Characterization and Pharmacological Inhibition of the Pore-Forming Clostridioides difficile CDTb Toxin

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
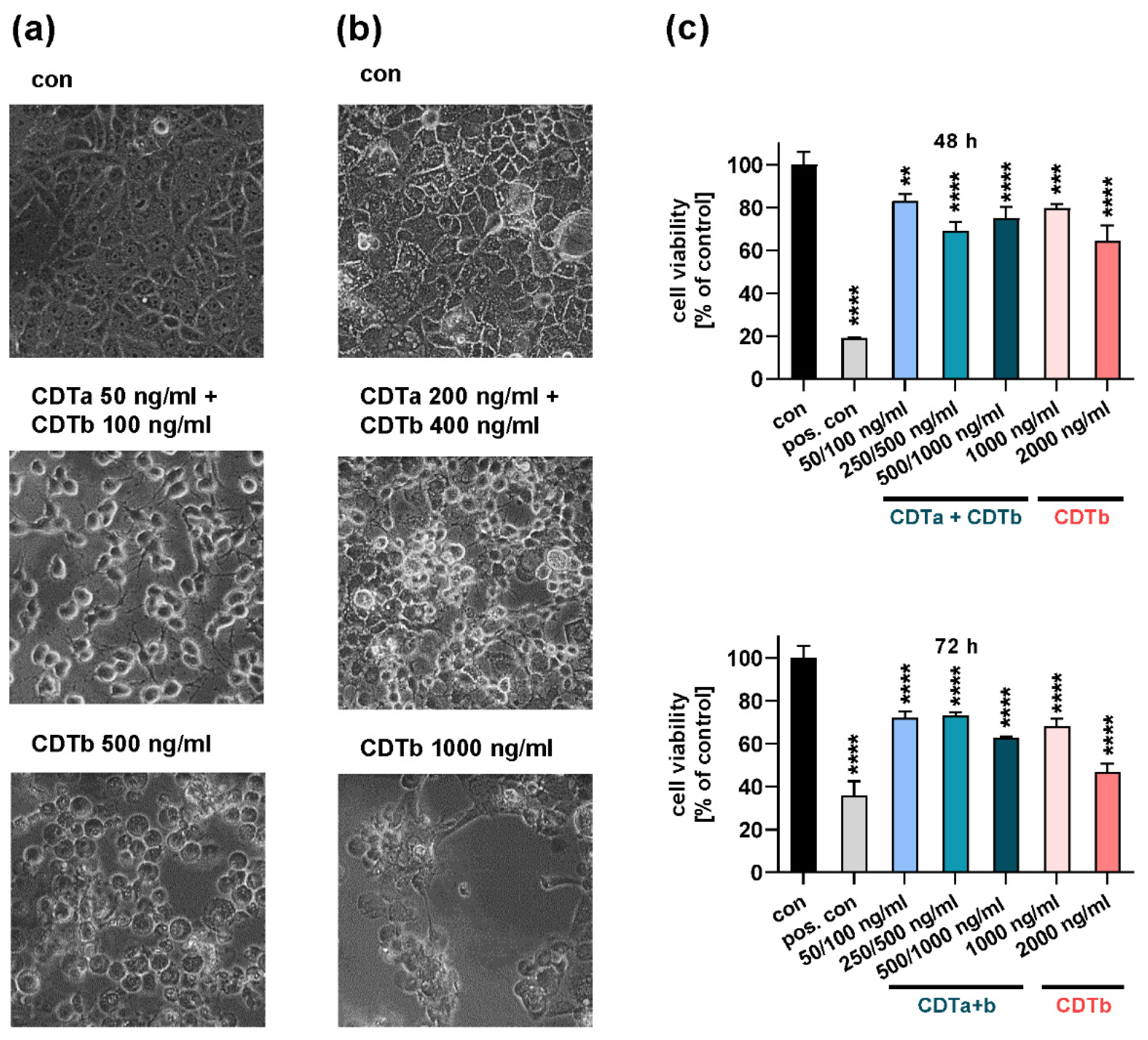
2.1. CDTb Impairs Cell Viability of CaCo-2 Cells
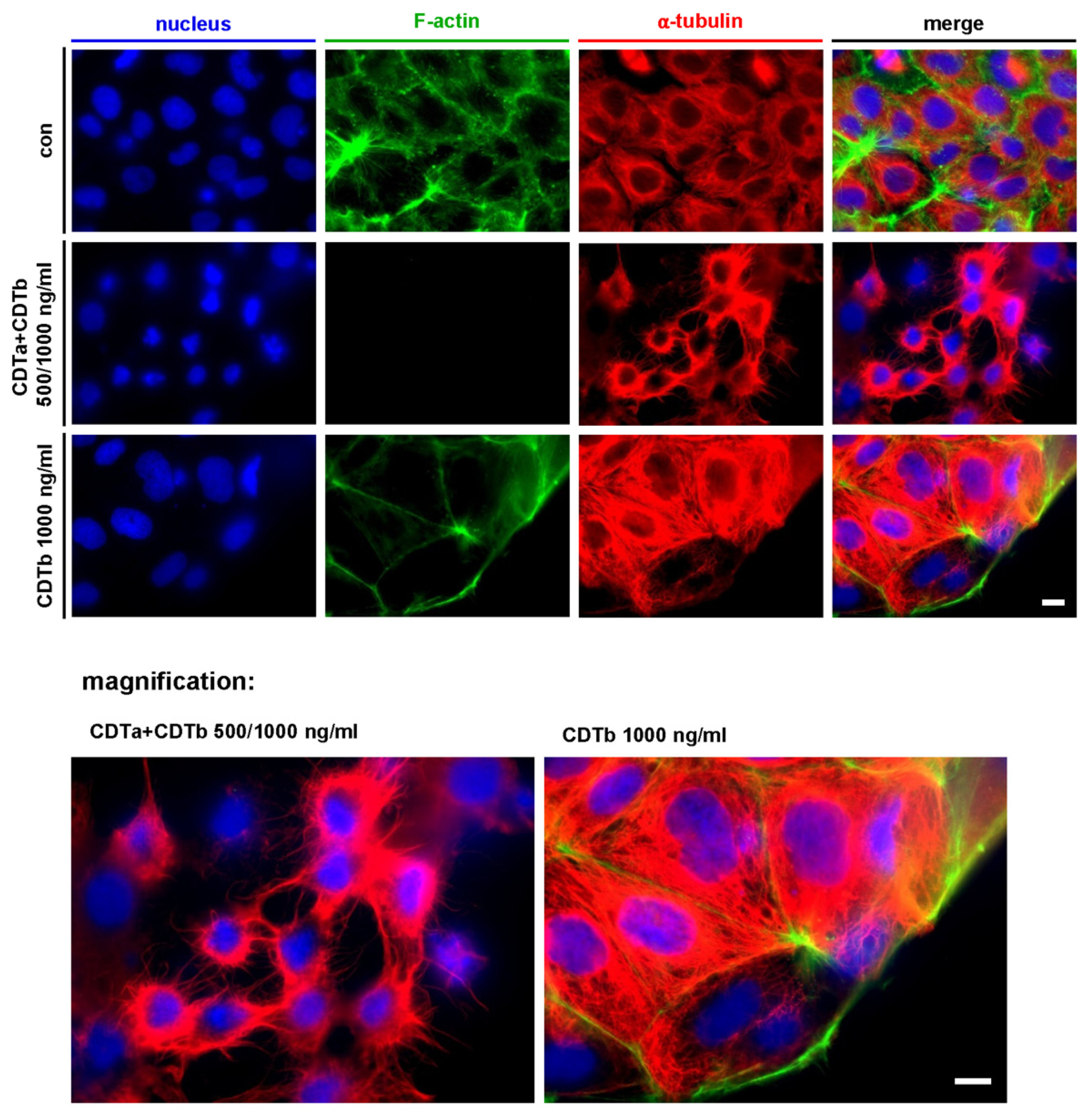
