Rapid Differential Detection of Abrin Isoforms by an Acetonitrile- and Ultrasound-Assisted On-Bead Trypsin Digestion Coupled with LC-MS/MS Analysis

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
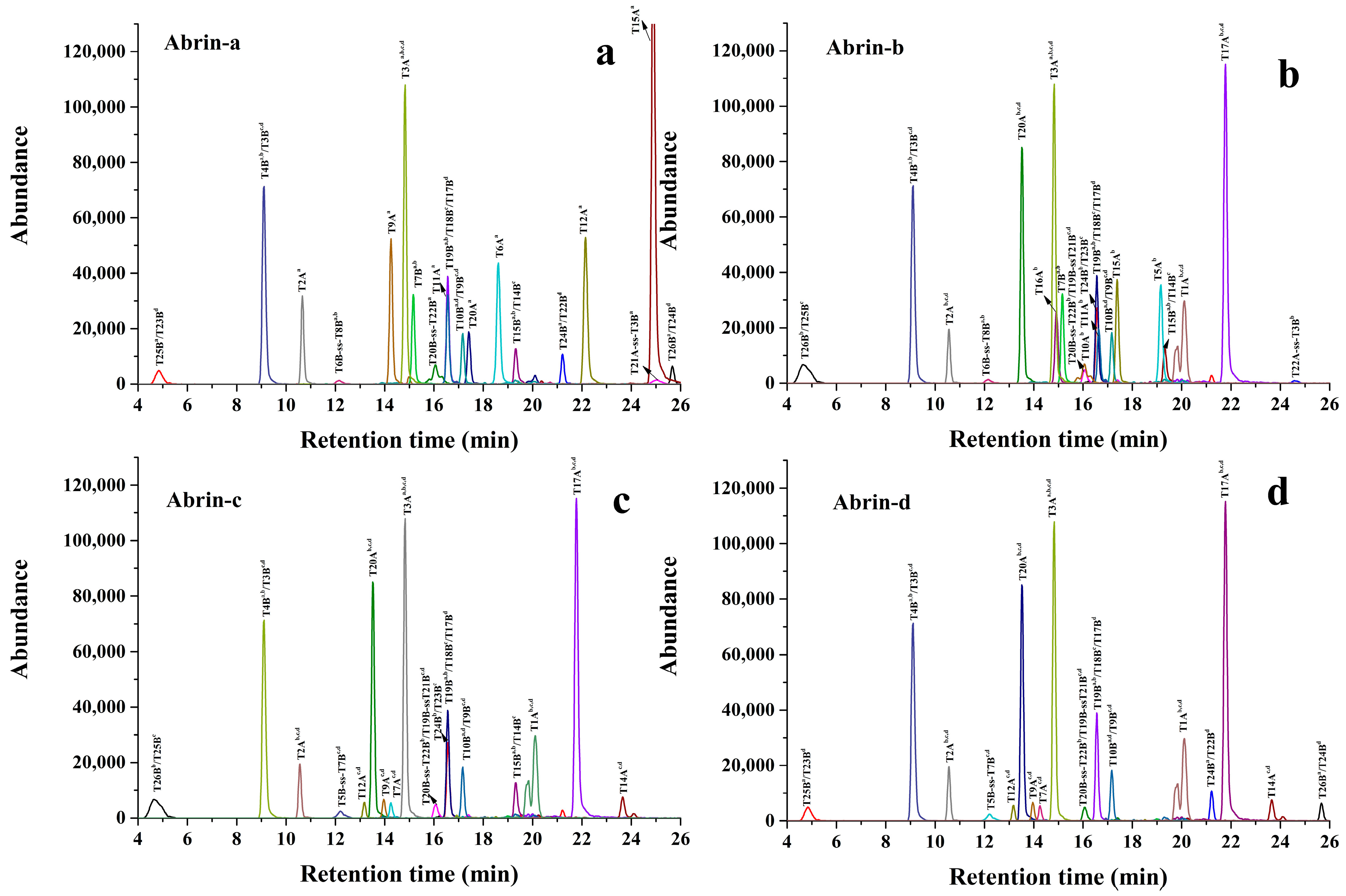
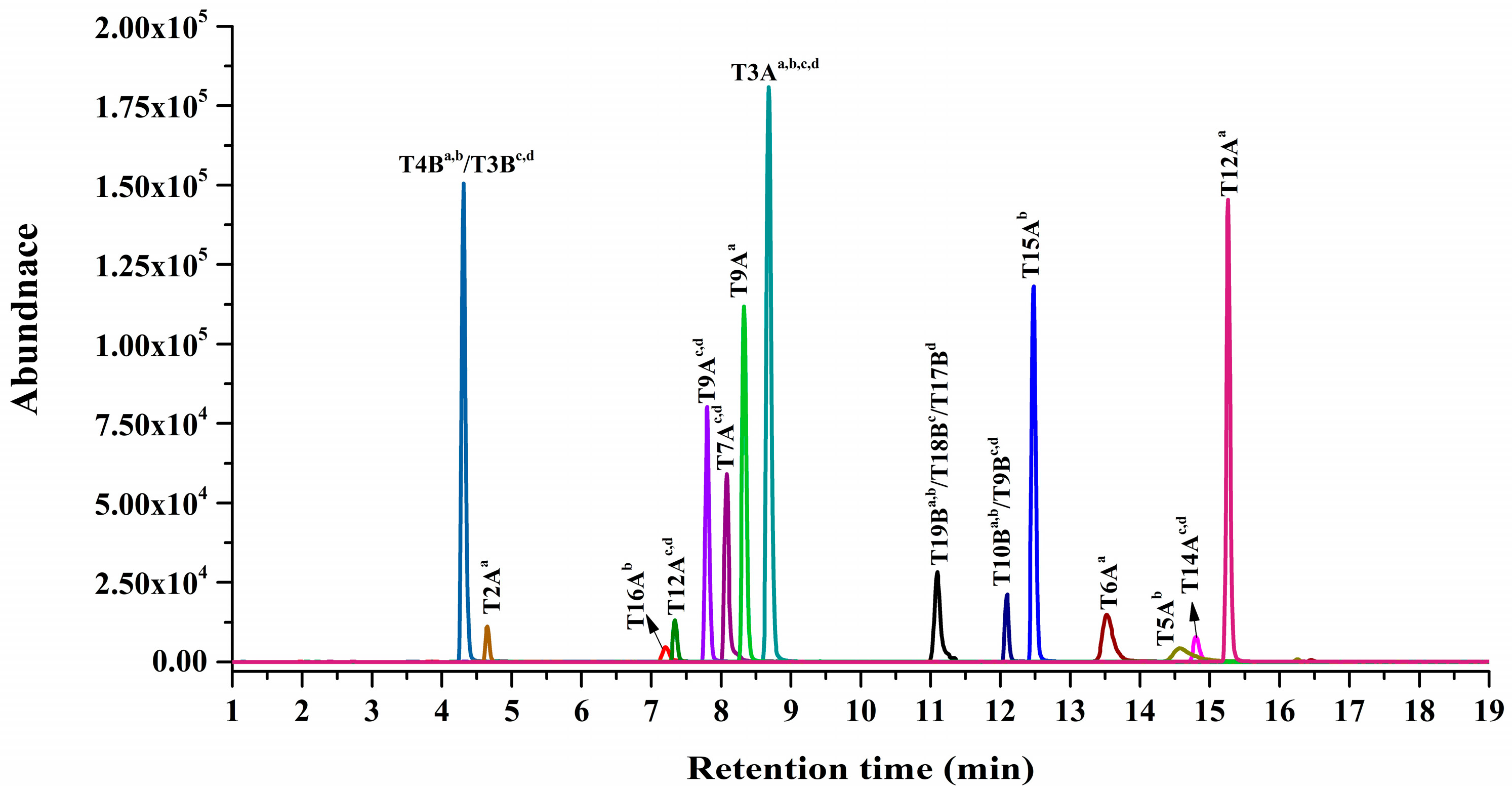
2.1. Screening and Identification of Specific Marker Peptides for Each Abrin Isoform
2.2. Enrichment and Purification of Abrin in Complex Matrix by Immunocapture
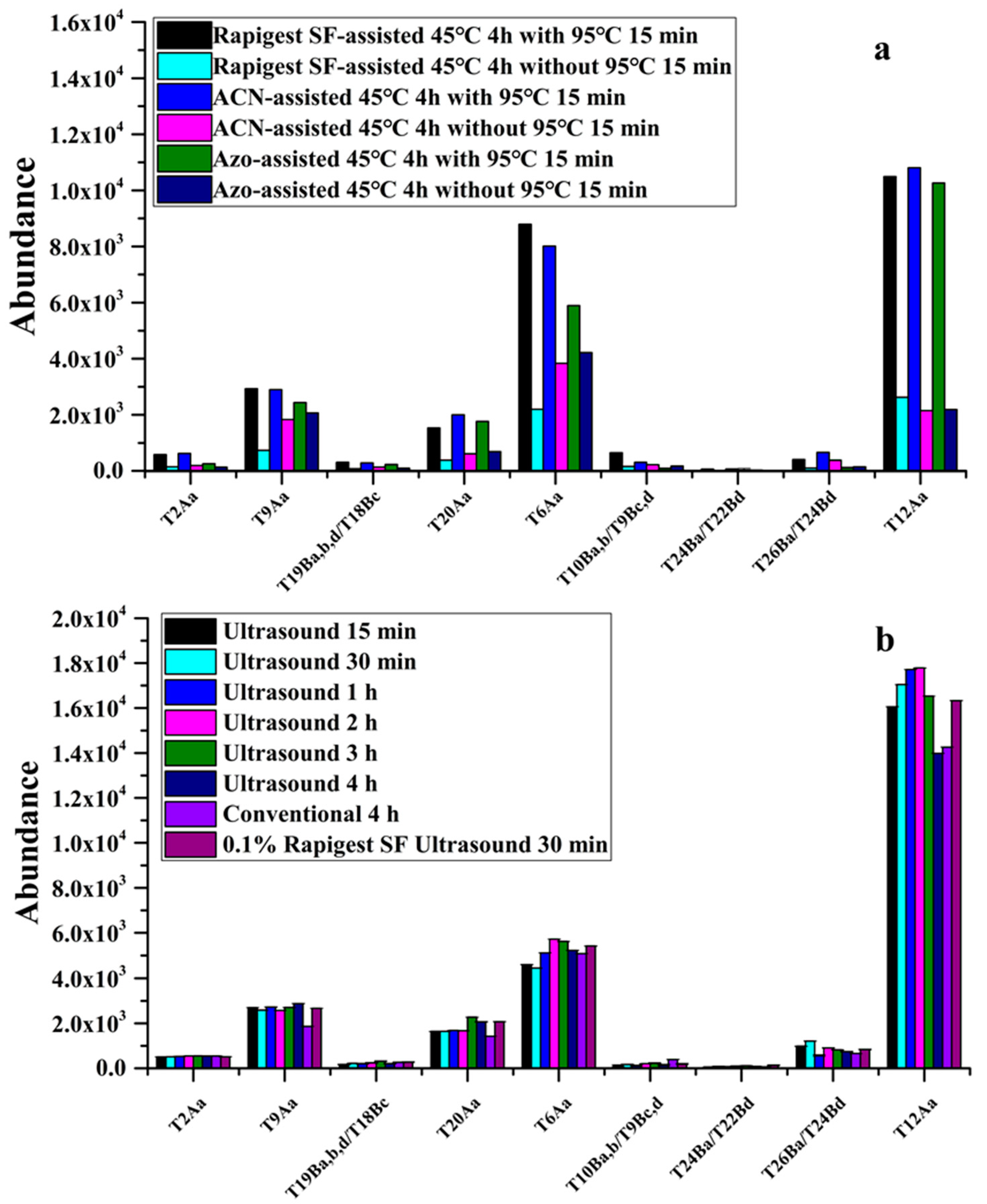
2.3. Development of ACN- and Ultrasound-Assisted On-Bead Trypsin Digestion of Abrin
2.4. HPLC-MS/MS (MRM) Method Development
2.5. Method Validation
2.5.1. Specificity
2.5.2. Linearity, Sensitivity, Accuracy and Precision (RSD)
