Innovative and Highly Sensitive Detection of Clostridium perfringens Enterotoxin Based on Receptor Interaction and Monoclonal Antibodies

 , , , , , , , , ,
, , , , , , , , ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Generation of High Affinity Monoclonal Antibodies (mAbs) Recognizing Distinct Epitopes
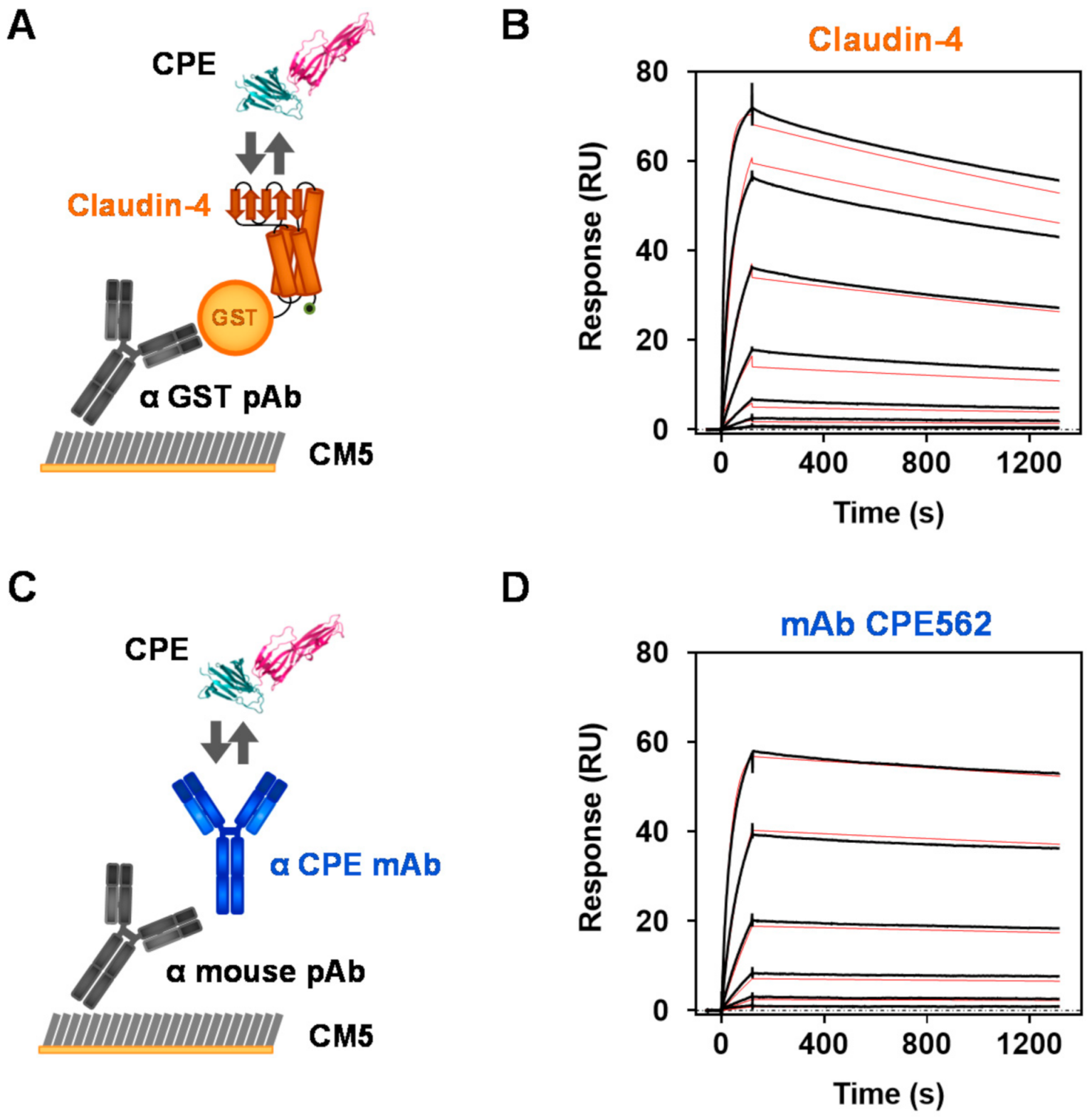
2.2. Comparing Affinities of the CPE Receptor Claudin-4 with a CPE-Specific Antibody
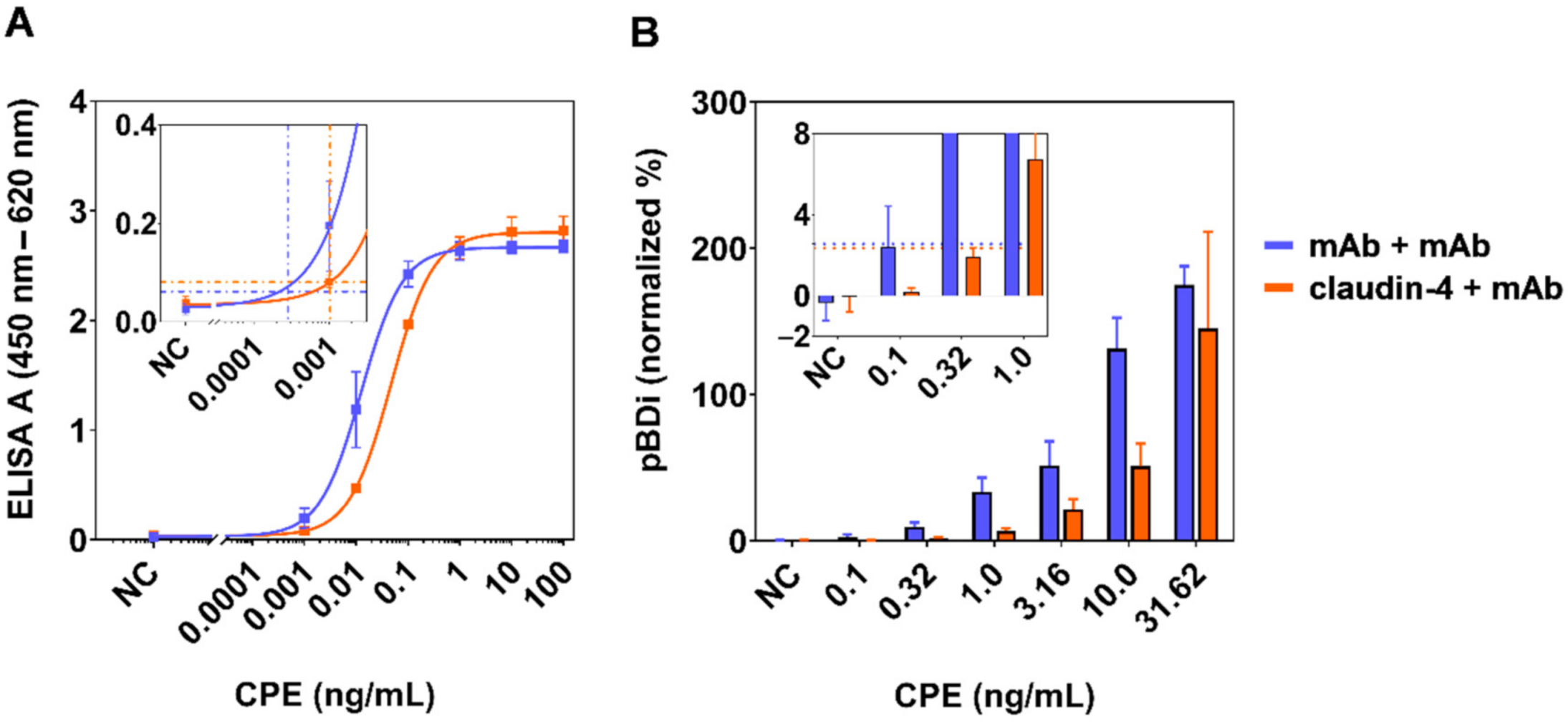
2.3. Development of Stationary and Mobile Sandwich ELISA Using High-Affinity mAbs and Receptor Interaction
2.4. Detection of CPE from Spiked Fecal Samples
2.5. Evaluation of Established ELISAs by Testing a Comprehensive Panel of C. perfringens Supernatants
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animal Experiments
5.2. Recombinant Expression of CPE Proteins and GST-Claudin-4 Fusion Protein
5.3. Generation of Monoclonal Antibodies
5.4. Indirect ELISA
5.5. Antibody- and Receptor-Based Sandwich ELISA
5.6. Surface Plasmon Resonance (SPR) Measurements: Binding Kinetics
5.7. SPR Measurements: Epitope Binning
5.8. SDS-PAGE and Western Blots
5.9. Neutralizing CPE in a Cell-Based Cytotoxicity Assay
5.10. On-Site Detection Based on an Electrochemical Biochip Platform (pBDi)
5.11. Preparation of Spiked Fecal Samples
5.12. Preparation of Supernatants from Broth Cultures
5.13. Gene Toxinotyping by qPCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Awad, M.M.; Ellemor, D.M.; Boyd, R.L.; Emmins, J.J.; Rood, J.I. Synergistic effects of alpha-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect. Immun. 2001, 69, 7904–7910. [Google Scholar] [CrossRef] [Green Version]
- Grass, J.E.; Gould, L.H.; Mahon, B.E. Epidemiology of foodborne disease outbreaks caused by Clostridium perfringens, United States, 1998–2010. Foodborne Pathog. Dis. 2013, 10, 131–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzal, F.A.; Freedman, J.C.; Shrestha, A.; Theoret, J.R.; Garcia, J.; Awad, M.M.; Adams, V.; Moore, R.J.; Rood, J.I.; McClane, B.A. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 2014, 9, 361–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Paredes-Sabja, D.; Sarker, M.R.; McClane, B.A. Clostridium perfringens sporulation and sporulation-associated toxin production. Microbiol. Spectr. 2016, 4, 331–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fohler, S.; Klein, G.; Hoedemaker, M.; Scheu, T.; Seyboldt, C.; Campe, A.; Jensen, K.C.; Abdulmawjood, A. Diversity of Clostridium perfringens toxin-genotypes from dairy farms. BMC Microbiol. 2016, 16, 199. [Google Scholar] [CrossRef] [Green Version]
- Kiu, R.; Hall, L.J. An update on the human and animal enteric pathogen Clostridium perfringens. Emerg. Microbes Infect. 2018, 7, 141. [Google Scholar] [CrossRef] [Green Version]
- Rood, J.I.; Adams, V.; Lacey, J.; Lyras, D.; McClane, B.A.; Melville, S.B.; Moore, R.J.; Popoff, M.R.; Sarker, M.R.; Songer, J.G.; et al. Expansion of the Clostridium perfringens toxin-based typing scheme. Anaerobe 2018, 53, 5–10. [Google Scholar] [CrossRef]
- Sakurai, J.; Nagahama, M.; Oda, M. Clostridium perfringens alpha-toxin: Characterization and mode of action. J. Biochem. 2004, 136, 569–574. [Google Scholar] [CrossRef] [Green Version]
- Stiles, B.G.; Wigelsworth, D.J.; Popoff, M.R.; Barth, H. Clostridial binary toxins: Iota and C2 family portraits. Front. Cell. Infect. Microbiol. 2011, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Knapp, O.; Maier, E.; Waltenberger, E.; Mazuet, C.; Benz, R.; Popoff, M.R. Residues involved in the pore-forming activity of the Clostridium perfringens iota toxin. Cell. Microbiol. 2015, 17, 288–302. [Google Scholar] [CrossRef]
