Volumetric Absorptive Microsampling as an Alternative Tool for Biomonitoring of Multi-Mycotoxin Exposure in Resource-Limited Areas




Abstract
:1. Introduction
2. Results
2.1. Sample Preparation
2.2. Optimization of the LC-MS/MS Conditions
2.3. Method Validation
2.4. Specificity and Calibration Curve
2.5. Limit of Detection and Limit of Quantification
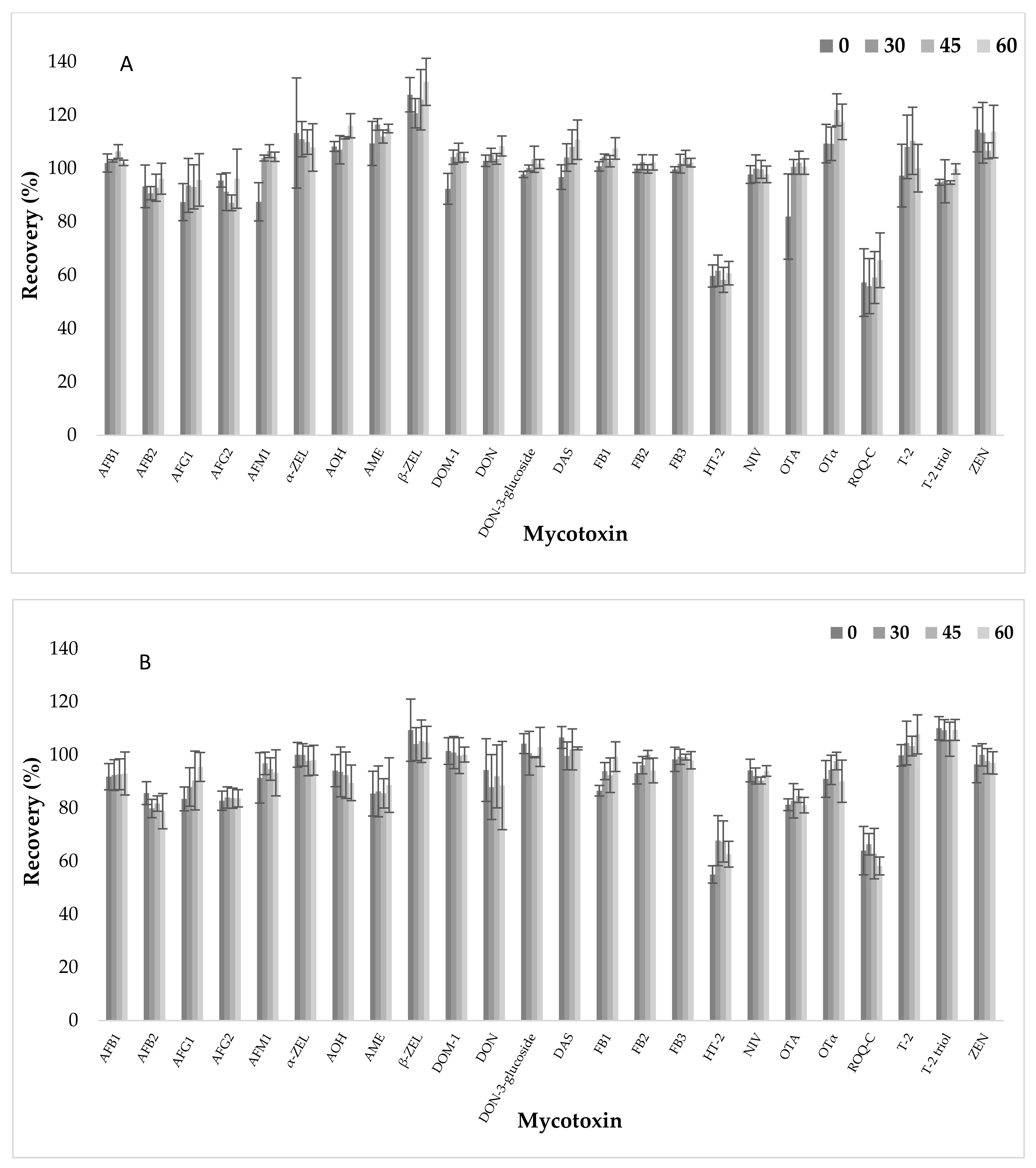
2.6. Apparent Recovery, Intraday Precision (Repeatability), Interday Precision (Reproducibility) and Measurement Uncertainty
2.7. Matrix Effect
2.8. Stability
2.9. Hematocrit Assessment
2.10. Application to Real Samples
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Sample Collection, Sample Preparation and Extraction Procedure
4.3. UPLC-MS/MS Analysis
4.4. Method Validation
4.5. Specificity
4.6. Calibration Curves
4.7. Limit of Detection and Lower Limit of Quantification
4.8. Apparent Recovery
4.9. Intraday Precision (Repeatability (RSDr)) and Interday Precision Intra-Laboratory (Reproducibility (RSDR))
4.10. Measurement Uncertainty (U)
4.11. Matrix Effect
4.12. Stability Trial
4.13. Hematocrit Level
4.14. Clinical Study
4.15. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Marin, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef] [PubMed]
- De Boevre, M.; Di Mavungu, J.D.; Landschoot, S.; Audenaert, K.; Eeckhout, M.; Maene, P.; Haesaert, G.; De Saeger, S. Natural occurrence of mycotoxins and their masked forms in food and feed products. World Mycotoxin J. 2012, 5, 207–219. [Google Scholar] [CrossRef]
- Pitt, J.I.; Basilico, J.C.; Abarca, M.L.; Lopez, C. Mycotoxins and toxigenic fungi. Med. Mycol. 2000, 38, 41–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, H.S.; Brasel, J.M. Toxicity, metabolism, and impact of mycotoxins on humans and animals. Toxicology 2001, 167, 101–134. [Google Scholar] [CrossRef]
- Bennet, J.; Klich, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 26, 497–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, A.; Mengelers, M.; Yang, S.; De Saeger, S.; De Boevre, M. Mycotoxin biomarkers of exposure: A comprehensive review. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1127–1155. [Google Scholar] [CrossRef] [Green Version]
- Stove, C.P.; Ingels, A.S.M.E.; De Kesel, P.M.M.; Lambert, W.E. Dried blood spots in toxicology: From the cradle to the grave? Crit. Rev. Toxicol. 2012, 42, 230–243. [Google Scholar] [CrossRef] [Green Version]
- Delahaye, L.; Janssens, B.; Stove, C. Alternative sampling strategies for the assessment of biomarkers of exposure. Curr. Opin. Toxicol. 2017, 4, 43–51. [Google Scholar] [CrossRef]
- Ssepuuya, G.; Van Poucke, C.; Ediage, E.N.; Mulholland, C.; Tritscher, A.; Verger, P.; Kenny, M.; Bessy, C.; De Saeger, S. Mycotoxin contamination of sorghum and its contribution to human dietary exposure in four sub-Saharan countries. Food Addit. Contam. Part A 2018, 1–10. [Google Scholar] [CrossRef]