2.5.3. Immunocapture Recovery and Matrix Effects
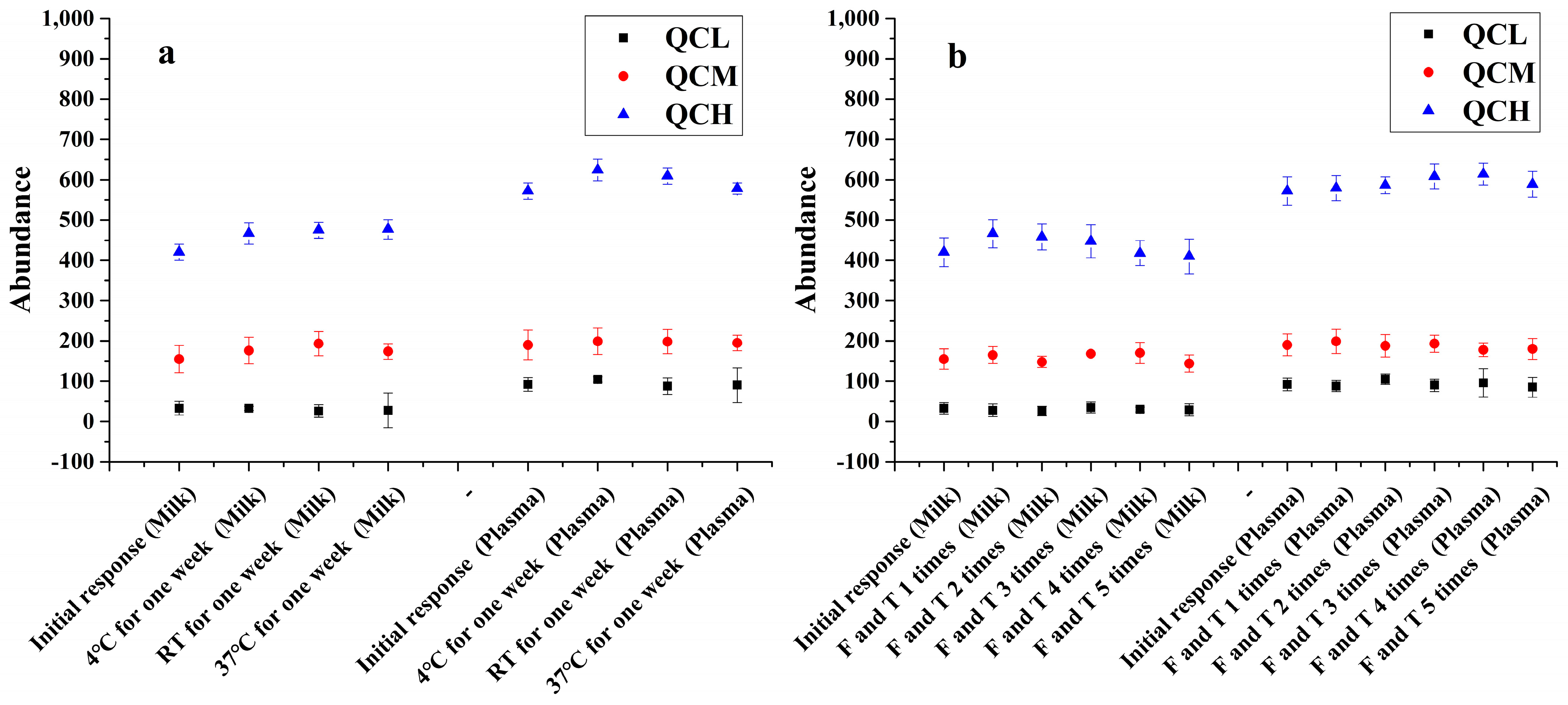
2.5.4. Stability
2.6. Method Application
2.6.1. Characterization of the Purified Abrin from Gel Filtration Chromatography
2.6.2. Identification of Abrin from the Samples of Biotoxin Exercises
3. Conclusions
4. Materials and Methods
4.1. Safety Precaution
4.2. Chemicals and Reagents
4.3. Samples for Calibration and Quality Control
4.3.1. Milk and Plasma Samples for Calibration and Quality Control
4.3.2. Abrin Extracts and Fraction Samples from the Fine Purification
4.4. Immunocapture and Ultrasound-Assisted on-Bead Trypsin Digestion