- Bruggisser, J.; Tarek, B.; Wyder, M.; Müller, P.; von Ballmoos, C.; Witz, G.; Enzmann, G.; Deutsch, U.; Engelhardt, B.; Posthaus, H. CD31 (PECAM-1) Serves as the Endothelial Cell-Specific Receptor of Clostridium perfringens β-Toxin. Cell Host Microbe 2020, 28, 69–78.e66. [Google Scholar] [CrossRef]
- Savva, C.G.; Fernandes da Costa, S.P.; Bokori-Brown, M.; Naylor, C.E.; Cole, A.R.; Moss, D.S.; Titball, R.W.; Basak, A.K. Molecular architecture and functional analysis of NetB, a pore-forming toxin from Clostridium perfringens. J. Biol. Chem. 2013, 288, 3512–3522. [Google Scholar] [CrossRef] [Green Version]
- Savva, C.G.; Clark, A.R.; Naylor, C.E.; Popoff, M.R.; Moss, D.S.; Basak, A.K.; Titball, R.W.; Bokori-Brown, M. The pore structure of Clostridium perfringens epsilon toxin. Nat. Commun. 2019, 10, 2641. [Google Scholar] [CrossRef]
- Briggs, D.C.; Naylor, C.E.; Smedley, J.G., 3rd; Lukoyanova, N.; Robertson, S.; Moss, D.S.; McClane, B.A.; Basak, A.K. Structure of the food-poisoning Clostridium perfringens enterotoxin reveals similarity to the aerolysin-like pore-forming toxins. J. Mol. Biol. 2011, 413, 138–149. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, K.; Yumine, N.; Mimura, K.; Nagahama, M.; Li, J.; McClane, B.A.; Akimoto, S. Identification of novel Clostridium perfringens type E strains that carry an iota toxin plasmid with a functional enterotoxin gene. PLoS ONE 2011, 6, e20376. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Miyamoto, K.; Sayeed, S.; McClane, B.A. Organization of the cpe locus in CPE-positive Clostridium perfringens type C and D isolates. PLoS ONE 2010, 5, e10932. [Google Scholar] [CrossRef] [Green Version]
- Sarker, M.R.; Carman, R.J.; McClane, B.A. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol. Microbiol. 1999, 33, 946–958. [Google Scholar] [CrossRef]
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States—Major pathogens. Emerg. Infect. Dis. 2011, 17, 7–15. [Google Scholar] [CrossRef]
- Lin, Y.T.; Labbe, R. Enterotoxigenicity and genetic relatedness of Clostridium perfringens isolates from retail foods in the United States. Appl. Environ. Microbiol. 2003, 69, 1642–1646. [Google Scholar] [CrossRef] [Green Version]
- Asha, N.J.; Tompkins, D.; Wilcox, M.H. Comparative analysis of prevalence, risk factors, and molecular epidemiology of antibiotic-associated diarrhea due to Clostridium difficile, Clostridium perfringens, and Staphylococcus aureus. J. Clin. Microbiol. 2006, 44, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Asha, N.J.; Wilcox, M.H. Laboratory diagnosis of Clostridium perfringens antibiotic-associated diarrhoea. J. Med. Microbiol. 2002, 51, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Mpamugo, O.; Donovan, T.; Brett, M.M. Enterotoxigenic Clostridium perfringens as a cause of sporadic cases of diarrhoea. J. Med. Microbiol. 1995, 43, 442–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Hitomi, S.; Sawahata, T. Nosocomial diarrhea caused by Clostridium perfringens in the Tsukuba-Tsuchiura district, Japan. J. Infect. Chemother. 2008, 14, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Skjelkvåle, R.; Uemura, T. Experimental Diarrhoea in human volunteers following oral administration of Clostridium perfringens enterotoxin. J. Appl. Bacteriol. 1977, 43, 281–286. [Google Scholar] [CrossRef]
- DuPont, H.L. Clinical practice. Bacterial diarrhea. N. Engl. J. Med. 2009, 361, 1560–1569. [Google Scholar] [CrossRef]