- Abdallah, M.F.; De Boevre, M.; Audenaert, K.; Haesaert, G.; De Saeger, S. Highlight report: Mycotoxins as food contaminants in Africa—challenges and perspectives. Arch. Toxicol. 2018, 92, 2151–2152. [Google Scholar] [CrossRef] [Green Version]
- Osteresch, B.; Viegas, S.; Cramer, B.; Humpf, H.U. Multi-mycotoxin analysis using dried blood spots and dried serum spots. Anal. Bioanal. Chem. 2017, 409, 3369–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kip, A.E.; Kiers, K.C.; Rosing, H.; Schellens, J.H.M.; Beijnen, J.H.; Dorlo, T.P.C. Volumetric absorptive microsampling (VAMS) as an alternative to conventional dried blood spots in the quantification of miltefosine in dried blood samples. J. Pharm. Biomed. Anal. 2017, 135, 160–166. [Google Scholar] [CrossRef]
- De Kesel, P.M.M.; Lambert, W.E.; Stove, C.P. Does volumetric absorptive microsampling eliminate the hematocrit bias for caffeine and paraxanthine in dried blood samples? A comparative study. Anal. Chim. Acta 2015, 881, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Denniff, P.; Spooner, N. Volumetric absorptive microsampling: A dried sample collection technique for quantitative bioanalysis. Anal. Chem. 2014, 86, 8489–8495. [Google Scholar] [CrossRef] [PubMed]
- Verougstraete, N.; Lapauw, B.; Van Aken, S.; Delanghe, J.; Stove, C.; Stove, V. Volumetric absorptive microsampling at home as an alternative tool for the monitoring of HbA1c in diabetes patients. Clin. Chem. Lab. Med. 2017, 55, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Kok, M.G.M.; Fillet, M. Volumetric absorptive microsampling: Current advances and applications. J. Pharm. Biomed. Anal. 2018, 147, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Verougstraete, N.; Stove, V.; Stove, C. Wet absorptive microsampling at home for HbA1c monitoring in diabetic children. Clin. Chem. Lab. Med. 2018. [Google Scholar] [CrossRef]
- Andersen, I.K.L.; Rosting, C.; Gjelstad, A.; Halvorsen, T.G. Volumetric absorptive MicroSampling vs. other blood sampling materials in LC–MS-based protein analysis—Preliminary investigations. J. Pharm. Biomed. Anal. 2018, 156, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Kita, K.; Mano, Y. Application of volumetric absorptive microsampling device for quantification of tacrolimus in human blood as a model drug of high blood cell partition. J. Pharm. Biomed. Anal. 2017, 143, 168–175. [Google Scholar] [CrossRef]
- Koponen, J.; Rudge, J.; Kushon, S.; Kiviranta, H. Novel volumetric adsorptive microsampling technique for determination of perfluorinated compounds in blood. Anal. Biochem. 2018, 545, 49–53. [Google Scholar] [CrossRef] [PubMed]
- 2002/657/EC. Commission Decision 2002/657/EC Implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Communities 2002, L221, 8–36.
- US Department of Health and Human Services. Bioanalytical Method Validation. Guidance for Industry. 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 20 April 2021).
