blood plasma and urine from subjects with chronic renal failure: Identification of trimethylamine-N-oxide. Biochim. Biophys. Acta 1991, 1096, 101–107. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Z. Impact of trimethylamine N-oxide (TMAO) metaorganismal pathway on cardiovascular disease. J. Lab. Precis. Med. 2020, 5. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Haghikia, A.; Li, X.S.; Liman, T.G.; Bledau, N.; Schmidt, D.; Zimmermann, F.; Kränkel, N.; Widera, C.; Sonnenschein, K.; Haghikia, A.; et al. Gut Microbiota-Dependent Trimethylamine N-Oxide Predicts Risk of Cardiovascular Events in Patients With Stroke and Is Related to Proinflammatory Monocytes. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2225–2235. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Wang, Z.; Tang, W.H.W.; Hazen, S.L. Gut Microbe-Generated Trimethylamine N-Oxide From Dietary Choline Is Prothrombotic in Subjects. Circulation 2017, 135, 1671–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craciun, S.; Balskus, E.P. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl. Acad. Sci. USA 2012, 109, 21307–21312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Gua, C.; Wu, B.; Chen, Y. Increased circulating trimethylamine N-oxide contributes to endothelial dysfunction in a rat model of chronic kidney disease. Biochem. Biophys. Res. Commun. 2018, 495, 2071–2077. [Google Scholar] [CrossRef]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-κB. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Pan, B.; Chen, Y.; Guo, C.; Zhao, M.; Zheng, L.; Chen, B. Trimethylamine N-oxide in atherogenesis: Impairing endothelial self-repair capacity and enhancing monocyte adhesion. BioSci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Chen, Y.; Gua, C.; Li, X. Elevated Circulating Trimethylamine N-Oxide Levels Contribute to Endothelial Dysfunction in Aged Rats through Vascular Inflammation and Oxidative Stress. Front. Physiol. 2017, 8, 350. [Google Scholar] [CrossRef]
- Boini, K.M.; Hussain, T.; Li, P.L.; Koka, S. Trimethylamine-N-Oxide Instigates NLRP3 Inflammasome Activation and Endothelial Dysfunction. Cell. Physiol. Biochem. 2017, 44, 152–162. [Google Scholar] [CrossRef]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.F.; Yu, T.; Chu, X.M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Hashiguchi, K.; Yamamoto, H.; Tagami, M. Dietary Apigenin Reduces Induction of LOX-1 and NLRP3 Expression, Leukocyte Adhesion, and Acetylated Low-Density Lipoprotein Uptake in Human Endothelial Cells Exposed to Trimethylamine-N-Oxide. J. Cardiovasc. Pharmacol. 2019, 74, 558–565. [Google Scholar] [CrossRef]
- Cheng, X.; Qiu, X.; Liu, Y.; Yuan, C.; Yang, X. Trimethylamine N-oxide promotes tissue factor expression and activity in vascular endothelial cells: A new link between trimethylamine N-oxide and atherosclerotic thrombosis. Thromb. Res. 2019, 177, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Fletcher, C. Trimethylamine N-oxide: Breathe new life. Br. J. Pharmacol. 2018, 175, 1344–1353. [Google Scholar] [CrossRef]
- Roberts, A.B.; Gu, X.; Buffa, J.A.; Hurd, A.G.; Wang, Z.; Zhu, W.; Gupta, N.; Skye, S.M.; Cody, D.B.; Levison, B.S.; et al. Development of a gut microbe-targeted nonlethal therapeutic to inhibit thrombosis potential. Nat. Med. 2018, 24, 1407–1417. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Zhao, Y.Y.; Pahl, M.V. Altered intestinal microbial flora and impaired epithelial barrier structure and function in CKD: The nature, mechanisms, consequences and potential treatment. Nephrol. Dial. Transpl. 2016, 31, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Vanholder, R.; Glorieux, G. The intestine and the kidneys: A bad marriage can be hazardous. Clin. Kidney J. 2015, 8, 168–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieffer, D.A.; Piccolo, B.D.; Vaziri, N.D.; Liu, S.; Lau, W.L.; Khazaeli, M.; Nazertehrani, S.; Moore, M.E.; Marco, M.L.; Martin, R.J.; et al. Resistant starch alters gut microbiome and metabolomic profiles concurrent with amelioration of chronic kidney disease in rats. Am. J. Physiol. Renal Physiol. 2016, 310, F857–F871. [Google Scholar] [CrossRef] [Green Version]