- Kitadokoro, K.; Nishimura, K.; Kamitani, S.; Fukui-Miyazaki, A.; Toshima, H.; Abe, H.; Kamata, Y.; Sugita-Konishi, Y.; Yamamoto, S.; Karatani, H.; et al. Crystal structure of Clostridium perfringens enterotoxin displays features of b-pore-forming toxins. J. Biol. Chem. 2011, 286, 19549–19555. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.R.; Gibert, M.; Popoff, M.; Moss, D.S.; Titball, R.W.; Basak, A.K. Clostridium perfringens ε-toxin shows structural similarity to the pore-forming toxin aerolysin. Nat. Struct. Mol. Biol. 2004, 11, 797–798. [Google Scholar] [CrossRef]
- Robertson, S.L.; Smedley, J.G., 3rd; Singh, U.; Chakrabarti, G.; Van Itallie, C.M.; Anderson, J.M.; McClane, B.A. Compositional and stoichiometric analysis of Clostridium perfringens enterotoxin complexes in Caco-2 cells and claudin 4 fibroblast transfectants. Cell. Microbiol. 2007, 9, 2734–2755. [Google Scholar] [CrossRef]
- Smedley, J.G., 3rd; Uzal, F.A.; McClane, B.A. Identification of a prepore large-complex stage in the mechanism of action of Clostridium perfringens enterotoxin. Infect. Immun. 2007, 75, 2381–2390. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Theoret, J.R.; Shrestha, A.; Smedley, J.G., 3rd; McClane, B.A. Cysteine-scanning mutagenesis supports the importance of Clostridium perfringens enterotoxin amino acids 80 to 106 for membrane insertion and pore formation. Infect. Immun. 2012, 80, 4078–4088. [Google Scholar] [CrossRef] [Green Version]
- McClane, B.A.; McDonel, J.L. Characterization of membrane permeability alterations induced in Vero cells by Clostridium perfringens enterotoxin. Biochim. Biophys. Acta 1980, 600, 974–985. [Google Scholar] [CrossRef]
- Hardy, S.P.; Denmead, M.; Parekh, N.; Granum, P.E. Cationic currents induced by Clostridium perfringens type A enterotoxin in human intestinal CaCO-2 cells. J. Med. Microbiol. 1999, 48, 235–243. [Google Scholar] [CrossRef]
- McClane, B.A.; Wnek, A.P.; Hulkower, K.I.; Hanna, P.C. Divalent cation involvement in the action of Clostridium perfringens type A enterotoxin. Early events in enterotoxin action are divalent cation-independent. J. Biol. Chem. 1988, 263, 2423–2435. [Google Scholar] [CrossRef]
- Matsuda, M.; Ozutsumi, K.; Iwahashi, H.; Sugimoto, N. Primary action of Clostridium perfringens type A enterotoxin on HeLa and Vero cells in the absence of extracellular calcium: Rapid and characteristic changes in membrane permeability. Biochem. Biophys. Res. Commun. 1986, 141, 704–710. [Google Scholar] [CrossRef]
- Krause, G.; Winkler, L.; Mueller, S.L.; Haseloff, R.F.; Piontek, J.; Blasig, I.E. Structure and function of claudins. Biochim. Biophys. Acta 2008, 1778, 631–645. [Google Scholar] [CrossRef] [Green Version]
- Mineta, K.; Yamamoto, Y.; Yamazaki, Y.; Tanaka, H.; Tada, Y.; Saito, K.; Tamura, A.; Igarashi, M.; Endo, T.; Takeuchi, K.; et al. Predicted expansion of the claudin multigene family. FEBS Lett. 2011, 585, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Shinoda, T.; Shinya, N.; Ito, K.; Ohsawa, N.; Terada, T.; Hirata, K.; Kawano, Y.; Yamamoto, M.; Kimura-Someya, T.; Yokoyama, S.; et al. Structural basis for disruption of claudin assembly in tight junctions by an enterotoxin. Sci. Rep. 2016, 6, 33632. [Google Scholar] [CrossRef]
- Veshnyakova, A.; Protze, J.; Rossa, J.; Blasig, I.E.; Krause, G.; Piontek, J. On the interaction of Clostridium perfringens enterotoxin with claudins. Toxins 2010, 2, 1336–1356. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, Y.; Suzuki, H.; Tani, K.; Nishikawa, K.; Irie, K.; Ogura, Y.; Tamura, A.; Tsukita, S.; Fujiyoshi, Y. Tight junctions. Structural insight into tight junction disassembly by Clostridium perfringens enterotoxin. Science 2015, 347, 775–778. [Google Scholar] [CrossRef]