- Degen, G.H.; Ali, N.; Gundert-Remy, U. Preliminary data on citrinin kinetics in humans and their use to estimate citrinin exposure based on biomarkers. Toxicol. Lett. 2018, 282, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.; Claeys, L.; Mengelers, M.; Vanhoorne, V.; Vervaet, C.; Huybrechts, B.; De Saeger, S.; De Boevre, M. Humans significantly metabolize and excrete the mycotoxin deoxynivalenol and its modified form deoxynivalenol-3-glucoside within 24 hours. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ruyck, K.; Huybrechts, I.; Yang, S.; Arcella, D.; Claeys, L.; Abbeddou, S.; De Keyzer, W.; De Vries, J.; Ocke, M.C.; Ruprich, J.; et al. Mycotoxin Exposure Assessments in a Multi-Center European Validation Study by 24-Hour Dietary Recall and Biological Fluid Sampling. Environ. Int. 2020, 137, 105539. [Google Scholar] [CrossRef] [PubMed]
- Groopman, J.D.; Dematos, P.; Egner, P.A.; Lovehunt, A.; Kensler, T.W. Molecular Dosimetry Of Urinary Aflatoxin-N7-Guanine And Serum Aflatoxin Albumin Adducts Predicts Chemoprotection By 1,2-Dithiole-3-Thione In Rats. Carcinogenesis 1992, 13, 101–106. [Google Scholar] [CrossRef]
- Fraeyman, S.; Croubels, S.; Devreese, M.; Antonissen, G. Emerging fusarium and alternaria mycotoxins: Occurrence, toxicity and toxicokinetics. Toxins 2017, 9, 228. [Google Scholar] [CrossRef] [Green Version]
- Velghe, S.; Stove, C.P. Volumetric absorptive microsampling as an alternative tool for therapeutic drug monitoring of first-generation anti-epileptic drugs. Anal. Bioanal. Chem. 2018, 410, 2331–2341. [Google Scholar] [CrossRef] [Green Version]
- Protti, M.; Mandrioli, R.; Mercolini, L. Tutorial: Volumetric absorptive microsampling (VAMS). Anal. Chim. Acta 2018. [Google Scholar] [CrossRef] [PubMed]
- Denniff, P.; Parry, S.; Dopson, W.; Spooner, N. Quantitative bioanalysis of paracetamol in rats using volumetric absorptive microsampling (VAMS)tVolumetric absorptive microsampling. J. Pharm. Biomed. Anal. 2015, 108, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Tanna, S.; Alalaqi, A.; Bernieh, D.; Lawson, G. Volumetric absorptive microsampling (VAMS) coupled with high-resolution, accurate-mass (HRAM) mass spectrometry as a simplified alternative to dried blood spot (DBS) analysis for therapeutic drug monitoring of cardiovascular drugs. Clin. Mass Spectrom. 2018, 10, 1–8. [Google Scholar] [CrossRef]
- Kim, Y.; Jeon, J.Y.; Han, S.H.; Ha, N.; Jang, K.; Kim, M.G. Quantitative analysis of acetylsalicylic acid in human blood using volumetric absorptive microsampling. Transl. Clin. Pharmacol. 2018, 26, 32–38. [Google Scholar] [CrossRef]
- Barco, S.; Castagnola, E.; Moscatelli, A.; Rudge, J.; Tripodi, G.; Cangemi, G. Volumetric adsorptive microsampling-liquid chromatography tandem mass spectrometry assay for the simultaneous quantification of four antibiotics in human blood: Method development, validation and comparison with dried blood spot. J. Pharm. Biomed. Anal. 2017, 145, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Kita, K.; Noritake, K.; Mano, Y. Application of a Volumetric Absorptive Microsampling Device to a Pharmacokinetic Study of Tacrolimus in Rats: Comparison with Wet Blood and Plasma. Eur. J. Drug Metab. Pharmacokinet. 2018, 1–12. [Google Scholar] [CrossRef]
- Schaarschmidt, S.; Fauhl-Hassek, C. The Fate of Mycotoxins During the Processing of Wheat for Human Consumption. Compr. Rev. Food Sci. Food Saf. 2018. [Google Scholar] [CrossRef] [Green Version]
- Vidal, A.; Sanchis, V.; Ramos, A.J.; Marín, S. Thermal stability and kinetics of degradation of deoxynivalenol, deoxynivalenol conjugates and ochratoxin A during baking of wheat bakery products. Food Chem. 2015, 178, 276–286. [Google Scholar] [CrossRef] [Green Version]
- Herzallah, S.; Alshawabkeh, K.; Al Fataftah, A. Aflatoxin decontamination of artificially contaminated feeds by sunlight, γ-radiation, and microwave heating. J. Appl. Poult. Res. 2008, 17, 515–521. [Google Scholar] [CrossRef]
