Dysbiosis-Related Advanced Glycation Endproducts and Trimethylamine N-Oxide in Chronic Kidney Disease


Abstract
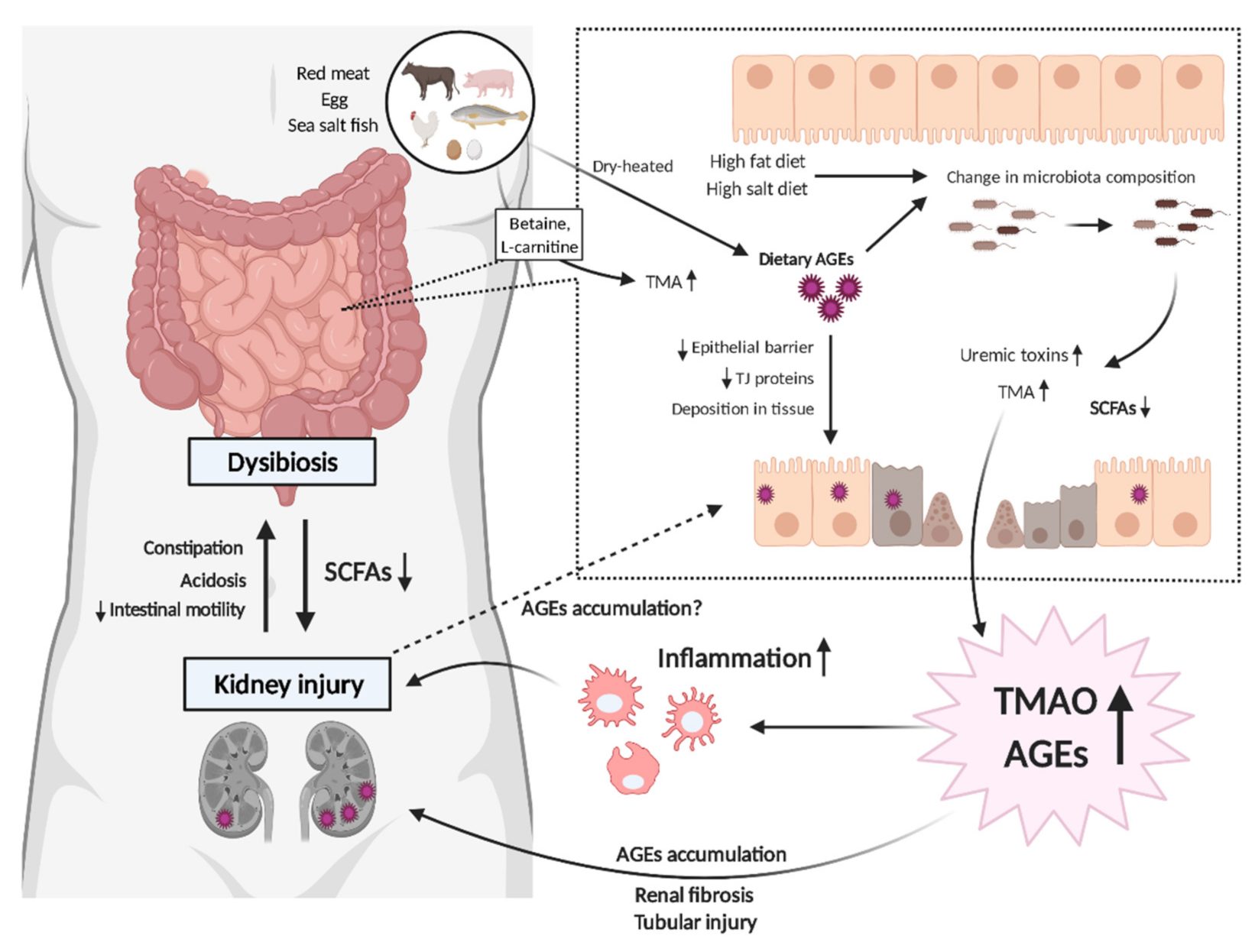
:1. Introduction
2. Gut Microbiota, AGEs, TMAO, and Inflammation
2.1. Dysbiosis in CKD
2.2. Disruption of Gut Epithelial Barrier in CKD
2.3. Gut Dysbiosis and Inflammation
2.4. AGEs–RAGE Axis and Inflammation
2.5. TMAO and Inflammation
3. Possible Interplay between Gut Microbiota and Uremic Toxins in CKD
3.1. AGEs and Dysbiosis in CKD
3.2. AGEs–RAGE Coordinates with TMAO to Progress CKD
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GFR | Glomerular filtration rate |
| CKD | Chronic kidney disease |
| ESRD | End-stage renal disease |
| AKI | Acute kidney injury |
| DKD | Diabetic kidney disease |
| SLE | Systemic lupus erythematosus |
| PTECs | Proximal tubular epithelial cells |
| PBMCs | Peripheral blood mononuclear cells |
| AGEs | Advanced glycation endproducts |
| RAGE | Receptor for advanced glycation endproducts |
| sRAGE | Soluble RAGE |
| N(ε)-CML | N(ε)-carboxymethyl lysine |
| TMAO | Trimethylamine N-oxide |
| TMA | Trimethylamine |
| IS | Indoxyl sulfate |
| PCS | p-cresol sulfate |
| LPS | Lipopolysaccharide |
| ROS | Reactive oxygen species |
| NF-ĸB | Nuclear factor-ĸB |
| TNF-α | Tumor necrosis factor-α |
| MCP-1 | Monocyte chemoattractant protein-1 |
| INF-1α | Interferon-1α |
| VEGF | Vascular endothelial growth factor |
| sICAM-1 | Soluble forms of intracellular adhesion molecule-1 |
| sVCAM-1 | Soluble forms of vascular cell adhesion molecule-1 |
| MDA | Malondialdehyde |
| TLR | Toll-like receptor |
| PAMPs | Pathogen-associated molecular patterns |
| DAMPs | Damage-associated molecular patterns |
| SCFAs | Short-chain fatty acids |
| DMB | 3:3-Dimethyl-1-butanol |
| HFD | High-fat diet |
| OAT | Organic anion transporter |
References
- Thompson, S.; James, M.; Wiebe, N.; Hemmelgarn, B.; Manns, B.; Klarenbach, S.; Tonelli, M. Cause of Death in Patients with Reduced Kidney Function. J. Am. Soc. Nephrol. 2015, 26, 2504–2511. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Arici, M.; Collins, A.J.; Garcia, G.G.; Hemmelgarn, B.R.; Jafar, T.H.; Pecoits-Filho, R.; Sola, L.; Swanepoel, C.R.; Tchokhonelidze, I.; et al. Understanding kidney care needs and implementation strategies in low- and middle-income countries: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2016, 90, 1164–1174. [Google Scholar] [CrossRef] [Green Version]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.-Y. Chronic Kidney Disease and the Risks of Death, Cardiovascular Events, and Hospitalization. N. Eng. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Seliger, S.L.; Gillen, D.L.; Longstreth, W.; Kestenbaum, B.; Stehman-Breen, C.O. Elevated risk of stroke among patients with end-stage renal disease. Kidney Int. 2003, 64, 603–609. [Google Scholar] [CrossRef] [Green Version]
- Stengel, B. Chronic kidney disease and cancer: A troubling connection. J. Nephrol. 2010, 23, 253–262. [Google Scholar] [PubMed]
- Tamura, K.M.; Xie, D.; Yaffe, K.; Cohen, D.L.; Teal, V.; Kasner, S.E.; Messe, S.R.; Sehgal, A.R.; Kusek, J.; De Salvo, K.; et al. Vascular risk factors and cognitive impairment in chronic kidney disease: The Chronic Renal Insufficiency Cohort (CRIC) study. Clin. J. Am. Soc. Nephrol. 2011, 6, 248–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonelli, M.; Wiebe, N.; Culleton, B.; House, A.; Rabbat, C.; Fok, M.; McAlister, F.; Garg, A.X. Chronic Kidney Disease and Mortality Risk: A Systematic Review. J. Am. Soc. Nephrol. 2006, 17, 2034–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldanha da Silva, A.A.; Prestes, T.R.R.; Lauar, A.O.; Finotti, B.B.; Silva, A.C.S. Renin Angiotensin System and Cytokines in Chronic Kidney Disease: Clinical and Experimental Evidence. Protein Pept. Lett. 2017, 24, 799–808. [Google Scholar] [CrossRef]
- Grassi, G.; Quarti-Trevano, F.; Seravalle, G.; Arenare, F.; Volpe, M.; Furiani, S.; Dell’Oro, R.; Mancia, G. Early Sympathetic Activation in the Initial Clinical Stages of Chronic Renal Failure. Hypertension 2011, 57, 846–851. [Google Scholar] [CrossRef] [Green Version]
- Colombo, P.C.; Ganda, A.; Lin, J.; Onat, D.; Harxhi, A.; Iyasere, J.E.; Uriel, N.; Cotter, G. Inflammatory activation: Cardiac, renal, and cardio-renal interactions in patients with the cardiorenal syndrome. Heart Fail. Rev. 2012, 17, 177–190. [Google Scholar] [CrossRef]
- Jazani, N.H.; Savoj, J.; Lustgarten, M.; Lau, W.L.; Vaziri, N.D. Impact of Gut Dysbiosis on Neurohormonal Pathways in Chronic. Kidney Dis. 2019, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Elias, B.C.; Brooks, C.R.; Ueda, S.; Fukami, K. Uremic Toxin–Targeting as a Therapeutic Strategy for Preventing Cardiorenal Syndrome. Circ. J. 2019, 84, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Lisowska-Myjak, B. Uremic Toxins and Their Effects on Multiple Organ Systems. Nephron 2014, 128, 303–311. [Google Scholar] [CrossRef]