- Mitchell, L.A.; Koval, M. Specificity of interaction between Clostridium perfringens enterotoxin and claudin-family tight junction proteins. Toxins 2010, 2, 1595–1611. [Google Scholar] [CrossRef] [Green Version]
- International Organization for Standardization. EN ISO 7937 Microbiology of Food and Animal Feeding Stuffs—Horizontal Method for the Enumeration of Clostridium Perfringens—Colony-Count Technique; International Organization for Standardization: Geneva, Switzerland, 2004. [Google Scholar]
- Veloo, A.C.; Knoester, M.; Degener, J.E.; Kuijper, E.J. Comparison of two matrix-assisted laser desorption ionisation-time of flight mass spectrometry methods for the identification of clinically relevant anaerobic bacteria. Clin. Microbiol. Infect. 2011, 17, 1501–1506. [Google Scholar] [CrossRef] [Green Version]
- Coltella, L.; Mancinelli, L.; Onori, M.; Lucignano, B.; Menichella, D.; Sorge, R.; Raponi, M.; Mancini, R.; Russo, C. Advancement in the routine identification of anaerobic bacteria by MALDI-TOF mass spectrometry. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 1183–1192. [Google Scholar] [CrossRef]
- Meer, R.R.; Songer, J.G. Multiplex polymerase chain reaction assay for genotyping Clostridium perfringens. Am. J. Vet. Res. 1997, 58, 702–705. [Google Scholar]
- Miyamoto, K.; Wen, Q.; McClane, B.A. Multiplex PCR genotyping assay that distinguishes between isolates of Clostridium perfringens type A carrying a chromosomal enterotoxin gene (cpe) locus, a plasmid cpe locus with an IS1470-like sequence, or a plasmid cpe locus with an IS1151 sequence. J. Clin. Microbiol. 2004, 42, 1552–1558. [Google Scholar] [CrossRef] [Green Version]
- Wen, Q.; Miyamoto, K.; McClane, B.A. Development of a duplex PCR genotyping assay for distinguishing Clostridium perfringens type A isolates carrying chromosomal enterotoxin (cpe) genes from those carrying plasmid-borne enterotoxin (cpe) genes. J. Clin. Microbiol. 2003, 41, 1494–1498. [Google Scholar] [CrossRef] [Green Version]
- Duncan, C.L. Time of enterotoxin formation and release during sporulation of Clostridium perfringens type A. J. Bacteriol. 1973, 113, 932–936. [Google Scholar] [CrossRef] [Green Version]
- McClane, B.A.; Snyder, J.T. Development and preliminary evaluation of a slide latex agglutination assay for detection of Clostridium perfringens type A enterotoxin. J. Immunol. Methods 1987, 100, 131–136. [Google Scholar] [CrossRef]
- McClane, B.A.; Strouse, R.J. Rapid detection of Clostridium perfringens type A enterotoxin by enzyme-linked immunosorbent assay. J. Clin. Microbiol. 1984, 19, 112–115. [Google Scholar] [CrossRef] [Green Version]
- Uemura, T.; Yoshitake, S.; Hu, D.; Kajikawa, T. A highly sensitive enzyme-linked immunosorbent assay for Clostridium perfringens enterotoxin. Lett. Appl. Microbiol. 1992, 15, 23–25. [Google Scholar] [CrossRef]
- Mehta, R.; Narayan, K.G.; Notermans, S. DOT-enzyme linked immunosorbent assay for detection of Clostridium perfringens type A enterotoxin. Int. J. Food Microbiol. 1989, 9, 45–50. [Google Scholar] [CrossRef]
- Pöhlmann, C.; Elßner, T. Multiplex immunoassay techniques for on-site detection of security sensitive toxins. Toxins 2020, 12, 727. [Google Scholar] [CrossRef] [PubMed]