- Delahaye, L.; Dhont, E.; De Cock, P.; De Paepe, P.; Stove, C.P. Volumetric absorptive microsampling as an alternative sampling strategy for the determination of paracetamol in blood and cerebrospinal fluid. Anal. Bioanal. Chem. 2019, 411, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.; Marín, S.; Sanchis, V.; De Saeger, S.; De Boevre, M. Hydrolysers of modified mycotoxins in maize: α-Amylase and cellulase induce an underestimation of the total aflatoxin content. Food Chem. 2018, 248, 86–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyo-Manzanares, N.; de Ruyck, K.; Uka, V.; Gámiz-Gracia, L.; García-Campaña, A.M.; de Saeger, S.; Diana Di Mavungu, J. In-house validation of a rapid and efficient procedure for simultaneous determination of ergot alkaloids and other mycotoxins in wheat and maize. Anal. Bioanal. Chem. 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Veprikova, Z.; Vaclavikova, M.; Lacina, O.; Dzuman, Z.; Zachariasova, M.; Hajslova, J. Occurrence of mono- and di-glycosylated conjugates of T-2 and HT-2 toxins in naturally contaminated cereals. World Mycotoxin J. 2012, 5, 231–240. [Google Scholar] [CrossRef]
- Heyndrickx, E.; Sioen, I.; Huybrechts, B.; Callebaut, A.; De Henauw, S.; De Saeger, S. Human biomonitoring of multiple mycotoxins in the Belgian population: Results of the BIOMYCO study. Environ. Int. 2015, 84, 82–89. [Google Scholar] [CrossRef]
- De Boevre, M.; Landschoot, S.; Audenaert, K.; Maene, P.; Di Mavungu, D.; Eeckhout, M.; Haesaert, G.; De Saeger, S. Occurrence and within field variability of fusarium mycotoxins and their masked forms in maize crops in Belgium. World Mycotoxin J. 2014, 7, 91–102. [Google Scholar] [CrossRef]
- Slobodchikova, I.; Vuckovic, D. Liquid chromatography—High resolution mass spectrometry method for monitoring of 17 mycotoxins in human plasma for exposure studies. J. Chromatogr. A 2018, 1548, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueberschär, K.H.; Brezina, U.; Dänicke, S. Zearalenone (ZEN) and ZEN metabolites in feed, urine and bile of sows: Analysis, determination of the metabolic profile and evaluation of the binding forms. Landbauforsch. Volkenrode 2016, 66, 21–28. [Google Scholar] [CrossRef]
- Drzymala, S.S.; Binder, J.; Brodehl, A.; Penkert, M.; Rosowski, M.; Garbe, L.A.; Koch, M. Estrogenicity of novel phase I and phase II metabolites of zearalenone and cis-zearalenone. Toxicon 2015, 105, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, J.M.; Real, M.; Jimenez-Diaz, I.; Belhassen, H.; Hedhili, A.; Torné, P.; Fernández, M.F.; Olea, N. Assessment of estrogenic and anti-androgenic activities of the mycotoxin zearalenone and its metabolites using in vitro receptor-specific bioassays. Food Chem. Toxicol. 2014, 74, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Spanjer, M.C.; Rensen, P.M.; Scholten, J.M. LC-MS/MS multi-method for mycotoxins after single extraction, with validation data for peanut, pistachio, wheat, maize, cornflakes, raisins and figs. Food Addit. Contam. 2008, 25, 472–489. [Google Scholar] [CrossRef] [PubMed]
- Young, J.C. Formation of Sodium Bisulfite Addition Products with Trichothecenones and Alkaline Hydrolysis of Deoxynivalenol and Its Sulfonate. J. Agric. Food Chem. 1986, 34, 919–923. [Google Scholar] [CrossRef]
- Broekaert, N.; Devreese, M.; De Mil, T.; Fraeyman, S.; Antonissen, G.; De Baere, S.; De Backer, P.; Vermeulen, A.; Croubels, S. Oral Bioavailability, Hydrolysis, and Comparative Toxicokinetics of 3-Acetyldeoxynivalenol and 15-Acetyldeoxynivalenol in Broiler Chickens and Pigs. J. Agric. Food Chem. 2015, 63, 8734–8742. [Google Scholar] [CrossRef]
- Sulyok, M.; Krska, R.; Schuhmacher, R. Application of a liquid chromatography-tandem mass spectrometric method to multi-mycotoxin determination in raw cereals and evaluation of matrix effects. Food Addit. Contam. 2007, 24, 1184–1195. [Google Scholar] [CrossRef]