- Chung, A.C.; Zhang, H.; Kong, Y.Z.; Tan, J.J.; Huang, X.R.; Kopp, J.B.; Lan, H.Y. Advanced glycation end-products induce tubular CTGF via TGF-beta-independent Smad3 signaling. J Am. Soc. Nephrol. 2010, 21, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Koeth, R.; Wang, Z.; Levison, B.S.; A Buffa, J.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.W.; Wang, Z.; Shrestha, K.; Borowski, A.G.; Wu, Y.; Troughton, R.; Klein, A.L.; Hazen, S.L. Intestinal Microbiota-Dependent Phosphatidylcholine Metabolites, Diastolic Dysfunction, and Adverse Clinical Outcomes in Chronic Systolic Heart Failure. J. Card. Fail. 2015, 21, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.W.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut Microbiota-Dependent TrimethylamineN-Oxide (TMAO) Pathway Contributes to Both Development of Renal Insufficiency and Mortality Risk in Chronic Kidney Disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.; Rodriguez, V.; Vermali, R.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut Dysbiosis Is Linked to Hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [Green Version]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalar, B.; Haslberger, A.; Peterlin, B. The Role of Microbiota in Depression—A brief review. Psychiatr. Danub. 2018, 30, 136–141. [Google Scholar] [CrossRef]
- Bansilal, S.; Castellano, J.M.; Fuster, V. Withdrawn: Global burden of CVD: Focus on secondary prevention of cardiovascular disease. Int. J. Cardiol. 2016, 201, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.-H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of Urease- and Uricase-Containing, Indole- and p-Cresol-Forming and Contraction of Short-Chain Fatty Acid-Producing Intestinal Microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Barba, C.; Soulage, C.O.; Caggiano, G.; Glorieux, G.; Fouque, D.; Koppe, L. Effects of Fecal Microbiota Transplantation on Composition in Mice with CKD. Toxins 2020, 12, 741. [Google Scholar] [CrossRef]
- Li, D.Y.; Tang, W.W. Contributory Role of Gut Microbiota and Their Metabolites Toward Cardiovascular Complications in Chronic Kidney Disease. Semin. Nephrol. 2018, 38, 193–205. [Google Scholar] [CrossRef]
- Yacoub, R.; Nugent, M.; Cai, W.; Nadkarni, G.N.; Chaves, L.D.; Abyad, S.; Honan, A.M.; Thomas, S.A.; Zheng, W.; Valiyaparambil, S.A.; et al. Advanced glycation end products dietary restriction effects on bacterial gut microbiota in peritoneal dialysis patients; a randomized open label controlled trial. PLoS ONE 2017, 12, e0184789. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Zhao, J.; Qu, W.; Zhang, Y.; Jia, B.; Fan, Z.; He, Q.; Li, J. Accumulation and effects of dietary advanced glycation end products on the gastrointestinal tract in rats. Int. J. Food Sci. Technol. 2018, 53, 2273–2281. [Google Scholar] [CrossRef]
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the Gut Microbiome in Uremia: A Potential Therapeutic Target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.; Macfarlane, G. Formation of Phenolic and Indolic Compounds by Anaerobic Bacteria in the Human Large Intestine. Microb. Ecol. 1997, 33, 180–188. [Google Scholar] [CrossRef]
- Koppe, L.; Mafra, D.; Fouque, D. Probiotics and chronic kidney disease. Kidney Int. 2015, 88, 958–966. [Google Scholar] [CrossRef] [Green Version]
- Fishbane, S.; Mathew, A.; Vaziri, N.D. Iron toxicity: Relevance for dialysis patients. Nephrol. Dial. Transplant. 2013, 29, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Iguchi, A.; Yamamoto, S.; Oda, A.; Tanaka, K.; Kazama, J.J.; Saeki, T.; Yamazaki, H.; Ishioka, K.; Suzutani, T.; Narita, I. Effect of sucroferric oxyhydroxide on gastrointestinal microbiome and uremic toxins in patients with chronic kidney disease undergoing hemodialysis. Clin. Exp. Nephrol. 2020, 24, 725–733. [Google Scholar] [CrossRef]
- Hviid, A.; Svanström, H.; Frisch, M. Antibiotic use and inflammatory bowel diseases in childhood. Gut 2010, 60, 49–54. [Google Scholar] [CrossRef]
- Imhann, F.; Vila, A.V.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; Van Dullemen, H.M.; et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef]
- Wu, M.-J.; Chang, C.-S.; Cheng, C.-H.; Chen, C.-H.; Lee, W.-C.; Hsu, Y.-H.; Shu, K.-H.; Tang, M.-J. Colonic transit time in long-term dialysis patients. Am. J. Kidney Dis. 2004, 44, 322–327. [Google Scholar] [CrossRef]