4.5. In Silico Digestion and Uniqueness Identification of Digested Peptides
4.6. LC-QTOF MS Analysis for Identification
4.7. HPLC-ESI MS/MS Analysis for Quantification
4.8. Method Validation
4.9. Calculation of Relative Content of Different Abrin Isoforms
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stirpe, F. Ribosome-inactivating proteins. Toxicon 2004, 44, 371–383. [Google Scholar] [CrossRef]
- Worbs, S.; Skiba, M.; Bender, J.; Zeleny, R.; Schimmel, H.; Luginbühl, W.; Dorner, B. An International Proficiency Test to Detect, Identify and Quantify Ricin in Complex Matrices. Toxins 2015, 7, 4987–5010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stirpe, F.; Battelli, M.G. Ribosome-inactivating proteins: Progress and problems. Cell. Mol. Life Sci. 2006, 63, 1850–1866. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Lee, T.C.; Hu, S.T.; Tung, T.C. Isolation of four isotoxic proteins and one agglutinin from jequiriti bean (Abrus precatorius). Toxicon 1981, 19, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Dickers, K.J.; Bradberry, S.M.; Rice, P.; Griffiths, G.D.; Vale, J.A. Abrin poisoning. Toxicol Rev. 2003, 22, 137–142. [Google Scholar] [CrossRef] [PubMed]
- OPCW-Organisation for the Prohibition of Chemical Weapons. Ricin Fact Sheet. 2014. Available online: https://www.opcw.org/sites/default/files/documents/SAB/en/sab-21-wp05_e_.pdf (accessed on 14 February 2021).