- Lauwers, M.; Croubels, S.; De Baere, S.; Sevastiyanova, M.; Sierra, E.M.R.; Letor, B.; Gougoulias, C.; Devreese, M. Assessment of dried blood spots for multi-mycotoxin biomarker analysis in pigs and broiler chickens. Toxins 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, S.B.; Schwartz-Zimmermann, H.E.; Varga, E.; Bichl, G.; Michlmayr, H.; Adam, G.; Berthiller, F. Metabolism of zearalenone and its major modified forms in pigs. Toxins 2017, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillan, A.; Renaud, J.B.; Burgess, K.M.N.; Orimadegun, A.E.; Akinyinka, O.O.; Allen, S.J.; Miller, J.D.; Reid, G.; Sumarah, M.W. Aflatoxin exposure in Nigerian children with severe acute malnutrition. Food Chem. Toxicol. 2018, 111, 356–362. [Google Scholar] [CrossRef]
- Capiau, S.; Veenhof, H.; Koster, R.A.; Bergqvist, Y.; Boettcher, M.; Halmingh, O.; Keevil, B.G.; Koch, B.C.P.; Linden, R.; Pistos, C.; et al. Official International Association for Therapeutic Drug Monitoring and Clinical Toxicology Guideline: Development and Validation of Dried Blood Spot-Based Methods for Therapeutic Drug Monitoring. Ther. Drug Monit. 2019, 41, 409–430. [Google Scholar] [CrossRef]
- De Kesel, P.M.M.; Sadones, N.; Capiau, S.; Lambert, W.E.; Stove, C.P. Hemato-critical issues in quantitative analysis of dried blood spots: Challenges and solutions. Bioanalysis 2013, 5, 2023–2041. [Google Scholar] [CrossRef] [PubMed]
- Velghe, S.; Delahaye, L.; Stove, C.P. Is the hematocrit still an issue in quantitative dried blood spot analysis? J. Pharm. Biomed. Anal. 2019, 163, 188–196. [Google Scholar] [CrossRef]
- Capiau, S.; Bolea-Fernandez, E.; Balcaen, L.; Van Der Straeten, C.; Verstraete, A.G.; Vanhaecke, F.; Stove, C.P. Development, validation and application of an inductively coupled plasma—Mass spectrometry method to determine cobalt in metal-on-metal prosthesis patients using volumetric absorptive microsampling. Talanta 2019. [Google Scholar] [CrossRef]
- Lauwers, M.; De Baere, S.; Letor, B.; Rychlik, M.; Croubels, S.; Devreese, M. Multi LC-MS/MS and LC-HRMS methods for determination of 24 mycotoxins including major phase I and II biomarker metabolites in biological matrices from pigs and broiler chickens. Toxins 2019, 11, 171. [Google Scholar] [CrossRef] [Green Version]
- Osteresch, B.; Cramer, B.; Humpf, H.U. Analysis of ochratoxin A in dried blood spots—Correlation between venous and finger-prick blood, the influence of hematocrit and spotted volume. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1020, 158–164. [Google Scholar] [CrossRef]
- Cramer, B.; Osteresch, B.; Muñoz, K.A.; Hillmann, H.; Sibrowski, W.; Humpf, H.U. Biomonitoring using dried blood spots: Detection of ochratoxin A and its degradation product 2’R-ochratoxin A in blood from coffee drinkers*. Mol. Nutr. Food Res. 2015, 59, 1837–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, A.; Cano-Sancho, G.; Marin, S.; Ramos, A.J.; Sanchis, V. Multidetection of urinary ochratoxin A, deoxynivalenol and its metabolites: Pilot time-course study and risk assessment in Catalonia, Spain. World Mycotoxin J. 2016, 9, 597–612. [Google Scholar] [CrossRef]
- Studer-Rohr, I.; Schlatter, J.; Dietrich, D.R. Kinetic parameters and intraindividual fluctuations of ochratoxin A plasma levels in humans. Arch. Toxicol. 2000, 74, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Muñoz, K.; Degen, G.H. Ochratoxin A and its metabolites in urines of German adults—An assessment of variables in biomarker analysis. Toxicol. Lett. 2017, 275, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.; Vidal, A.; De Boevre, M.; De Saeger, S.; Nunes, C.; Torres, D.; Goios, A.; Lopes, C.; Assunção, R.; Alvito, P. Exposure assessment of Portuguese population to multiple mycotoxins: The human biomonitoring approach. Int. J. Hyg. Environ. Health 2019, 222, 913–925. [Google Scholar] [CrossRef] [PubMed]
- WHO, IARC. IARC monographs on the evaluation of carcinogenic risks to humans. Some Ind. Chem. 1994, 60, 389–433. [Google Scholar]


{kind=link}
| Mycotoxin | Precursor Ion (m/z) | Product Ions (m/z) Quantifier/Qualifier | CE (eV) | CV (v) | Retention Time (min) |