- Wang, X.; Yang, S.; Li, S.; Zhao, L.; Hao, Y.; Qin, J.; Zhang, L.; Zhang, C.; Bian, W.; Zuo, L.; et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 2020, 69, 2131–2142. [Google Scholar] [CrossRef]
- Zheng, H.J.; Guo, J.; Wang, Q.; Wang, L.; Wang, Y.; Zhang, F.; Huang, W.-J.; Zhang, W.; Liu, W.J.; Wang, Y. Probiotics, prebiotics, and synbiotics for the improvement of metabolic profiles in patients with chronic kidney disease: A systematic review and meta-analysis of randomized controlled trials. Crit. Rev. Food Sci. Nutr. 2021, 61, 577–598. [Google Scholar] [CrossRef]
- Bakhtiary, M.; Morvaridzadeh, M.; Agah, S.; Rahimlou, M.; Christopher, E.; Zadro, J.R.; Heshmati, J. Effect of Probiotic, Prebiotic, and Synbiotic Supplementation on Cardiometabolic and Oxidative Stress Parameters in Patients With Chronic Kidney Disease: A Systematic Review and Meta-analysis. Clin. Ther. 2021, 43, e71–e96. [Google Scholar] [CrossRef]
- Rossi, M.; Johnson, D.W.; Morrison, M.; Pascoe, E.M.; Coombes, J.S.; Forbes, J.M.; Szeto, C.-C.; McWhinney, B.C.; Ungerer, J.P.; Campbell, K.L. Synbiotics Easing Renal Failure by Improving Gut Microbiology (SYNERGY): A Randomized Trial. Clin. J. Am. Soc. Nephrol. 2016, 11, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Madero, M.; Cano, K.B.; Campos, I.; Tao, X.; Maheshwari, V.; Brown, J.; Cornejo, B.; Handelman, G.; Thijssen, S.; Kotanko, P. Removal of Protein-Bound Uremic Toxins during Hemodialysis Using a Binding Competitor. Clin. J. Am. Soc. Nephrol. 2019, 14, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Etinger, A.; Kumar, S.R.; Ackley, W.; Soiefer, L.; Chun, J.; Singh, P.; Grossman, E.; Matalon, A.; Holzman, R.; Meijers, B. The effect of isohydric hemodialysis on the binding and removal of uremic retention solutes. PLoS ONE 2018, 13, e0192770. [Google Scholar]
- Lee, S.; Sirich, T.L.; Meyer, T.W. Improving Clearance for Renal Replacement Therapy. Kidney360 2021, 10, 340670002922021. [Google Scholar] [CrossRef]
- Anders, H.-J.; Andersen, K.; Stecher, B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013, 83, 1010–1016. [Google Scholar] [CrossRef] [Green Version]
- Lano, G.; Burtey, S.; Sallée, M. Indoxyl Sulfate, a Uremic Endotheliotoxin. Toxins 2020, 12, 229. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, K.; Saigusa, D.; Kanemitsu, Y.; Matsumoto, Y.; Thanai, P.; Suzuki, N.; Mise, K.; Yamaguchi, H.; Nakamura, T.; Asaji, K.; et al. Gut microbiome-derived phenyl sulfate contributes to albuminuria in diabetic kidney disease. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Hung, S.; Kuo, K.; Wu, C.; Tarng, D. Indoxyl Sulfate: A Novel Cardiovascular Risk Factor in Chronic Kidney Disease. J. Am. Heart Assoc. 2017, 6, e005022. [Google Scholar] [CrossRef] [Green Version]
- Fujii, H.; Yonekura, Y.; Yamashita, Y.; Kono, K.; Nakai, K.; Goto, S.; Sugano, M.; Goto, S.; Fujieda, A.; Ito, Y.; et al. Anti-oxidative effect of AST-120 on kidney injury after myocardial infarction. Br. J. Pharmacol. 2016, 173, 1302–1313. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-J.; Wu, V.; Wu, P.-C.; Wu, C.-J. Meta-Analysis of the Associations of p-Cresyl Sulfate (PCS) and Indoxyl Sulfate (IS) with Cardiovascular Events and All-Cause Mortality in Patients with Chronic Renal Failure. PLoS ONE 2015, 10, e0132589. [Google Scholar] [CrossRef]
- Poesen, R.; Claes, K.; Evenepoel, P.; De Loor, H.; Augustijns, P.; Kuypers, D.; Meijers, B. Microbiota-Derived Phenylacetylglutamine Associates with Overall Mortality and Cardiovascular Disease in Patients with CKD. J. Am. Soc. Nephrol. 2016, 27, 3479–3487. [Google Scholar] [CrossRef] [Green Version]
- Villard, A.; Boursier, J.; Andriantsitohaina, R. Bacterial and eukaryotic extracellular vesicles and non-alcoholic fatty liver disease: New players in the gut-liver axis? Am. J. Physiol. Liver Physiol. 2021, 320, 485. [Google Scholar] [CrossRef]
- McIntyre, C.W.; Harrison, L.E.; Eldehni, M.T.; Jefferies, H.J.; Szeto, C.-C.; John, S.G.; Sigrist, M.K.; Burton, J.O.; Hothi, D.; Korsheed, S.; et al. Circulating Endotoxemia: A Novel Factor in Systemic Inflammation and Cardiovascular Disease in Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2010, 6, 133–141. [Google Scholar] [CrossRef]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef]