- Smedley, J.G., 3rd; McClane, B.A. Fine mapping of the N-terminal cytotoxicity region of Clostridium perfringens enterotoxin by site-directed mutagenesis. Infect. Immun. 2004, 72, 6914–6923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinoda, T.; Shinya, N.; Ito, K.; Ishizuka-Katsura, Y.; Ohsawa, N.; Terada, T.; Hirata, K.; Kawano, Y.; Yamamoto, M.; Tomita, T.; et al. Cell-free methods to produce structurally intact mammalian membrane proteins. Sci. Rep. 2016, 6, 30442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisemann, J.; Mahrhold, S.; Krüger, M.; Neumann, T.; Krez, N.; Dorner, B.G.; Rummel, A. Functionalization of the protein receptor claudin-4 for detection of Clostridium perfringens enterotoxin. Manuscript in preparation Clostridium erfringens enterotoxin. Manuscript in preparation.
- Elsholz, B.; Wörl, R.; Blohm, L.; Albers, J.; Feucht, H.; Grunwald, T.; Jürgen, B.; Schweder, T.; Hintsche, R. Automated detection and quantitation of bacterial RNA by using electrical microarrays. Anal. Chem. 2006, 78, 4794–4802. [Google Scholar] [CrossRef]
- Pauly, D.; Kirchner, S.; Stoermann, B.; Schreiber, T.; Kaulfuss, S.; Schade, R.; Zbinden, R.; Avondet, M.A.; Dorner, M.B.; Dorner, B.G. Simultaneous quantification of five bacterial and plant toxins from complex matrices using a multiplexed fluorescent magnetic suspension assay. Analyst 2009, 134, 2028–2039. [Google Scholar] [CrossRef] [Green Version]
- Stern, D.; Weisemann, J.; Le Blanc, A.; von Berg, L.; Mahrhold, S.; Piesker, J.; Laue, M.; Luppa, P.B.; Dorner, M.B.; Dorner, B.G.; et al. A lipid-binding loop of botulinum neurotoxin serotypes B, DC and G is an essential feature to confer their exquisite potency. PLoS Pathog. 2018, 14, e1007048. [Google Scholar] [CrossRef] [Green Version]
- Da Silva Felício, M.T.; Hald, T.; Liebana, E.; Allende, A.; Hugas, M.; Nguyen-The, C.; Johannessen, G.S.; Niskanen, T.; Uyttendaele, M.; McLauchlin, J. Risk ranking of pathogens in ready-to-eat unprocessed foods of non-animal origin (FoNAO) in the EU: Initial evaluation using outbreak data (2007–2011). Int. J. Food Microbiol. 2015, 195, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Bos, J.; Smithee, L.; McClane, B.; Distefano, R.F.; Uzal, F.; Songer, J.G.; Mallonee, S.; Crutcher, J.M. Fatal necrotizing colitis following a foodborne outbreak of enterotoxigenic Clostridium perfringens type A infection. Clin. Infect. Dis. 2005, 40, e78–e83. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Katahira, J.; Horiguchi, Y.; Sonoda, N.; Furuse, M.; Tsukita, S. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction integral membrane protein. FEBS Lett. 2000, 476, 258–261. [Google Scholar] [CrossRef] [Green Version]
- Ling, J.; Liao, H.; Clark, R.; Wong, M.S.; Lo, D.D. Structural constraints for the binding of short peptides to claudin-4 revealed by surface plasmon resonance. J. Biol. Chem. 2008, 283, 30585–30595. [Google Scholar] [CrossRef] [Green Version]
- Sonoda, N.; Furuse, M.; Sasaki, H.; Yonemura, S.; Katahira, J.; Horiguchi, Y.; Tsukita, S. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: Evidence for direct involvement of claudins in tight junction barrier. J. Cell Biol. 1999, 147, 195–204. [Google Scholar] [CrossRef]
- Harada, M.; Kondoh, M.; Ebihara, C.; Takahashi, A.; Komiya, E.; Fujii, M.; Mizuguchi, H.; Tsunoda, S.; Horiguchi, Y.; Yagi, K.; et al. Role of tyrosine residues in modulation of claudin-4 by the C-terminal fragment of Clostridium perfringens enterotoxin. Biochem. Pharmacol. 2007, 73, 206–214. [Google Scholar] [CrossRef]