- Bagaria, A.; Surendranath, K.; Ramagopal, U.A.; Ramakumar, S.; Karande, A.A. Structure-Function Analysis and Insights into the Reduced Toxicity of Abrus precatorius Agglutinin I in Relation to Abrin. J. Biol. Chem. 2006, 281, 34465–34474. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.Y.; Lee, T.C.; Tung, T.C. Isolation of antitumor proteins abrin-A and abrin-B from Abrus precatorius. Int. J. Pept. Protein Res. 1978, 12, 311–317. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Patfield, S.; Cheng, L.W.; Stanker, L.H.; Rasooly, R.; McKeon, T.A.; Zhang, Y.; Brandon, D.L. Detection of Abrin Holotoxin Using Novel Monoclonal Antibodies. Toxins 2017, 9, 386. [Google Scholar] [CrossRef] [Green Version]
- Li, X.B.; Yang, W.; Zhang, Y.; Zhang, Z.G.; Kong, T.; Li, D.N.; Tang, J.J.; Liu, L.; Liu, G.W.; Wang, Z. Preparation and Identification of Monoclonal Antibody against Abrin-a. J. Agric. Food. Chem. 2011, 59, 9796–9799. [Google Scholar] [CrossRef]
- Antoine, M.D.; Hagan, N.A.; Lin, J.S.; Feldman, A.B.; Demirev, P.A. Rapid detection of ribosome inactivating protein toxins by mass-spectrometry-based functional assays. Int. J. Mass Spectrom. 2012, 312, 41–44. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, X.P.; Kahn, J.N.; Tumer, N.E. Functional Assays for Measuring the Catalytic Activity of Ribosome Inactivating Proteins. Toxins 2018, 10, 240. [Google Scholar] [CrossRef] [Green Version]
- Fredriksson, S.-Å.; Artursson, E.; Bergström, T.; Östin, A.; Nilsson, C.; Åstot, C. Identification of RIP-II toxins by affinity enrichment, enzymatic digestion and LC-MS. Anal. Chem. 2015, 87, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Dupré, M.; Gilquin, B.; Fenaille, F.; Feraudet-Tarisse, C.; Dano, J.; Ferro, M.; Simon, S.; Junot, C.; Brun, V.; Becher, F. Multiplex Quantification of Protein Toxins in Human Biofluids and Food Matrices Using Immunoextraction and High-Resolution Targeted Mass Spectrometry. Anal. Chem. 2015, 87, 8473–8480. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.H.; Cheng, X.; Yu, H.L.; Yang, Y.; Mu, X.H.; Chen, B.; Li, X.S.; Wu, J.N.; Yan, L.; Liu, C.C.; et al. Quantitative detection of ricin in beverages using trypsin/Glu-C tandem digestion coupled with ultra-high-pressure liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Tang, J.; Li, C.; Liu, Q.; Chen, J.; Li, H.; Guo, L.; Xie, J. Identification and quantification of ricin in biomedical samples by magnetic immunocapture enrichment and liquid chromatography electrospray ionization tandem mass spectrometry. Anal. Bioanal. Chem. 2014, 406, 5147–5155. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Liang, L.H.; Yang, Y.; Yu, H.L.; Yan, L.; Li, X.S.; Chen, B.; Liu, S.L.; Xi, H.L. Direct Acetonitrile-Assisted Trypsin Digestion Method Combined with LC-MS/MS-Targeted Peptide Analysis for Unambiguous Identification of Intact Ricin. J. Proteome Res. 2020. [Google Scholar] [CrossRef]
- Hansbauer, E.M.; Worbs, S.; Volland, H.; Simon, S.; Junot, C.; Fenaille, F.; Dorner, B.G.; Becher, F. Rapid Detection of Abrin Toxin and Its Isoforms in Complex Matrices by Immuno-Extraction and Quantitative High Resolution Targeted Mass Spectrometry. Anal. Chem. 2017, 89, 11719–11727. [Google Scholar] [CrossRef]