- Ortega, A.; Fernández, A.; Arenas, M.I.; López-Luna, P.; Muñóz-Moreno, C.; Arribas, I.; Olea, N.; García-Bermejo, L.; Lucio-Cazana, J.; Bosch, R.J. Outcome of Acute Renal Injury in Diabetic Mice with Experimental Endotoxemia: Role of Hypoxia-Inducible Factor-1α. J. Diabetes Res. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Nakano, D.; Kitada, K.; Wan, N.; Zhang, Y.; Wiig, H.; Wararat, K.; Yanagita, M.; Lee, S.; Jia, L.; Titze, J.M.; et al. Lipopolysaccharide induces filtrate leakage from renal tubular lumina into the interstitial space via a proximal tubular Toll-like receptor 4–dependent pathway and limits sensitivity to fluid therapy in mice. Kidney Int. 2020, 97, 904–912. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.K.; Masilamani, M.; Li, X.-M.; Sampson, H.A. The false alarm hypothesis: Food allergy is associated with high dietary advanced glycation end-products and proglycating dietary sugars that mimic alarmins. J. Allergy Clin. Immunol. 2017, 139, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Nakabayashi, I.; Nakamura, M.; Kawakami, K.; Ohta, T.; Kato, I.; Uchida, K.; Yoshida, M. Effects of synbiotic treatment on serum level of p-cresol in haemodialysis patients: A preliminary study. Nephrol. Dial. Transplant. 2011, 26, 1094–1098. [Google Scholar] [CrossRef] [Green Version]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. Association between Albuminuria, Kidney Function, and Inflammatory Biomarker Profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol. 2012, 7, 1938–1946. [Google Scholar] [CrossRef] [Green Version]
- Akchurin, O.M.; Kaskel, F. Update on Inflammation in Chronic Kidney Disease. Blood Purif. 2015, 39, 84–92. [Google Scholar] [CrossRef]
- Bellocq, A.; Suberville, S.; Philippe, C.; Bertrand, F.; Perez, J.; Fouqueray, B.; Cherqui, G.; Baud, L. Low Environmental pH Is Responsible for the Induction of Nitric-oxide Synthase in Macrophages. J. Biol. Chem. 1998, 273, 5086–5092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesias, P.; Díez, J.J. Adipose tissue in renal disease: Clinical significance and prognostic implications. Nephrol. Dial. Transplant. 2010, 25, 2066–2077. [Google Scholar] [CrossRef] [PubMed]
- Lustgarten, M.S. The Kidney–Gut–Muscle Axis in End-Stage Renal Disease is Similarly Represented in Older Adults. Nutrients 2019, 12, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaney, L.M.; Davies, O.G.; Selby, N.M. Gut microbial metabolites as mediators of renal disease: Do short-chain fatty acids offer some hope? Future Sci. 2019, 5, FSO384. [Google Scholar] [CrossRef] [Green Version]
- Rabb, H.; Griffin, M.; McKay, D.B.; Swaminathan, S.; Pickkers, P.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Inflammation in AKI: Current Understanding, Key Questions, and Knowledge Gaps. J. Am. Soc. Nephrol. 2015, 27, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Mishima, E.; Fukuda, S.; Mukawa, C.; Yuri, A.; Kanemitsu, Y.; Matsumoto, Y.; Akiyama, Y.; Fukuda, N.N.; Tsukamoto, H.; Asaji, K.; et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS–based metabolomics approach. Kidney Int. 2017, 92, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Andrade-Oliveira, V.; Amano, M.T.; Correa-Costa, M.; Castoldi, A.; Felizardo, R.J.; De Almeida, D.C.; Bassi, E.J.; Moraes-Vieira, P.M.; Hiyane, M.I.; Rodas, A.C.; et al. Gut Bacteria Products Prevent AKI Induced by Ischemia-Reperfusion. J. Am. Soc. Nephrol. 2015, 26, 1877–1888. [Google Scholar] [CrossRef]
- Lobel, L.; Cao, Y.G.; Fenn, K.; Glickman, J.N.; Garrett, W.S. Diet posttranslationally modifies the mouse gut microbial proteome to modulate renal function. Science 2020, 369, 1518–1524. [Google Scholar] [CrossRef]
- Snelson, M.; Tan, S.; Sourris, K.; Thallas-Bonke, V.; Ziemann, M.; El-Osta, S.; Cooper, M.; Coughlan, M. SAT-301 Resistant Starch Ameliorates Advanced Glycation Endproduct-Induced Gut Dysbiosis and Albuminuria in a Mouse Model of Type 2 Diabetes. Kidney Int. Rep. 2019, 4, S134. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Liu, S.-M.; Lau, W.L.; Khazaeli, M.; Nazertehrani, S.; Farzaneh, S.H.; Kieffer, D.A.; Adams, S.H.; Martin, R.J. High Amylose Resistant Starch Diet Ameliorates Oxidative Stress, Inflammation, and Progression of Chronic Kidney Disease. PLoS ONE 2014, 9, e114881. [Google Scholar] [CrossRef]
- Mishima, E.; Fukuda, S.; Shima, H.; Hirayama, A.; Akiyama, Y.; Takeuchi, Y.; Fukuda, N.N.; Suzuki, T.; Suzuki, C.; Yuri, A.; et al. Alteration of the Intestinal Environment by Lubiprostone Is Associated with Amelioration of Adenine-Induced CKD. J. Am. Soc. Nephrol. 2015, 26, 1787–1794. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.R.; Gandolfo, M.T.; Ko, G.J.; Satpute, S.; Racusen, L.; Rabb, H. Early exposure to germs modifies kidney damage and inflammation after experimental ischemia-reperfusion injury. Am. J. Physiol. Physiol. 2009, 297, F1457–F1465. [Google Scholar] [CrossRef] [Green Version]
- Emal, D.; Rampanelli, E.; Stroo, I.; Butter, L.M.; Teske, G.J.; Claessen, N.; Stokman, G.; Florquin, S.; Leemans, J.C.; Dessing, M.C. Depletion of Gut Microbiota Protects against Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2016, 28, 1450–1461. [Google Scholar] [CrossRef] [Green Version]