- Forti, K.; Ferroni, L.; Pellegrini, M.; Cruciani, D.; De Giuseppe, A.; Crotti, S.; Papa, P.; Maresca, C.; Severi, G.; Marenzoni, M.L.; et al. Molecular characterization of Clostridium perfringens strains isolated in Italy. Toxins 2020, 12, 650. [Google Scholar] [CrossRef]
- Mahamat Abdelrahim, A.; Radomski, N.; Delannoy, S.; Djellal, S.; Le Négrate, M.; Hadjab, K.; Fach, P.; Hennekinne, J.A.; Mistou, M.Y.; Firmesse, O. Large-scale genomic analyses and toxinotyping of Clostridium perfringens implicated in foodborne outbreaks in France. Front. Microbiol. 2019, 10, 777. [Google Scholar] [CrossRef] [Green Version]
- Legros, N.; Pohlentz, G.; Steil, D.; Müthing, J. Shiga toxin-glycosphingolipid interaction: Status quo of research with focus on primary human brain and kidney endothelial cells. Int. J. Med. Microbiol. 2018, 308, 1073–1084. [Google Scholar] [CrossRef]
- Bartholomew, B.A.; Stringer, M.F.; Watson, G.N.; Gilbert, R.J. Development and application of an enzyme linked immunosorbent assay for Clostridium perfringens type A enterotoxin. J. Clin. Pathol. 1985, 38, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Majumder, S.; Nag, M.; Kingston, J.J. A sandwich duplex immuno PCR for rapid and sensitive identification of Clostridium perfringens alpha and enterotoxin. Anaerobe 2019, 57, 63–74. [Google Scholar] [CrossRef]
- RIDASCREEN®. Clostridium Perfringens Enterotoxin ELISA. Available online: https://clinical.r-biopharm.com/products/ridascreen-clostridium-perfringens-enterotoxin/ (accessed on 19 February 2021).
- Wimsatt, J.C.; Harmon, S.M.; Shah, D.B. Detection of Clostridium perfringens enterotoxin in stool specimens and culture supernatants by enzyme-linked immunosorbent assay. Diagn. Microbiol. Infect. Dis. 1986, 4, 307–313. [Google Scholar] [CrossRef]
- Berry, P.R.; Rodhouse, J.C.; Hughes, S.; Bartholomew, B.A.; Gilbert, R.J. Evaluation of ELISA, RPLA, and Vero cell assays for detecting Clostridium perfringens enterotoxin in faecal specimens. J. Clin. Pathol. 1988, 41, 458–461. [Google Scholar] [CrossRef]
- Minamoto, Y.; Dhanani, N.; Markel, M.E.; Steiner, J.M.; Suchodolski, J.S. Prevalence of Clostridium perfringens, Clostridium perfringens enterotoxin and dysbiosis in fecal samples of dogs with diarrhea. Vet. Microbiol. 2014, 174, 463–473. [Google Scholar] [CrossRef]
- Kyne, L.; Warny, M.; Qamar, A.; Kelly, C.P. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet 2001, 357, 189–193. [Google Scholar] [CrossRef]
- Skjelkvålé, R.; Duncan, C.L. Characterization of enterotoxin purified from Clostridium perfringens type C. Infect. Immun. 1975, 11, 1061–1068. [Google Scholar] [CrossRef] [Green Version]
- Dorner, B.G.; Steinbach, S.; Hüser, M.B.; Kroczek, R.A.; Scheffold, A. Single-cell analysis of the murine chemokines MIP-1a, MIP-1b, RANTES and ATAC/lymphotactin by flow cytometry. J. Immunol. Methods 2003, 274, 83–91. [Google Scholar] [CrossRef]
- Worbs, S.; Skiba, M.; Söderström, M.; Rapinoja, M.-L.; Zeleny, R.; Russmann, H.; Schimmel, H.; Vanninen, P.; Fredriksson, S.-Å.; Dorner, B.G. Characterization of ricin and R. communis agglutinin reference materials. Toxins 2015, 7, 4906–4934. [Google Scholar] [CrossRef] [Green Version]
- Stern, D.; Pauly, D.; Zydek, M.; Müller, C.; Avondet, M.A.; Worbs, S.; Lisdat, F.; Dorner, M.B.; Dorner, B.G. Simultaneous differentiation and quantification of ricin and agglutinin by an antibody-sandwich surface plasmon resonance sensor. Biosens. Bioelectron. 2016, 78, 111–117. [Google Scholar] [CrossRef]