- Fredriksson, S.-Å.; Hulst, A.G.; Artursson, E.; Jong, A.L.; Nilsson, C.; Baar, B.L.M. Forensic Identification of Neat Ricin and of Ricin from Crude Castor Bean Extracts by Mass Spectrometry. Anal. Chem. 2005, 77, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, O.K.; Roen, B.T.; Enger, S.; Lundanes, E.; Wilson, S.R. Multichannel Open Tubular Enzyme Reactor Online Coupled with Mass Spectrometry for Detecting Ricin. Anal. Chem. 2017, 89, 8667–8673. [Google Scholar] [CrossRef]
- Ostin, A.; Bergstrom, T.; Fredriksson, S.-A.; Nilsson, C. Solvent-Assisted Trypsin Digestion of Ricin for Forensic Identification by LC-ESI MS/MS. Anal. Chem. 2007, 79, 6271–6278. [Google Scholar] [CrossRef]
- Brown, K.A.; Chen, B.; Guardado-Alvarez, T.M.; Lin, Z.; Hwang, L.; Ayaz-Guner, S.; Jin, S.; Ge, Y. A photocleavable surfactant for top-down proteomics. Nat. Methods 2019, 16, 417–420. [Google Scholar] [CrossRef]
- Chen, D.; Bryden, W.A.; Fenselau, C. Rapid analysis of ricin using hot acid digestion and MALDI-TOF mass spectrometry. J. Mass Spectrom. 2018, 53, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Dycka, F.; Bobal, P.; Mazanec, K.; Bobalova, J. Rapid and efficient protein enzymatic digestion: An experimental comparison. Electrophoresis 2012, 33, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Hovde, B.T.; Daligault, H.E.; Hanschen, E.R.; Kunde, Y.A.; Johnson, M.B.; Starkenburg, S.R.; Johnson, S.L. Detection of abrin-like and prepropulchellin-like toxin genes and transcripts using whole genome sequencing and full-length transcript sequencing of abrus precatorius. Toxins 2019, 11, 691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OPCW Technical Secretariat. Guidelines for the fourth biotoxin sample analysis exercise. 2019. Available online: https://www.opcw.org/resources/documents/technical-secretariat/2019 (accessed on 14 February 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Amino Acid Sequences | Precursor Ion (m/z) | Product Ions (m/z) | Fragmentor (V) | Collision Energy (V) |
|---|---|---|---|---|---|
| T3Aa,b,c,d | QFIEALR | 438.9 (2+) | 601.4, 488.7, 429.6 | 90 | 15, 10, 8 |
| T2Aa | FSTEGATSQSYK | 653.4 (2+) | 841.4, 713.3, 527.3 | 170 | 20, 20, 20 |
| T6Aa | GGLIHDIPVLPDPTTLQER | 691.2 (3+) | 1015.5, 902.5, 844.4 | 120 | 15, 20, 25 |
| T9Aa | AGTQSYFLR | 521.8 (2+) | 685.4, 598.3, 512.8 | 120 | 20, 10, 10 |
| T12Aa | QQIPLGLQALTHGISFFR | 676.2 (3+) | 829.4, 670.2, 553.4 | 160 | 15, 15, 15 |
| T5Ab | LTGGLIHGIPVLPDPTTLQER | 743.2 (3+) | 862.5, 844.4, 528.8 | 120 | 25, 30, 15 |
| T15Ab | QQIPLGLQALR | 619.0 (2+) | 867.6, 610.0, 257.2 | 140 | 20, 15, 20 |
| T16Ab | HAISFLQSGTDDQEIAR | 630.2 (3+) | 1091.6, 556.4, 86.2 | 130 | 15, 20, 30 |
| T7Ac,d | YITVELSNSER | 656.0 (2+) | 1034.6, 834.5, 249.2 | 150 | 20, 20, 25 |
| T9Ac,d | AGSQSYFLR | 514.8 (2+) | 685.4, 505.8, 441.8 | 110 | 15, 10, 15 |
| T12Ac,d | FDGSYGDLER | 579.8 (2+) | 896.8, 752.4, 589.4 | 130 | 20, 20, 20 |
| T14Ac,d | EEISLGLQALTHAISFLR | 666.9 (3+) | 814.1, 770.6, 714.4 | 140 | 15, 15, 15 |
| T4Ba,b/T3Bc,d | YEPTVR | 382.8 (2+) | 472.3, 265.2, 236.7 | 90 | 8, 10, 8 |
| T10Ba,b/T9Bc,d | LEENQLWTLK | 637.4 (2+) | 361.2, 243.1, 215.1 | 160 | 20, 20, 25 |
| T19Ba,b/T18Bc/T17Bd | EQQWALYTDGSIR | 783.9 (2+) | 924.4, 811.4, 368.2 | 170 | 25, 25, 30 |
| T3Aa,b,c,d* | QFIEALR* | 443.9 (2+) | 611.4, 498.3, 434.6 | 90 | 15, 12, 8 |
| T12A a* | QQIPLGLQALTHGISFFR* | 679.5 (3+) | 834.0, 673.5, 556.4 | 150 | 18, 15, 15 |