- Henning, C.; Glomb, M.A. Pathways of the Maillard reaction under physiological conditions. Glycoconj. J. 2016, 33, 499–512. [Google Scholar] [CrossRef]
- de Oliveira, F.C.; Coimbra, J.S.; Oliveira, E.B.; Zuniga, A.D.G.; Rojas, E.E.R. Food Protein-polysaccharide Conjugates Obtained via the Maillard Reaction: A Review. Crit. Rev. Food Sci. Nutr. 2016, 56, 1108–1125. [Google Scholar] [CrossRef]
- Bunn, H.F.; Briehl, R.W. The interaction of 2,3-diphosphoglycerate with various human hemoglobins. J. Clin. Investig. 1970, 49, 1088–1095. [Google Scholar] [CrossRef] [Green Version]
- Bunn, H.F.; Haney, D.N.; Kamin, S.; Gabbay, K.H.; Gallop, P.M. The biosynthesis of human hemoglobin A1c. Slow glycosylation of hemoglobin in vivo. J. Clin. Investig. 1976, 57, 1652–1659. [Google Scholar] [CrossRef] [Green Version]
- Verbeke, P.; Perichon, M.; Borot–Laloi, C.; Schaeverbeke, J.; Bakala, H. Accumulation of Advanced Glycation Endproducts in the Rat Nephron: Link with Circulating AGEs During Aging. J. Histochem. Cytochem. 1997, 45, 1059–1068. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-H.; Lin, X.; Bu, C.; Zhang, X. Role of advanced glycation end products in mobility and considerations in possible dietary and nutritional intervention strategies. Nutr. Metab. 2018, 15, 72. [Google Scholar] [CrossRef]
- Nagai, R.; Unno, Y.; Hayashi, M.C.; Masuda, S.; Hayase, F.; Kinae, N.; Horiuchi, S. Peroxynitrite induces formation of N( epsilon )-(carboxymethyl) lysine by the cleavage of Amadori product and generation of glucosone and glyoxal from glucose: Novel pathways for protein modification by peroxynitrite. Diabetes 2002, 51, 2833–2839. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Yamagishi, S.-I.; Yokoro, M.; Ito, S.; Kodama, G.; Kaida, Y.; Nakayama, Y.; Ando, R.; Yamada-Obara, N.; Asanuma, K.; et al. RAGE-aptamer attenuates deoxycorticosterone acetate/salt-induced renal injury in mice. Sci. Rep. 2018, 8, 2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, N.; Taguchi, K.; Miyazono, Y.; Uemura, K.; Koike, K.; Kurokawa, Y.; Nakayama, Y.; Kaida, Y.; Shibata, R.; Tsuchimoto, A.; et al. AGEs–RAGE overexpression in a patient with smoking-related idiopathic nodular glomerulosclerosis. CEN Case Rep. 2018, 7, 48–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanji, N.; Markowitz, G.S.; Fu, C.; Kislinger, T.; Taguchi, A.; Pischetsrieder, M.; Stern, D.; Schmidt, A.M.; D’Agati, V.D. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J. Am. Soc. Nephrol. 2000, 11, 1656–1666. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R.; Yamamoto, Y.; Nangaku, M.; Usuda, N.; Okamato, H.; Kurokawa, K.; Strihou, C.V.Y.D.; Yamamoto, H.; Miyata, T. A Severe Diabetic Nephropathy Model with Early Development of Nodule-Like Lesions Induced by Megsin Overexpression in RAGE/iNOS Transgenic Mice. Diabetes 2006, 55, 356–366. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, C.S.; Jeansson, M.; Quaggin, S.E. Vascular Growth Factors and Glomerular Disease. Annu. Rev. Physiol. 2016, 78, 437–461. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Higashimoto, Y.; Nishino, Y.; Nakamura, N.; Fukami, K.; Yamagishi, S.-I. RAGE-Aptamer Blocks the Development and Progression of Experimental Diabetic Nephropathy. Diabetes 2017, 66, 1683–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kierdorf, K.; Fritz, G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yao, T.; Zhou, Z.; Zhu, J.; Zhang, S.; Hu, W.; Shen, C. Advanced Glycation End Products Enhance Macrophages Polarization into M1 Phenotype through Activating RAGE/NF-κB Pathway. Biomed. Res. Int. 2015, 2015, 732450. [Google Scholar] [CrossRef] [Green Version]
- Akirav, E.M.; Preston-Hurlburt, P.; Garyu, J.; Henegariu, O.; Clynes, R.; Schmidt, A.M.; Herold, K.C. RAGE Expression in Human T Cells: A Link between Environmental Factors and Adaptive Immune Responses. PLoS ONE 2012, 7, e34698. [Google Scholar] [CrossRef]
- Avalos, A.M.; Kiefer, K.; Tian, J.; Christensen, S.; Shlomchik, M.; Coyle, A.J.; Marshak-Rothstein, A. RAGE-independent autoreactive B cell activation in response to chromatin and HMGB1/DNA immune complexes. Autoimmun. Rev. 2009, 43, 103–110. [Google Scholar] [CrossRef]
- Abdulahad, D.; Westra, J.; Limburg, P.; Kallenberg, C.; Bijl, M. HMGB1 in systemic lupus Erythematosus: Its role in cutaneous lesions development. Autoimmun. Rev. 2010, 9, 661–665. [Google Scholar] [CrossRef]
- Bucciarelli, L.G.; Wendt, T.; Rong, L.; Lalla, E.; Hofmann, M.A.; Goova, M.T.; Taguchi, A.; Yan, S.F.; Stern, D.M.; Schmidt, A.M. RAGE is a multiligand receptor of the immunoglobulin superfamily: Implications for homeostasis and chronic disease. Cell. Mol. Life Sci. 2002, 59, 1117–1128. [Google Scholar] [CrossRef]