|---|---|---|---|---|---|
| AFB1 | 313.0 | 241.1/285.0 | 32/20 | 65 | 6.93 |
| AFB2 | 315.0 | 259.0/287.0 | 28/24 | 25 | 6.65 |
| AFG1 | 329.0 | 243.0/311.0 | 24/20 | 50 | 6.28 |
| AFG2 | 331.0 | 285.0/313.0 | 28/24 | 40 | 5.97 |
| AFM1 | 329.1 | 259.1/273.1 | 25/22 | 30 | 6.02 |
| α-ZEL | 321.1 | 175.1/177.0 | 22/17 | 30 | 9.29 |
| AOH | 258.9 | 185.1/213.1 | 30/26 | 40 | 8.12 |
| AME | 272.9 | 199.3/258.2 | 30/29 | 57 | 10.25 |
| β-ZEL | 321.5 | 177.3/189.1 | 15/20 | 30 | 8.50 |
| DOM-1 | 281.1 | 215.1/233.1 | 9/9 | 40 | 4.43 |
| DON | 297.0 | 231.0/249.0 | 9/9 | 40 | 3.21 |
| DON-3-glucoside | 476.1 | 249.0/297.0 | 16/12 | 15 | 3.08 |
| DAS | 384.1 | 247.1/307.1 | 12/9 | 35 | 7.11 |
| FB1 | 722.1 | 334.2/352.1 | 36/32 | 40 | 8.28 |
| FB2 | 706.1 | 336.2/354.2 | 36/30 | 70 | 10.59 |
| FB3 | 706.2 | 354.3/530.2 | 30/28 | 70 | 9.67 |
| HT-2 | 447.0 | 285.0/345.0 | 20/18 | 40 | 7.98 |
| NIV | 313.0 | 175.0/177.0 | 21/16 | 30 | 2.55 |
| OTA | 403.9 | 358.0/238.9 | 12/20 | 40 | 9.33 |
| OTα | 257.0 | 221.1/239.1 | 20/10 | 30 | 5.83 |
| ROQ-C | 390.1 | 193.0/322.0 | 24/24 | 25 | 8.35 |
| T-2 | 484.3 | 215.2/305.2 | 18/12 | 40 | 8.82 |
| T-2 triol | 405.2 | 125.2/303.1 | 14/14 | 30 | 7.17 |
| ZEN | 319.2 | 283.1/301.1 | 12/10 | 40 | 9.64 |
| 13C17 –AFB1 | 330.0 | 285.0/301.0 | 26/22 | 40 | 6.94 |
| 13C15 –DON | 311.9 | 103.4/245.2 | 10/10 | 30 | 3.21 |
| 13C34 –FB1 | 756.4 | 356.2/374.2 | 40/36 | 40 | 8.28 |
| 13C18 –ZEN | 337.3 | 199.1/214.9 | 22/22 | 40 | 9.65 |
| Mycotoxin | LOD (ng/mL) | LLOQ (ng/mL) | Absolute Matrix Effect (%) | Concentration (ng/mL) | Rapp (%) | U (%) | |
|---|---|---|---|---|---|---|---|
| Analyte | IS Compensated | ||||||
| AFB1 | 0.04 | 0.07 | 15.3 | 102 | 0.50 | 104 | 24.0 |
| 1.25 | 96.1 | 22.1 | |||||
| 2.50 | 84.6 | 32.4 | |||||
| 5.00 | 91.3 | 35.6 | |||||
| 12.5 | 88.9 | 39.9 | |||||
| AFB2 | 0.13 | 0.26 | 15.1 | 97.9 | 0.50 | 76.9 | 41.9 |
| 1.25 | 88.4 | 22.8 | |||||
| 2.50 | 90.0 | 34.2 | |||||
| 5.00 | 95.8 | 28.4 | |||||
| 12.5 | 76.3 | 37.8 | |||||
| AFG1 | 0.12 | 0.24 | 13.8 | 101 | 0.50 | 70.1 | 12.1 |
| 1.25 | 89.8 | 18.2 | |||||
| 2.50 | 86.3 | 27.9 | |||||
| 5.00 | 92.1 | 19.4 | |||||
| 12.5 | 82.8 | 24.6 | |||||
| AFG2 | 0.15 | 0.30 | 12.9 | 92.1 | 0.50 | 81.0 | 26.3 |
| 1.25 | 95.1 | 25.6 | |||||
| 2.50 | 87.2 | 19.5 | |||||
| 5.00 | 94.3 | 12.3 | |||||
| 12.5 | 79.9 | 17.6 | |||||
| AFM1 | 0.13 | 0.26 | 15.2 | 100 | 0.50 | 113 | 13.9 |
| 1.25 | 110 | 19.8 | |||||
| 2.50 | 100 | 25.3 | |||||
| 5.00 | 109 | 17.9 | |||||
| 12.5 | 92.5 | 19.3 | |||||
| α-ZEL | 2.64 | 5.30 | 15.5 | 107 | 10.0 | 148 | 35.3 |
| 25.0 | 110 | 24.1 | |||||
| 50.0 | 114 | 13.3 | |||||
| 100 | 111 | 12.7 | |||||
| 250 | 106 | 14.1 | |||||
| AOH | 1.37 | 2.74 | 13.2 | 105 | 10.0 | 136 | 49.1 |
| 25.0 | 124 | 43.1 | |||||
| 50.0 | 138 | 42.8 | |||||
| 100 | 127 | 48.2 | |||||
| 250 | 92.8 | 30.1 | |||||
| AME | 1.86 | 3.72 | 10.4 | 83.1 | 10.0 | 124 | 57.2 |
| 25.0 | 124 | 42.1 | |||||
| 50.0 | 134 | 40.7 | |||||
| 100 | 118 | 42.1 | |||||
| 250 | 89.3 | 32.7 | |||||
| β-ZEL | 6.76 | 13.52 | 14.0 | 106 | 10.0 | 181 | 79.2 |
| 25.0 | 122 | 73.9 | |||||
| 50.0 | 149 | 40.4 | |||||
| 100 | 151 | 45.7 | |||||
| 250 | 115 | 33.5 | |||||
| DOM-1 | 0.57 | 1.14 | 42.5 | 94.5 | 2.50 | 100 | 14.9 |
| 6.25 | 101 | 12.9 | |||||
| 12.5 | 110 | 16.5 | |||||
| 25.0 | 113 | 14.6 | |||||
| 62.5 | 101 | 15.2 | |||||
| DON | 0.39 | 0.78 | 47.9 | 96.2 | 2.50 | 113 | 15.9 |
| 6.25 | 93.4 | 15.6 | |||||
| 12.5 | 106 | 18.0 | |||||
| 25.0 | 105 | 14.1 | |||||
| 62.5 | 98.9 | 18.5 | |||||
| DON-3-glucoside | 0.85 | 1.70 | 50.2 | 97.4 | 2.50 | 126 | 54.6 |
| 6.25 | 118 | 18.5 | |||||
| 12.5 | 113 | 19.9 | |||||
| 25.0 | 115 | 18.0 | |||||
| 62.5 | 102 | 15.6 | |||||
| DAS | 0.85 | 1.71 | 16.6 | 109 | 2.50 | 109 | 29.3 |
| 6.25 | 103 | 21.3 | |||||
| 12.5 | 104 | 20.6 | |||||
| 25.0 | 113 | 18.0 | |||||
| 62.5 | 101 | 17.7 | |||||
| FB1 | 1.54 | 3.09 | 10.0 | 103 | 10.0 | 111 | 19.8 |