| T15A b* | QQIPLGLQALR* | 624.0 (2+) | 877.5, 614.9, 439.3 | 120 | 35, 15, 15 |
| T9A c,d* | AGSQSYFLR* | 519.9 (2+) | 695.4, 510.9, 446.8 | 100 | 15, 812 |
| Peptide | Matrices | Calibration Curve | R2 | LOD (ng/mL) | LOQ (ng/mL) | Range (ng/mL) |
|---|---|---|---|---|---|---|
| T3Aa,b,c,d | Milk | Y = 0.3103X + 1.4980 | 0.998 | 0.50 | 1.00 | 1.00–500 |
| T12Aa | Milk | Y = 0.0851X − 0.1942 | 0.998 | 0.47 | 2.35 | 2.35–235 |
| T15Ab | Milk | Y = 0.0041X + 0.2192 | 0.996 | 1.71 | 3.42 | 3.42–513 |
| T9Ac,d | Milk | Y = 0.0041X + 0.4499 | 0.998 | 5.64 | 9.40 | 9.40–376 |
| T3Aa,b,c,d | Plasma | Y = 0.037X + 1.6101 | 0.999 | 0.50 | 1.00 | 1.00–500 |
| T12Aa | Plasma | Y = 0.0879X + 0.4292 | 0.999 | 0.47 | 2.35 | 2.35–235 |
| T15Ab | Plasma | Y = 0.0045X + 0.2651 | 0.996 | 1.71 | 3.42 | 3.42–513 |
| T9Ac,d | Plasma | Y = 0.0045X + 0.4597 | 0.995 | 5.64 | 9.40 | 9.40–376 |
| Sample | Quantitative Peptides | Matrices | Spiking Concentration (ng/mL) | Intra-Day (n = 3) | Inter-Day (n = 8) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Calculated Concentration (ng/mL) | Accuracy (%) | RSD (%) | Calculated Concentration (ng/mL) | Accuracy (%) | RSD (%) | ||||
| QCL | T3Aa,b,c,d | Milk | 10.0 (total abrin) | 9.76 ± 0.14 | 97.6 | 6.3 | 9.45 ± 0.19 | 94.5 | 11.4 |
| QCL | T12Aa | Milk | 4.70 (abrin-a) | 4.38 ± 0.04 | 94.7 | 8.8 | 4.07 ± 0.10 | 86.5 | 7.5 |
| QCL | T15Ab | Milk | 3.42 (abrin-b) | 3.67 ± 0.02 | 107.4 | 7.6 | 3.18 ± 0.16 | 93.1 | 7.8 |
| QCL | T9Ac,d | Milk | 1.88 (abrin-c/d) | - | - | - | - | - | - |
| QCM | T3Aa,b,c,d | Milk | 150 (total abrin) | 150.2 ± 3.90 | 100.1 | 3.7 | 142.0 ± 0.81 | 94.7 | 9.2 |
| QCM | T12Aa | Milk | 70.5 (abrin-a) | 67.4 ± 0.02 | 95.6 | 3.3 | 64.9 ± 0.11 | 92.1 | 8.2 |
| QCM | T15Ab | Milk | 51.3 (abrin-b) | 48.5 ± 1.04 | 94.6 | 2.7 | 46.1 ± 0.15 | 88.9 | 6.9 |
| QCM | T9Ac,d | Milk | 28.2 (abrin-c/d) | 24.4 ± 0.04 | 86.7 | 2.9 | 24.3 ± 0.08 | 86.1 | 4.9 |
| QCH | T3Aa,b,c,d | Milk | 300 (total abrin) | 288.0 ± 0.18 | 96.0 | 1.7 | 279.5 ± 8.71 | 93.2 | 5.5 |
| QCH | T12Aa | Milk | 141 (abrin-a) | 129.9 ± 0.95 | 92.1 | 2.9 | 123.1 ± 6.32 | 87.3 | 5.3 |
| QCH | T15Ab | Milk | 102.6 (abrin-b) | 97.8 ± 1.23 | 95.3 | 2.2 | 92.1 ± 4.83 | 89.8 | 2.9 |
| QCH | T9Ac,d | Milk | 56.4 (abrin-c/d) | 49.2 ± 1.74 | 87.2 | 3.8 | 47.1 ± 5.71 | 83.5 | 4.5 |
| QCL | T3Aa,b,c,d | Plasma | 10.0 (total abrin) | 11.2 ± 0.18 | 112.0 | 8.5 | 10.9 ± 0.23 | 109.0 | 9.9 |
| QCL | T12Aa | Plasma | 4.70 (abrin-a) | 6.88 ± 1.01 | 102.7 | 5.3 | 6.32 ± 0.88 | 94.0 | 8.8 |
| QCL | T15Ab | Plasma | 3.42 (abrin-b) | 3.02 ± 0.53 | 111.1 | 4.9 | 2.50 ± 0.58 | 92.9 | 11.3 |
| QCL | T9Ac,d | Plasma | 1.88 (abrin-c/d) | - | - | - | - | - | - |
| QCM | T3Aa,b,c,d | Plasma | 150 (total abrin) | 148.6 ± 3.3 | 99.1 | 3.2 | 142.6 ± 4.8 | 94.9 | 6.9 |
| QCM | T12Aa | Plasma | 70.5 (abrin-a) | 67.8 ± 5.6 | 96.2 | 4.8 | 64.9 ± 4.6 | 92.0 | 8.6 |
| QCM | T15Ab | Plasma | 51.3 (abrin-b) | 48.9 ± 1.8 | 95.3 | 2.9 | 42.6 ± 2.1 | 83.0 | 6.2 |
| QCM | T9Ac,d | Plasma | 28.2 (abrin-c/d) | 24.3 ± 0.8 | 86.2 | 5.6 | 24.9 ± 1.9 | 88.3 | 8.9 |
| QCH | T3Aa,b,c,d | Plasma | 300 (total abrin) | 293.3 ± 1.92 | 97.8 | 1.8 | 289.5 ± 2.3 | 96.5 | 4.4 |
| QCH | T12Aa | Plasma | 141 (abrin-a) | 132.4 ± 4.13 | 93.9 | 1.7 | 126.1 ± 5.3 | 89.4 | 5.3 |
| QCH | T15Ab | Plasma | 102.6 (abrin-b) | 98.7 ± 3.2 | 96.2 | 2.8 | 94.7 ± 3.3 | 92.3 | 5.9 |