- Teissier, T.; Boulanger, E. The receptor for advanced glycation end-products (RAGE) is an important pattern recognition receptor (PRR) for inflammaging. Biogerontology 2019, 20, 279–301. [Google Scholar] [CrossRef]
- Li, L.-M.; Hou, D.-X.; Guo, Y.-L.; Yang, J.-W.; Liu, Y.; Zhang, C.-Y.; Zen, K. Role of MicroRNA-214–Targeting Phosphatase and Tensin Homolog in Advanced Glycation End Product-Induced Apoptosis Delay in Monocytes. J. Immunol. 2011, 186, 2552–2560. [Google Scholar] [CrossRef]
- Weinhage, T.; Wirth, T.; Schütz, P.; Becker, P.; Lueken, A.; Skryabin, B.V.; Wittkowski, H.; Foell, D. The Receptor for Advanced Glycation Endproducts (RAGE) Contributes to Severe Inflammatory Liver Injury in Mice. Front. Immunol. 2020, 11, 1157. [Google Scholar] [CrossRef]
- Sadik, N.A.; Mohamed, W.A.; Ahmed, M.I. The association of receptor of advanced glycated end products and inflammatory mediators contributes to endothelial dysfunction in a prospective study of acute kidney injury patients with sepsis. Mol. Cell. Biochem. 2012, 359, 73–81. [Google Scholar] [CrossRef]
- Durning, S.P.; Preston-Hurlburt, P.; Clark, P.R.; Xu, D.; Herold, K.C.; Type 1 Diabetes TrialNet Study Group. The Receptor for Advanced Glycation Endproducts Drives T Cell Survival and Inflammation in Type 1 Diabetes Mellitus. J. Immunol. 2016, 197, 3076–3085. [Google Scholar] [CrossRef]
- Teissier, T.; Quersin, V.; Gnemmi, V.; Daroux, M.; Howsam, M.; Delguste, F.; Lemoine, C.; Fradin, C.; Schmidt, A.-M.; Cauffiez, C.; et al. Knockout of receptor for advanced glycation end-products attenuates age-related renal lesions. Aging Cell 2019, 18, e12850. [Google Scholar] [CrossRef]
- Hofmann, M.; Drury, S.; Hudson, B.; Gleason, M.R.; Qu, W.; Lu, Y.; Lalla, E.; Chitnis, S.; Monteiro, J.; Stickland, M.H.; et al. RAGE and arthritis: The G82S polymorphism amplifies the inflammatory response. Genes Immun. 2002, 3, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Avalos, A.M.; Mao, S.-Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.T.; Sirois, C.M.; et al. Toll-like receptor 9–dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef]
- Martens, H.; A Nienhuis, H.L.; Gross, S.; Van Der Steege, G.; Brouwer, E.; Berden, J.H.M.; De Sévaux, R.G.L.; Derksen, R.H.W.M.; E Voskuyl, A.; Berger, S.P.; et al. Receptor for advanced glycation end products (RAGE) polymorphisms are associated with systemic lupus erythematosus and disease severity in lupus nephritis. Lupus 2012, 21, 959–968. [Google Scholar] [CrossRef]
- Wiedeman, A.M.; Barr, S.I.; Green, T.J.; Xu, Z.; Innis, S.M.; Kitts, D.D. Dietary Choline Intake: Current State of Knowledge Across the Life Cycle. Nutrients 2018, 10, 1513. [Google Scholar] [CrossRef] [Green Version]
- Ueland, P.M. Choline and betaine in health and disease. J. Inherit. Metab. Dis. 2010, 34, 3–15. [Google Scholar] [CrossRef]
- Koc, H.; Mar, M.H.; Ranasinghe, A.; Swenberg, J.; Zeisel, S. Quantitation of choline and its metabolites in tissues and foods by liquid chromatography/electrospray ionization-isotope dilution mass spectrometry. Anal. Chem. 2002, 74, 4734–4740. [Google Scholar] [CrossRef]
- Rigault, C.; Mazué, F.; Bernard, A.; Demarquoy, J.; Le Borgne, F. Changes in l-carnitine content of fish and meat during domestic cooking. Meat Sci. 2008, 78, 331–335. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nat. Cell Biol. 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laxson, C.J.; Condon, N.E.; Drazen, J.; Yancey, P.H. Decreasing urearatiotrimethylamine N-oxide ratios with depth in chondrichthyes: A physiological depth limit? Physiol. Biochem. Zool. 2011, 84, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Evenepoel, P.; Poesen, R.; Meijers, B. The gut-kidney axis. Pediatr. Nephrol. 2017, 32, 2005–2014. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Gua, C.; Wu, B.; Chen, Y. Increased circulating trimethylamine N-oxide contributes to endothelial dysfunction in a rat model of chronic kidney disease. Biochem. Biophys. Res. Commun. 2018, 495, 2071–2077. [Google Scholar] [CrossRef]
- Bielinska, K.; Radkowski, M.; Grochowska, M.; Perlejewski, K.; Huc, T.; Jaworska, K.; Motooka, D.; Nakamura, S.; Ufnal, M. High salt intake increases plasma trimethylamine N-oxide (TMAO) concentration and produces gut dysbiosis in rats. Nutrition 2018, 54, 33–39. [Google Scholar] [CrossRef]
- Sun, G.; Yin, Z.; Liu, N.; Bian, X.; Yu, R.; Su, X.; Zhang, B.; Wang, Y. Gut microbial metabolite TMAO contributes to renal dysfunction in a mouse model of diet-induced obesity. Biochem. Biophys. Res. Commun. 2017, 493, 964–970. [Google Scholar] [CrossRef]