| 25.0 | 94.3 | 16.9 | |||||
| 50.0 | 87.5 | 12.5 | |||||
| 100 | 89.6 | 13.5 | |||||
| 250 | 88.7 | 13.1 | |||||
| FB2 | 0.97 | 1.94 | 78.1 | 106 | 10.0 | 91.6 | 37.3 |
| 25.0 | 90.7 | 31.0 | |||||
| 50.0 | 116 | 36.1 | |||||
| 100 | 95.8 | 17.9 | |||||
| 250 | 93.1 | 19.5 | |||||
| FB3 | 1.06 | 2.12 | 8.61 | 110 | 10.0 | 86.6 | 33.2 |
| 25.0 | 94.6 | 29.9 | |||||
| 50.0 | 118 | 30.1 | |||||
| 100 | 106 | 9.78 | |||||
| 250 | 100 | 18.1 | |||||
| HT-2 | 0.74 | 1.48 | 38.4 | 89.2 | 2.50 | 44.7 | 55.9 |
| 6.25 | 41.3 | 27.4 | |||||
| 12.5 | 49.2 | 21.7 | |||||
| 25.0 | 50.6 | 19.6 | |||||
| 62.5 | 42.5 | 19.7 | |||||
| NIV | 0.68 | 1.36 | 124 | 98.7 | 10.0 | 86.1 | 38.6 |
| 25.0 | 90.0 | 17.0 | |||||
| 50.0 | 99.2 | 12.9 | |||||
| 100 | 106 | 14.6 | |||||
| 250 | 93.6 | 12.3 | |||||
| OTA | 0.18 | 0.36 | 27.5 | 107 | 0.50 | 90.9 | 24.5 |
| 1.25 | 85.9 | 15.9 | |||||
| 2.50 | 91.3 | 13.9 | |||||
| 5.00 | 89.9 | 18.4 | |||||
| 12.5 | 84.5 | 6.97 | |||||
| OTα | 0.14 | 0.28 | 19.4 | 87.7 | 0.50 | 124 | 30.9 |
| 1.25 | 135 | 45.6 | |||||
| 2.50 | 86.9 | 35.3 | |||||
| 5.00 | 95.9 | 31.6 | |||||
| 12.5 | 79.5 | 28.7 | |||||
| ROQ-C | 1.57 | 3.14 | 9.63 | 76.7 | 10.0 | 46.1 | 21.8 |
| 25.0 | 59.6 | 19.7 | |||||
| 50.0 | 61.1 | 28.9 | |||||
| 100 | 58.8 | 24.1 | |||||
| 250 | 55.7 | 26.0 | |||||
| T-2 | 0.58 | 1.16 | 65.3 | 109 | 2.50 | 111 | 31.0 |
| 6.25 | 107 | 13.7 | |||||
| 12.5 | 114 | 11.7 | |||||
| 25.0 | 109 | 9.13 | |||||
| 62.5 | 98.6 | 21.9 | |||||
| T-2 triol | 1.26 | 2.52 | 47.2 | 92.1 | 10.0 | 80.4 | 46.4 |
| 25.0 | 97.0 | 13.8 | |||||
| 50.0 | 122 | 15.2 | |||||
| 100 | 129 | 11.1 | |||||
| 250 | 116 | 14.9 | |||||
| ZEN | 2.15 | 4.28 | 15.9 | 105 | 10.0 | 118 | 10.6 |
| 25.0 | 98.4 | 19.3 | |||||
| 50.0 | 108 | 7.70 | |||||
| 100 | 106 | 15.2 | |||||
| 250 | 104 | 14.9 | |||||
| Mycotoxin | Spiked Concentration (ng/mL) | 7 Days | 21 Days | ||
|---|---|---|---|---|---|
| Room Temperature | 4 °C | Room Temperature | 4 °C | ||
| AFB1 | 0.5 | 129 ± 20 | 101 ± 34 | 116 ± 2 | 120 ± 15 |
| 2.5 | 93.7 ± 8.7 | 98.8 ± 11.9 | 119 ± 5 | 121 ± 9 | |
| 12.5 | 99.5 ± 4.2 | 88.3 ± 10.5 | 86.3 ± 6.2 | 91.8 ± 4.9 | |
| AFB2 | 0.5 | 99.8 ± 20.2 | 138 ± 34 | 94.6 ± 15.6 | 118 ± 10 |
| 2.5 | 105 ± 1 | 98.7 ± 9.9 | 106 ± 7 | 119 ± 16 | |
| 12.5 | 93.5 ± 10.7 | 101 ± 2 | 101 ± 7 | 91.0 ± 10.9 | |
| AFG1 | 0.5 | 32 ± 29 * | 51.6 ± 15.3 * | 107 ± 37 | 100 ± 1 |
| 2.5 | 86.4 ± 1.4 | 91.8 ± 5.4 | 39 ± 25 * | 112 ± 19 | |
| 12.5 | 99.0 ± 2.5 | 101 ± 5 | 72 ± 21 | 92.3 ± 6.2 | |
| AFG2 | 0.5 | 106 ± 7 | 108 ± 7 | 85.9 ± 15.2 | 82.7 ± 11.4 |
| 2.5 | 92.1 ± 2.4 | 87.4 ± 2.0 | 105 ± 5 | 91.8 ± 10.3 | |
| 12.5 | 85.8 ± 2.0 | 88.8 ± 2.9 | 92.3 ± 10.9 | 112 ± 21 | |
| AFM1 | 0.5 | 109 ± 15 | 143 ± 17 | 110 ± 2 | 115 ± 15 |
| 2.5 | 103 ± 15 | 122 ± 2 | 104 ± 1 | 90.4 ± 10.9 | |
| 12.5 | 105 ± 4 | 114 ± 3 | 104 ± 4 | 90.9 ± 11.9 | |
| α-ZEL | 10 | 106 ± 36 | 120 ± 19 | 82.9 ± 17.4 | 101 ± 16 |
| 50 | 81.2 ± 9.9 | 90.5 ± 11.0 | 117 ± 14 | 94.5 ± 1.3 | |
| 250 | 121 ± 16 | 98.3 ± 11.8 | 120 ± 10 | 80.3 ± 2.4 | |
| AOH | 10 | 118 ± 17 | 94.2 ± 1.7 | 92.6 ± 10.3 | 91.0 ± 12.3 |
| 50 | 90.5 ± 2.9 | 83.3 ± 12.8 | 98.1 ± 18.7 | 90.5 ± 11.9 | |
| 250 | 78.7 ± 1.8 | 76.1 ± 3.3 | 96.8 ± 12.3 | 80.9 ± 12.8 | |
| AME | 10 | 89.3 ± 10.5 | 98.5 ± 27.8 | 83.6 ± 12.7 | 85.1 ± 13.4 |
| 50 | 83.6 ± 1.5 | 82.6 ± 2.1 | 82.6 ± 13.5 | 89.5 ± 11.3 | |
| 250 | 76.5 ± 3.8 | 77.8 ± 2.1 | 81.8 ± 4.9 | 98.7 ± 4.2 | |
| β-ZEL | 10 | 138 ± 22 | 108 ± 19 | 93 ± 22 | 121 ± 15 |
| 5 0 | 129 ± 9 | 99.5 ± 11.3 | 145 ± 18 | 110 ± 11 | |
| 250 | 122 ± 10 | 115 ± 12 | 142 ± 20 | 119 ± 24 | |
| DOM-1 | 2.5 | 106 ± 16 | 110 ± 10 | 101 ± 11 | 119 ± 9 |
| 12.5 | 108 ± 1 | 94.0 ± 10.9 | 85.6 ± 20.6 | 89.5 ± 20.1 | |
| 62.5 | 115 ± 11 | 114 ± 12 | 108 ± 4 | 81.3 ± 11.9 | |
| DON | 2.5 | 94.9 ± 13.6 | 120 ± 22 | 98.3 ± 35.7 | 109 ± 19 |
| 12.5 | 88.4 ± 13.3 | 106 ± 2 | 90.2 ± 3.5 | 99.5 ± 11.3 | |
| 62.5 | 99.9 ± 10.9 | 98.7 ± 7.4 | 94.8 ± 4.9 | 98.3 ± 2.4 | |