| QCH | T9Ac,d | Plasma | 56.4 (abrin-c/d) | 53.2 ± 1.2 | 94.3 | 6.1 | 51.8 ± 2.5 | 91.8 | 8.6 |
| Sample ID | Spiking Concentration (ng/mL) | Matrices | Extraction Recovery % | RSD % |
|---|---|---|---|---|
| QCL | 10.0 | Milk | 76.3 | 3.3 |
| QCM | 150 | Milk | 77.4 | 2.1 |
| QCH | 300 | Milk | 71.1 | 1.8 |
| QCL | 10.0 | Plasma | 80.9 | 4.9 |
| QCM | 150 | Plasma | 77.8 | 2.8 |
| QCH | 300 | Plasma | 73.4 | 1.2 |
| Sample ID | Matrices | Spiking Concertation of Peptides (ng/mL) | Matrices Effect of T3Aa,b,c,d % | Matrix Effect of T12Aa % | Matrix Effect of T15Ab % | Matrix Effect of T9Ac,d % |
|---|---|---|---|---|---|---|
| QC-L | Milk | 2.00 | 128.8 ± 2.8 | 110.8 ± 3.7 | 124.2 ± 5.6 | 106.9 ± 3.3 |
| QC-M | Milk | 20.0 | 106.2 ± 4.2 | 96.1 ± 4.1 | 96.5 ± 4.8 | 91.5 ± 2.8 |
| QC-H | Milk | 100 | 103.3 ± 1.1 | 94.3 ± 6.2 | 96.9 ± 3.1 | 102.7 ± 2.6 |
| QC-L | Plasma | 2.00 | 112.5 ± 2.6 | 113.0 ± 2.9 | 125.9 ± 2.5 | 110.9 ± 3.4 |
| QC-M | Plasma | 20.0 | 112.7 ± 3.6 | 97.3 ± 4.5 | 108.5 ± 2.6 | 98.1 ± 5.6 |
| QC-H | Plasma | 100 | 91.9 ± 2.9 | 92.8 ± 2.3 | 98.6 ± 3.7 | 96.5 ± 2.7 |
| Sample | Spiking Chemicals/(Nominal Concentration) | Matrices | Identification | Identified Peptide Markers |
|---|---|---|---|---|
| A172.1 | Blank | BSA 1 mg/mL in 5% AcOH | - | |
| A172.4 | Purified abrin (100 μg/mL) | BSA 1 mg/mL in 5% AcOH | Abrin | 15 (12 from A chain and 3 from B chain) |
| A172.5 | Purified ricin (100 μg/g) & purified abrin (1 mg/g) | Commercial Stevia powder | Abrin | 15 (12 from A chain and 3 from B chain) |
| BT18.1 | Purified * Abrin (150 μg/g) | Protein powder | Abrin | 11 (8 from A chain and 3 from B chain) |
| BT18.2 | Abrus Agglutinin * (300 μg/g) Purified Viscumin (50 μg/g) Purified Ricin (15 μg/g) | Protein powder | Abrin | 11 (8 from A chain and 3 from B chain) |
| BT18.4 | Blank | Protein powder | - | |
| BT18.7 | Blank | Saline solution | - | |
| BT19.1 | Blank (Contains BSA) | SDS-PAGE gel * | ||
| BT19.3 | Crude Castor Bean Extract (15 μg/spot) Purified Abrin (15 μg/spot) | SDS-PAGE gel | Abrin | 9 (6 from A chain and 3 from B chain) |
| BT19.5 | Blank | Spray buffer † | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, L.-H.; Yang, Y.; Geng, S.; Cheng, X.; Yu, H.-L.; Liu, C.-C.; Liu, S.-L. Rapid Differential Detection of Abrin Isoforms by an Acetonitrile- and Ultrasound-Assisted On-Bead Trypsin Digestion Coupled with LC-MS/MS Analysis. Toxins 2021, 13, 358. https://doi.org/10.3390/toxins13050358
Liang L-H, Yang Y, Geng S, Cheng X, Yu H-L, Liu C-C, Liu S-L. Rapid Differential Detection of Abrin Isoforms by an Acetonitrile- and Ultrasound-Assisted On-Bead Trypsin Digestion Coupled with LC-MS/MS Analysis. Toxins. 2021; 13(5):358. https://doi.org/10.3390/toxins13050358
Chicago/Turabian StyleLiang, Long-Hui, Yang Yang, Shu Geng, Xi Cheng, Hui-Lan Yu, Chang-Cai Liu, and Shi-Lei Liu. 2021. "Rapid Differential Detection of Abrin Isoforms by an Acetonitrile- and Ultrasound-Assisted On-Bead Trypsin Digestion Coupled with LC-MS/MS Analysis" Toxins 13, no. 5: 358. https://doi.org/10.3390/toxins13050358
APA StyleLiang, L. -H., Yang, Y., Geng, S., Cheng, X., Yu, H. -L., Liu, C. -C., & Liu, S. -L. (2021). Rapid Differential Detection of Abrin Isoforms by an Acetonitrile- and Ultrasound-Assisted On-Bead Trypsin Digestion Coupled with LC-MS/MS Analysis. Toxins, 13(5), 358. https://doi.org/10.3390/toxins13050358