- Wu, K.; Yuan, Y.; Yu, H.; Dai, X.; Wang, S.; Sun, Z.; Wang, F.; Fei, H.; Lin, Q.; Jiang, H. The gut microbial metabolite trimethylamine N-oxide aggravates GVHD by inducing M1 macrophage polarization in mice. Blood 2020, 136, 501–515. [Google Scholar] [CrossRef]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Yang, C.; Wang, B.; Zhang, X.; Hu, T.; Gu, Y.; Li, J. Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. Biomed. Pharmacother. 2018, 97, 941–947. [Google Scholar] [CrossRef]
- Li, D.Y.; Wang, Z.; Jia, X.; Yan, D.; Shih, D.M.; Hazen, S.L.; Lusis, A.J.; Tang, W.W. Loop Diuretics Inhibit Renal Excretion of Trimethylamine N-Oxide. JACC: Basic Transl. Sci. 2021, 6, 103–115. [Google Scholar] [CrossRef]
- Bush, K.T.; Singh, P.; Nigam, S.K. Gut-derived uremic toxin handling in vivo requires OAT-mediated tubular secretion in chronic kidney disease. JCI Insight 2020, 5, e133817. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, T.; Cai, W.; Peppa, M.; Dardaine, V.; Baliga, B.S.; Uribarri, J.; Vlassara, H. Advanced glycoxidation end products in commonly consumed foods. J. Am. Diet. Assoc. 2004, 104, 1287–1291. [Google Scholar] [CrossRef]
- Lee, T.C.; Kimiagar, M.; Pintauro, S.J.; Chichester, C. Physiological and safety aspects of Maillard browning of foods. Prog. Food Nutr. Sci. 1981, 5, 243–256. [Google Scholar]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced Glycation End Products in Foods and a Practical Guide to Their Reduction in the Diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e12. [Google Scholar] [CrossRef] [Green Version]
- Koschinsky, T.; He, C.-J.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, K.; Vlassara, H. Orally absorbed reactive glycation products (glycotoxins): An environmental risk factor in diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1997, 94, 6474–6479. [Google Scholar] [CrossRef] [Green Version]
- Münch, G.; Gerlach, M.; Sian, J.; Wong, A.; Riederer, P. Advanced glycation end products in neurodegeneration: More than early markers of oxidative stress? Ann. Neurol. 1998, 44, S85–S88. [Google Scholar] [CrossRef] [PubMed]
- Mitome, J.; Yamamoto, H.; Saito, M.; Yokoyama, K.; Marumo, K.; Hosoya, T. Nonenzymatic Cross-Linking Pentosidine Increase in Bone Collagen and Are Associated with Disorders of Bone Mineralization in Dialysis Patients. Calcif. Tissue Int. 2011, 88, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, J.; Ueda, S.; Yamagishi, S.-I.; Nohara, N.; Nagasawa, H.; Wakabayashi, K.; Matsui, T.; Yuichiro, H.; Kadoguchi, T.; Otsuka, T.; et al. Association of advanced glycation end products with sarcopenia and frailty in chronic kidney disease. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Collotta, D.; Gaudioso, G.; Le Berre, M.; Cento, A.; Ferreira, G.A.; Chiazza, F.; Verta, R.; Bertocchi, I.; Manig, F.; et al. Effects of Exogenous Dietary Advanced Glycation End Products on the Cross-Talk Mechanisms Linking Microbiota to Metabolic Inflammation. Nutrients 2020, 12, 2497. [Google Scholar] [CrossRef]
- Qu, W.; Yuan, X.; Zhao, J.; Zhang, Y.; Hu, J.; Wang, J.; Li, J. Dietary advanced glycation end products modify gut microbial composition and partially increase colon permeability in rats. Mol. Nutr. Food Res. 2017, 61, 1700118. [Google Scholar] [CrossRef]
- Ueda, S.; Yamagishi, S.-I.; Takeuchi, M.; Kohno, K.; Shibata, R.; Matsumoto, Y.; Kaneyuki, U.; Fujimura, T.; Hayashida, A.; Okuda, S. Oral Adsorbent AST-120 Decreases Serum Levels of AGEs in Patients with Chronic Renal Failure. Mol. Med. 2006, 12, 180–184. [Google Scholar] [CrossRef]
- Vlassara, H.; Uribarri, J.; Cai, W.; Goodman, S.; Pyzik, R.; Post, J.; Grosjean, F.; Woodward, M.; Striker, G.E. Effects of Sevelamer on HbA1c, Inflammation, and Advanced Glycation End Products in Diabetic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2012, 7, 934–942. [Google Scholar] [CrossRef]
- Kakuta, T.; Tanaka, R.; Hyodo, T.; Suzuki, H.; Kanai, G.; Nagaoka, M.; Takahashi, H.; Hirawa, N.; Oogushi, Y.; Miyata, T.; et al. Effect of Sevelamer and Calcium-Based Phosphate Binders on Coronary Artery Calcification and Accumulation of Circulating Advanced Glycation End Products in Hemodialysis Patients. Am. J. Kidney Dis. 2011, 57, 422–431. [Google Scholar] [CrossRef]
- Snelson, M.; Coughlan, M.T. Dietary Advanced Glycation End Products: Digestion, Metabolism and Modulation of Gut Microbial Ecology. Nutrients 2019, 11, 215. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.M.; Milan, A.M.; Mitchell, C.J.; Gillies, N.A.; D’Souza, R.F.; Zeng, N.; Ramzan, F.; Sharma, P.; O’Knowles, S.; Roy, N.C.; et al. Protein Intake at Twice the RDA in Older Men Increases Circulatory Concentrations of the Microbiome Metabolite Trimethylamine-N-Oxide (TMAO). Nutrients 2019, 11, 2207. [Google Scholar] [CrossRef] [Green Version]