- Furuse, S.U.; Ohse, T.; Jo-Watanabe, A.; Shigehisa, A.; Kawakami, K.; Matsuki, T.; Chonan, O.; Nangaku, M. Galacto-oligosaccharides attenuate renal injury with microbiota modification. Physiol. Rep. 2014, 2. [Google Scholar] [CrossRef]
- Meijers, B.K.; De Preter, V.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. p-Cresyl sulfate serum concentrations in haemodialysis patients are reduced by the prebiotic oligofructose-enriched inulin. Nephrol. Dial. Transpl. 2010, 25, 219–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, C.I.; Armani, R.G.; Canziani, M.E.F.; Dalboni, M.A.; Dolenga, C.J.R.; Nakao, L.S.; Campbell, K.L.; Cuppari, L. Effect of prebiotic (fructooligosaccharide) on uremic toxins of chronic kidney disease patients: A randomized controlled trial. Nephrol. Dial. Transpl. 2019, 34, 1876–1884. [Google Scholar] [CrossRef]
- Rossi, M.; Johnson, D.W.; Morrison, M.; Pascoe, E.M.; Coombes, J.S.; Forbes, J.M.; Szeto, C.C.; McWhinney, B.C.; Ungerer, J.P.; Campbell, K.L. Synbiotics Easing Renal Failure by Improving Gut Microbiology (SYNERGY): A Randomized Trial. Clin J. Am. Soc. Nephrol. 2016, 11, 223–231. [Google Scholar] [CrossRef]
- Gallè, F.; Valeriani, F.; Cattaruzza, M.S.; Ubaldi, F.; Romano Spica, V.; Liguori, G. Exploring the association between physical activity and gut microbiota composition: A review of current evidence. Ann Ig 2019, 31, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Hiramoto, T.; Nishino, R.; Aiba, Y.; Kimura, T.; Yoshihara, K.; Koga, Y.; Sudo, N. Critical role of gut microbiota in the production of biologically active, free catecholamines in the gut lumen of mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1288–G1295. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, O.; Cronin, O.; Clarke, S.F.; Murphy, E.F.; Molloy, M.G.; Shanahan, F.; Cotter, P.D. Exercise and the microbiota. Gut Microbes 2015, 6, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Headley, S.A.; Chapman, D.J.; Germain, M.J.; Evans, E.E.; Hutchinson, J.; Madsen, K.L.; Ikizler, T.A.; Miele, E.M.; Kirton, K.; O’Neill, E.; et al. The effects of 16-weeks of prebiotic supplementation and aerobic exercise training on inflammatory markers, oxidative stress, uremic toxins, and the microbiota in pre-dialysis kidney patients: A randomized controlled trial-protocol paper. BMC Nephrol. 2020, 21, 517. [Google Scholar] [CrossRef] [PubMed]
- Esgalhado, M.; Borges, N.A.; Mafra, D. Could physical exercise help modulate the gut microbiota in chronic kidney disease? Future Microbiol. 2016, 11, 699–707. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gut Microbiota | Enzyme Possessed by Bacteria | ||
|---|---|---|---|
| Phylum | Family | ||
| More abundant in ESRD | Actinobacteria | Cellulomonadaceae | urease |
| Dermabacteraceae | urease | ||
| Microccaceae | urease | ||
| Firmicutes | Clostridiaceae | urease; tryptophanase; p-cresol production enzymes | |
| Proteobacteria | Polyangiaceae | urease | |
| Alteromonadaceae | urease | ||
| Enterobacteriaceae | urease; tryptophanase; p-cresol production enzymes | ||
| Methylococcaceae | urease | ||
| Halomonadaceae | urease | ||
| Moraxellaceae | urease | ||
| Pseudomonadaceae | urease | ||
| Xanthomonadaceae | urease | ||
| Verrucomicrobia | Verrucomicrobiaceae | urease; tryptophanase | |
| Less abundant in ESRD | Bacteroidetes | Prevotellaceae | phosphotransbutyrylase; butyrate kinase |
| Firmicutes | Lactobacillaceae | Phosphotransbutyrylase; butyrate kinase | |
| Proteobacteria | Alcaligenaceae | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fryc, J.; Naumnik, B. Thrombolome and Its Emerging Role in Chronic Kidney Diseases. Toxins 2021, 13, 223. https://doi.org/10.3390/toxins13030223
Fryc J, Naumnik B. Thrombolome and Its Emerging Role in Chronic Kidney Diseases. Toxins. 2021; 13(3):223. https://doi.org/10.3390/toxins13030223
Chicago/Turabian StyleFryc, Justyna, and Beata Naumnik. 2021. "Thrombolome and Its Emerging Role in Chronic Kidney Diseases" Toxins 13, no. 3: 223. https://doi.org/10.3390/toxins13030223
APA StyleFryc, J., & Naumnik, B. (2021). Thrombolome and Its Emerging Role in Chronic Kidney Diseases. Toxins, 13(3), 223. https://doi.org/10.3390/toxins13030223