| DON-3-glucoside | 2.5 | 75.5 ± 15.2 | 87.6 ± 9.1 | 75.6 ± 25.2 | 81.8 ± 9.1 |
| 12.5 | 80.2 ± 10.5 | 91.5 ± 10.1 | 119 ± 30 | 90.9 ± 19.3 | |
| 62.5 | 85.7 ± 14.8 | 100 ± 16 | 128 ± 28 | 91.6 ± 15.1 | |
| DAS | 2.5 | 110 ± 16 | 90.2 ± 1.4 | 81.6 ± 10.7 | 84.1 ± 15.3 |
| 12.5 | 106 ± 12 | 110 ± 9 | 92.2 ± 13.5 | 90.6 ± 19.1 | |
| 62.5 | 109 ± 94 | 115 ± 12 | 104 ± 11 | 85.7 ± 0.2 | |
| FB1 | 10 | 77.5 ± 16.9 | 77.1 ± 16.5 | 81.3 ± 4.2 | 67.5 ± 6.1 |
| 50 | 99.5 ± 17.2 | 91.6 ± 10.6 | 89.8 ± 7.6 | 95.5 ± 4.6 | |
| 250 | 106 ± 21 | 103 ± 23 | 92.2 ± 2.7 | 93.0 ± 8.9 | |
| FB2 | 10 | 101 ± 16 | 115 ± 11 | 42.0 ± 20.9 | 48.1 ± 20.6 |
| 50 | 79.2 ± 13.0 | 112 ± 6 | 97.6 ± 30.6 | 91.8 ± 10.1 | |
| 250 | 80.8 ± 18.2 | 80.1 ± 9.6 | 77.6 ± 20.8 | 78.3 ± 12.2 | |
| FB3 | 10 | 113 ± 10 | 110 ± 19 | 101 ± 26 | 101 ± 9 |
| 50 | 120 ± 19 | 119 ± 20 | 55.6 ± 23.5 * | 69.5 ± 10.9 | |
| 250 | 120 ± 18 | 90.5 ± 15.4 | 53.2 ± 34.9 * | 70.1 ± 12.4 | |
| HT-2 | 2.5 | 35.7 ± 40.5 | 45.3 ± 5.7 | 45.6 ± 3.9 | 46.6 ± 9.9 |
| 12.5 | 42.8 ± 7.2 | 49.2 ± 9.5 | 39.6 ± 4.5 | 49.5 ± 1.3 | |
| 62.5 | 42.9 ± 5.5 | 50.2 ± 12.1 | 37.1 ± 4.3 | 48.3 ± 20.4 | |
| NIV | 10 | 80.2 ± 12.2 | 119 ± 14 | 97.6 ± 18.6 | 118 ± 5 |
| 50 | 81.1 ± 19.5 | 110 ± 20 | 80.6 ± 12.6 | 81.9 ± 1.0 | |
| 250 | 95.6 ± 16.2 | 103 ± 12 | 109 ± 16 | 100 ± 4 | |
| OTA | 0.5 | 117 ± 15 | 89.2 ± 14.2 | 106 ± 4 | 119 ± 1 |
| 2.5 | 109 ± 8 | 99.9 ± 6.6 | 109 ± 3 | 91.5 ± 11 | |
| 12.5 | 90.5 ± 3.1 | 88.1 ± 1.9 | 103 ± 2 | 90.1 ± 12.8 | |
| OTα | 0.5 | 128 ± 36 | 97.5 ± 4.6 | 116 ± 18 | 102 ± 14 |
| 2.5 | 89.2 ± 3.5 | 92.0 ± 8.6 | 82.6 ± 12.4 | 106 ± 19 | |
| 12.5 | 101 ± 5 | 91.6 ± 9.1 | 118 ± 11 | 93.2 ± 12.0 | |
| ROQ-C | 10 | 70.2 ± 22.9 | 70.6 ± 19.4 | 84.6 ± 19.5 | 81.1 ± 10.9 |
| 50 | 41.2 ± 20.2 | 95.5 ± 14.8 | 87.9 ± 10.6 | 81.5 ± 6.5 | |
| 250 | 103 ± 16 | 75.6 ± 16.2 | 111 ± 15 | 89.1 ± 4.1 | |
| T-2 | 2.5 | 40.0 ± 23.5 * | 45.8 ± 18.2 * | 90.5 ± 14.2 | 78.6 ± 15.1 |
| 12.5 | 89.9 ± 6.3 | 74.1 ± 7.0 * | 111 ± 7 | 82.6 ± 11.3 | |
| 62.5 | 85.8 ± 9.3 | 79.3 ± 6.5 | 116 ± 17 | 81.3 ± 9.9 | |
| T-2 triol | 10 | 81.7 ± 6.7 | 88.4 ± 11.5 | 82.6 ± 7.2 | 118 ± 12 |
| 50 | 104 ± 7 | 120 ± 7 | 107 ± 15 | 91.1 ± 8.2 | |
| 250 | 102 ± 8 | 111 ± 8.2 | 96.9 ± 10.4 | 97.9 ± 4.0 | |
| ZEN | 10 | 98.8 ± 3.6 | 98.8 ± 1.1 | 98.1 ± 1.2 | 116 ± 22 |
| 50 | 103 ± 2 | 101 ± 1 | 103. ± 3.5 | 80.4 ± 20.8 | |
| 250 | 104 ± 5 | 100 ± 5 | 101 ± 3 | 90.1 ± 17.9 | |
| Mycotoxin | VAMS | Liquid/Liquid | ||||
|---|---|---|---|---|---|---|
| Presence (%) | Average ± SD (ng/mL) | Max. (ng/mL) | Presence (%) | Average ± SD (ng/mL) | Max. (ng/mL) | |
| AFB1 | 10 | 0.10 ± 0.06 | 0.16 | 10 | 0.09 ± 0.08 | 0.12 |
| α-ZEL | 0 | n.a. | n.a. | 5 | 2.71 | n.a. |
| OTA | 60 | 0.56 ± 0.12 | 0.71 | 75 | 0.42 ± 0.18 | 0.76 |
| OTα | 80 | 0.83 ± 0.21 | 1.14 | 80 | 0.78 ± 0.29 | 1.28 |
| ZEN | 10 | 8.05 ± 5.02 | 14.02 | 10 | 7.68 ± 4.81 | 13.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidal, A.; Belova, L.; Stove, C.; De Boevre, M.; De Saeger, S. Volumetric Absorptive Microsampling as an Alternative Tool for Biomonitoring of Multi-Mycotoxin Exposure in Resource-Limited Areas. Toxins 2021, 13, 345. https://doi.org/10.3390/toxins13050345
Vidal A, Belova L, Stove C, De Boevre M, De Saeger S. Volumetric Absorptive Microsampling as an Alternative Tool for Biomonitoring of Multi-Mycotoxin Exposure in Resource-Limited Areas. Toxins. 2021; 13(5):345. https://doi.org/10.3390/toxins13050345
Chicago/Turabian StyleVidal, Arnau, Lidia Belova, Christophe Stove, Marthe De Boevre, and Sarah De Saeger. 2021. "Volumetric Absorptive Microsampling as an Alternative Tool for Biomonitoring of Multi-Mycotoxin Exposure in Resource-Limited Areas" Toxins 13, no. 5: 345. https://doi.org/10.3390/toxins13050345
APA StyleVidal, A., Belova, L., Stove, C., De Boevre, M., & De Saeger, S. (2021). Volumetric Absorptive Microsampling as an Alternative Tool for Biomonitoring of Multi-Mycotoxin Exposure in Resource-Limited Areas. Toxins, 13(5), 345. https://doi.org/10.3390/toxins13050345