- Brinkley, T.; Semba, R.D.; Kritchevsky, S.B.; Houston, D.K. Dietary protein intake and circulating advanced glycation end product/receptor for advanced glycation end product concentrations in the Health, Aging, and Body Composition Study. Am. J. Clin. Nutr. 2020, 112, 1558–1565. [Google Scholar] [CrossRef]
- Pitt, B.; Kober, L.; Ponikowski, P.; Gheorghiade, M.; Filippatos, G.; Krum, H.; Nowack, C.; Kolkhof, P.; Kim, S.-Y.; Zannad, F. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: A randomized, double-blind trial. Eur. Heart J. 2013, 34, 2453–2463. [Google Scholar] [CrossRef]
- Tahara, A.; Tahara, N.; Yamagishi, S.-I.; Honda, A.; Igata, S.; Nitta, Y.; Bekki, M.; Nakamura, T.; Sugiyama, Y.; Sun, J.; et al. Ratio of serum levels of AGEs to soluble RAGE is correlated with trimethylamine-N-oxide in non-diabetic subjects. Int. J. Food Sci. Nutr. 2017, 68, 1013–1020. [Google Scholar] [CrossRef]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.G.; Abedin, J.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 2003, 370, 1097–1109. [Google Scholar] [CrossRef]
- Hurot, J.-M.; Cucherat, M.; Haugh, M.; Fouque, D. Effects of L-carnitine supplementation in maintenance hemodialysis patients: A systematic review. J. Am. Soc. Nephrol. 2002, 13, 708–714. [Google Scholar] [CrossRef]
- Adachi, T.; Fukami, K.; Yamagishi, S.-I.; Kaida, Y.; Ando, R.; Sakai, K.; Adachi, H.; Otsuka, A.; Ueda, S.; Sugi, K.; et al. Decreased serum carnitine is independently correlated with increased tissue accumulation levels of advanced glycation end products in haemodialysis patients. Nephrology 2012, 17, 689–694. [Google Scholar] [CrossRef]
- Fukami, K.; Yamagishi, S.; Sakai, K.; Kaida, Y.; Adachi, K.; Ando, R.; Okuda, S. Potential inhibitory effects of L-carnitine supplementation on tissue advanced glycation end products in patients with hemodialysis. Rejuvenation Res. 2013, 16, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Fukami, K.; Yamagishi, S.-I.; Sakai, K.; Kaida, Y.; Yokoro, M.; Ueda, S.; Wada, Y.; Takeuchi, M.; Shimizu, M.; Yamazaki, H.; et al. Oral L-Carnitine Supplementation Increases Trimethylamine-N-oxide but Reduces Markers of Vascular Injury in Hemodialysis Patients. J. Cardiovasc. Pharmacol. 2015, 65, 289–295. [Google Scholar] [CrossRef]


{kind=link}
| Authors [Reference] (Number of Participants) | Intervention | Outcome |
|---|---|---|
| Mitchell et al. [130] (n = 20) | High protein diet (1.6 g/kgBW/day) | Serum TMAO↑ |
| Brinkley et al. [131] (n = 2439) | High protein diet (≥1.2 g/kgBW/day) | Serum Nε-CML↑, Serum sRAGE↑ |
| Yacoub et al. [28] (n = 20) | Restriction of dietary AGEs intake | Serum Nε-CML↓, Serum methylglyoxal-derivatives↓ Prevotella copri↓, Bifidobacterium animalis↓ Alistipes indistinctus↑, Clostridium citroniae↑ |
| Adachi et al. [136] (n = 204) | Observational study in healthy subjects (n = 75) and HD patients (n = 129) | Clostridium hathewayi↑, Ruminococcus gauvreauii↑ Serum-free carnitine inversely correlates with skin AGEs |
| Tahara et al. [133] | Observational study in non-diabetic subjects | Clostridium hathewayi↑, Ruminococcus gauvreauii↑ Serum-free carnitine inversely correlates with skin AGEs |
| Fukami et al. [137] (n = 102, HD patients) | Oral L-carnitine supplementation (900 mg/d), six months | Skin AGEs↓ Serum-free carnitine inversely correlates with the decrease in skin AGEs |
| Fukami et al. [138] (n = 31, HD patients) | Oral L-carnitine supplementation (900 mg/d), six months | Vascular injury markers (sICAM-1, sVCAM-1)↓ Oxidative stress marker (MDA)↓ Serum AGE tends to be decreased TMA↑, TMAO↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taguchi, K.; Fukami, K.; Elias, B.C.; Brooks, C.R. Dysbiosis-Related Advanced Glycation Endproducts and Trimethylamine N-Oxide in Chronic Kidney Disease. Toxins 2021, 13, 361. https://doi.org/10.3390/toxins13050361
Taguchi K, Fukami K, Elias BC, Brooks CR. Dysbiosis-Related Advanced Glycation Endproducts and Trimethylamine N-Oxide in Chronic Kidney Disease. Toxins. 2021; 13(5):361. https://doi.org/10.3390/toxins13050361
Chicago/Turabian StyleTaguchi, Kensei, Kei Fukami, Bertha C. Elias, and Craig R. Brooks. 2021. "Dysbiosis-Related Advanced Glycation Endproducts and Trimethylamine N-Oxide in Chronic Kidney Disease" Toxins 13, no. 5: 361. https://doi.org/10.3390/toxins13050361
APA StyleTaguchi, K., Fukami, K., Elias, B. C., & Brooks, C. R. (2021). Dysbiosis-Related Advanced Glycation Endproducts and Trimethylamine N-Oxide in Chronic Kidney Disease. Toxins, 13(5), 361. https://doi.org/10.3390/toxins13050361