- Myszka, D.G. Improving biosensor analysis. J. Mol. Recognit. 1999, 12, 279–284. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Pauly, D.; Worbs, S.; Kirchner, S.; Shatohina, O.; Dorner, M.B.; Dorner, B.G. Real-time cytotoxicity assay for rapid and sensitive detection of ricin from complex matrices. PLoS ONE 2012, 7, e35360. [Google Scholar] [CrossRef]
- Schulz, K.; Pöhlmann, C.; Dietrich, R.; Märtlbauer, E.; Elßner, T. Electrochemical Biochip Assays Based on Anti-idiotypic Antibodies for Rapid and Automated On-Site Detection of Low Molecular Weight Toxins. Front. Chem. 2019, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Keyburn, A.L.; Boyce, J.D.; Vaz, P.; Bannam, T.L.; Ford, M.E.; Parker, D.; Di Rubbo, A.; Rood, J.I.; Moore, R.J. NetB, a new toxin that is associated with avian necrotic enteritis caused by Clostridium perfringens. PLoS Pathog. 2008, 4, e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messelhäußer, U.; Ziegler, H.; Elmer-Englhard, D.; Busch, U.; Hörmansdorfer, S.; Kahlau, D.; Pudich, U.; Höller, C. Development of real-time PCR assays for detection and characterization of Clostridium perfringens in food samples. Arch. Lebensm. 2006, 57, 102–105. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Epitope a | Reagent | Source | Isotype | Specificity b | Neutralization c | kad (M−1 s−1) | kdd (s−1) | Affinity d KD (M) |
|---|---|---|---|---|---|---|---|---|
| 1 | mAb CPE1 | RKI | IgG1 | RBD | Yes | 1.0 × 106 | 5.4 × 10−5 | 5.3 × 10−11 |
| 1 | mAb CPE9 | CEA | IgG1 | RBD | Yes | 6.7 × 105 | 2.6 × 10−5 | 3.9 × 10−11 |
| 1 | mAb CPE58 | RKI | IgG1 | RBD | Yes | 6.9 × 105 | 1.6 × 10−3 | 2.3 × 10−9 |
| 1 | mAb CPE281 | RKI | IgG1 | RBD | Yes | 7.5 × 105 | 1.0 × 10−3 | 1.4 × 10−9 |
| 2 | mAb CPE639 | RKI | IgG1 | PFD | No | 4.1 × 105 | 8.6 × 10−4 | 2.1 × 10−9 |
| 3 | mAb CPE562 | RKI | IgG1 | PFD | Yes | 3.1 × 105 | 6.8 × 10−5 | 2.2 × 10−10 |
| 3/4 | mAb CPE18 | CEA | IgG1 | PFD | Yes | 4.6 × 105 | 2.0 × 10−4 | 4.4 × 10−10 |
| 4 | mAb CPE384 | RKI | IgG1 | PFD | Yes | 2.7 × 105 | 4.9 × 10−4 | 1.9 × 10−9 |
| 5/4 | mAb CPE1339 | RKI | IgG1 | RBD | Yes | 1.2 × 106 | 1.3 × 10−5 | 1.1 × 10−11 |
| 1 | rec. claudin-4 | MHH | − | RBD | n.t. | 5.6 × 105 | 2.1 × 10−4 | 3.8 × 10−10 |
| Domain Specificity | Capture | Detection | ||||
|---|---|---|---|---|---|---|
| CPE1 | CPE639 | CPE562 | CPE384 | CPE1339 | ||
| RBD | CPE1 | - | 0.20 | 0.04 | 0.26 | 0.08 |
| PFD | CPE639 | 0.11 | - | 0.61 | 0.22 | 0.13 |
| PFD | CPE562 | 0.06 | 3.14 | - | 0.16 | 0.11 |
| PFD | CPE384 | 0.15 | 0.28 | 0.11 | - | 1.17 |
| RBD | CPE1339 | 0.10 | 0.32 | 0.08 | 3.47 | - |
| RBD | Rec. claudin-4 | - | 0.53 | 0.16 | 1.81 | 0.63 |
| Capture | Detection | Platform | EC50 (pg/mL) a,b | LOD (pg/mL) c,d |
|---|---|---|---|---|
| mAb CPE1 | mAb CPE562 | ELISA | 13.7 ± 5.9 a | 0.28 ± 0.16 c |
| pBDi | 3160 b | 316 d | ||
| rec. claudin-4 | mAb CPE562 | ELISA | 47.2 ± 5.1 a | 1.03 ± 0.30 c |
| pBDi | 10000 b | 1000 d |
| Strain | Toxinotype a | Reference | PCR cpe | Sandwich ELISA b | |
|---|---|---|---|---|---|
| mAb/mAb | Claudin-4/mAb | ||||
| 61a | F | Miprolab c | + | +++ | +++ |
| 11-13136 | D | LGL d | − | − | − |
| 21638/07-L574 | D | LGL d | − | − | − |
| HF 2109 | A | LGL d | − | − | − |
| HF 2110-2 | A | LGL d | − | − | − |
| 11-44702 | D | LGL d | − | − | − |
| 10-70639 | D | LGL d | − | − | − |
| 3570/08 | A | LGL d | − | − | − |
| SO2253/1 | D | LGL d | − | − | − |
| 11-30379-03 | D | LGL d | − | − | − |
| KV3 29.05 | D | LGL d | − | − | − |
| 12-45390 | D | LGL d | − | − | − |
| 12-45681 | D | LGL d | + | +++ | +++ |
| SO 21002 | D | LGL d | − | − | − |
| 11.-2294 | E | LGL d | − | − | − |
| BA 204 | D | LGL d | − | − | − |
| PP 42138-15 | F | LGL d | + | +++ | +++ |
| 12-102988 | D | LGL d | − | − | − |
| SO 221 | A | LGL d | − | − | − |
| 15-0273682 | D | LGL d | + | +++ | +++ |
| 11-4999/1 | D | LGL d | − | − | − |
| 11-18210 | D | LGL d | − | − | − |
| S 726 | D | LGL d | − | − | − |
| E 728 | F | LGL d | + | +++ | +++ |
| L 443/05 | F | LGL d | + | +++ | +++ |
| 21439/07 - G 1144 | F | LGL d | + | +++ | +++ |
| 6466/08 | F | LGL d | + | +++ | +++ |
| 10-0058087-1 | F | LGL d | + | ++ | ++ |
| 6682/1 | F | LGL d | + | +++ | +++ |
| 10-0029262-001-01 L93 | A | LGL d | − | − | − |
| 10-70711/4 | F | LGL d | + | +++ | +++ |
| 11-162672 | D | LGL d | − | − | − |
| 12-45390 | D | LGL d | − | − | − |
| 12-105747 L362 | F | LGL d | + | +++ | ++ |
| P V4 8.7. | E | LGL d | − | − | − |
| A202 | E | LGL d | − | − | − |
| 14-130465 | F | LGL d | + | +++ | +++ |
| 17-52183-001 | F | LGL d | + | +++ | +++ |
| 17-52386-001 | F | LGL d | + | +++ | +++ |
| 12-73336_G737/1 | F | LGL d | + | +++ | + |
| E730 | F | LGL d | + | ++ | +++ |
| PS8150/07 | F | LGL d | + | ++ | + |
| 175-8/97 | F | LGL d | + | +++ | +++ |
| MB30 o.H. | F | LGL d | + | ++ | +++ |
| 11 1331 | F | LGL d | + | +++ | +++ |
| E732 | F | LGL d | + | ++ | +++ |
| 12-134928_L457 | F | LGL d | + | + | + |
| PS10950/07 | A | LGL d | − | − | − |
| F436 | A | LGL d | − | − | − |
| VA00249/12 | D | TLV e | + | +++ | +++ |
| VA00807/14 | D | TLV e | + | +++ | +++ |
| VA00084/19 | D | TLV e | + | +++ | +++ |
| 572c | D | Miprolab c | − | − | − |
| NCTC 8239-01 | F | NCTC f | + | +++ | ++ |
| NCTC 8798-01 | F | NCTC f | + | +++ | +++ |
| NCTC8346 | D | NCTC f | − | − | − |
| NCTC13110 | B | NCTC f | − | − | − |
| NCTC3110 | B | NCTC f | − | − | − |
| NCTC6121 | B | NCTC f | − | − | − |
| NCTC8084 | E | NCTC f | − | − | − |
| NCTC8238 | F | NCTC f | + | ++ | + |
| NCTC8504 | D | NCTC f | − | − | − |
| NCTC8533 | B | NCTC f | − | − | − |
| NCTC9851 | F | NCTC f | + | +++ | +++ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neumann, T.; Krüger, M.; Weisemann, J.; Mahrhold, S.; Stern, D.; Dorner, M.B.; Feraudet-Tarisse, C.; Pöhlmann, C.; Schulz, K.; Messelhäußer, U.; et al. Innovative and Highly Sensitive Detection of Clostridium perfringens Enterotoxin Based on Receptor Interaction and Monoclonal Antibodies. Toxins 2021, 13, 266. https://doi.org/10.3390/toxins13040266
Neumann T, Krüger M, Weisemann J, Mahrhold S, Stern D, Dorner MB, Feraudet-Tarisse C, Pöhlmann C, Schulz K, Messelhäußer U, et al. Innovative and Highly Sensitive Detection of Clostridium perfringens Enterotoxin Based on Receptor Interaction and Monoclonal Antibodies. Toxins. 2021; 13(4):266. https://doi.org/10.3390/toxins13040266
Chicago/Turabian StyleNeumann, Thea, Maren Krüger, Jasmin Weisemann, Stefan Mahrhold, Daniel Stern, Martin B. Dorner, Cécile Feraudet-Tarisse, Christopher Pöhlmann, Katharina Schulz, Ute Messelhäußer, and et al. 2021. "Innovative and Highly Sensitive Detection of Clostridium perfringens Enterotoxin Based on Receptor Interaction and Monoclonal Antibodies" Toxins 13, no. 4: 266. https://doi.org/10.3390/toxins13040266
APA StyleNeumann, T., Krüger, M., Weisemann, J., Mahrhold, S., Stern, D., Dorner, M. B., Feraudet-Tarisse, C., Pöhlmann, C., Schulz, K., Messelhäußer, U., Rimek, D., Gessler, F., Elßner, T., Simon, S., Rummel, A., & Dorner, B. G. (2021). Innovative and Highly Sensitive Detection of Clostridium perfringens Enterotoxin Based on Receptor Interaction and Monoclonal Antibodies. Toxins, 13(4), 266. https://doi.org/10.3390/toxins13040266