2.2. CDTb Leads to Redistribution of F-Actin in CaCo-2 cells
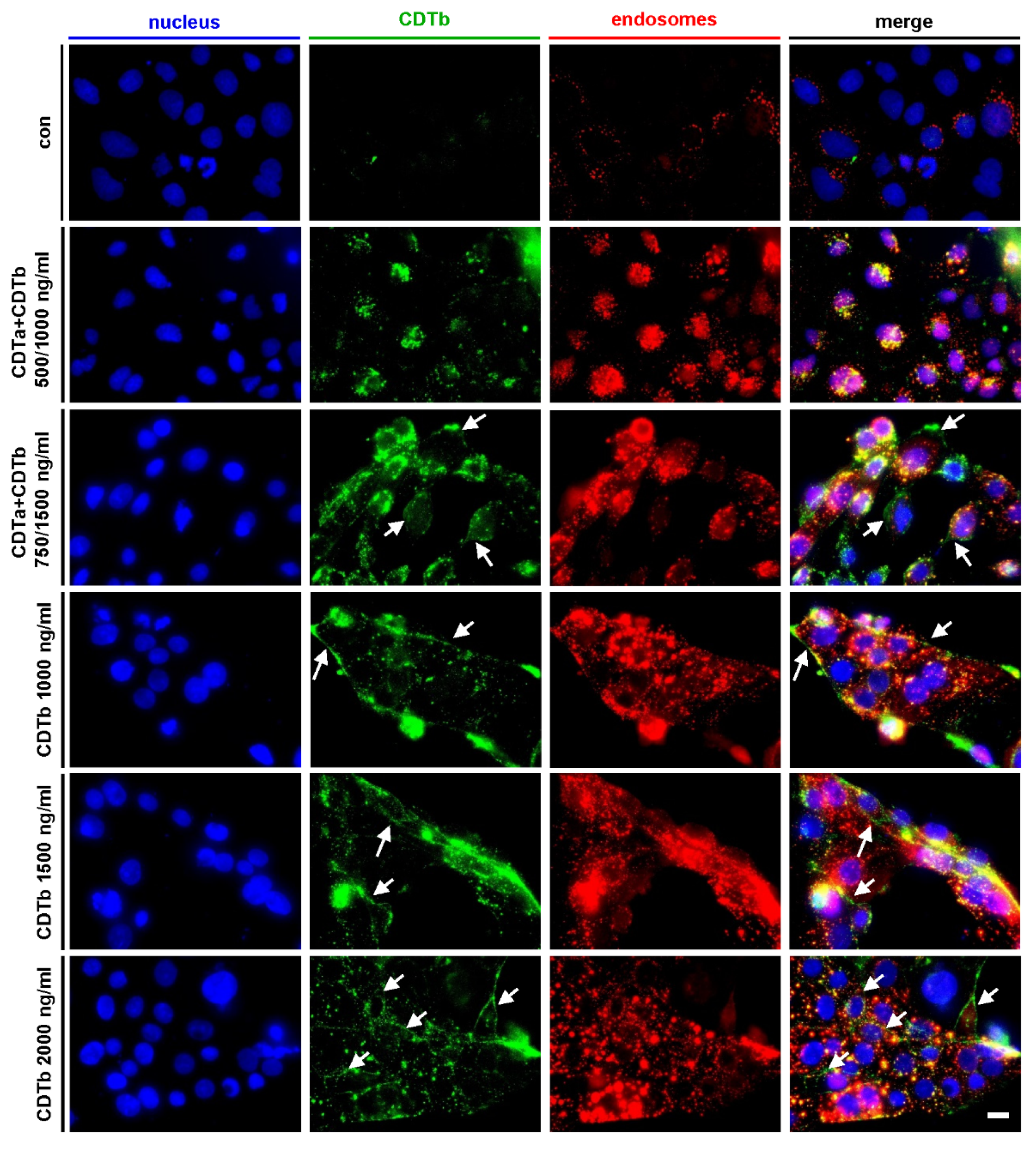
2.3. CDTb Is Partially Localized at the Cytoplasmic Membrane
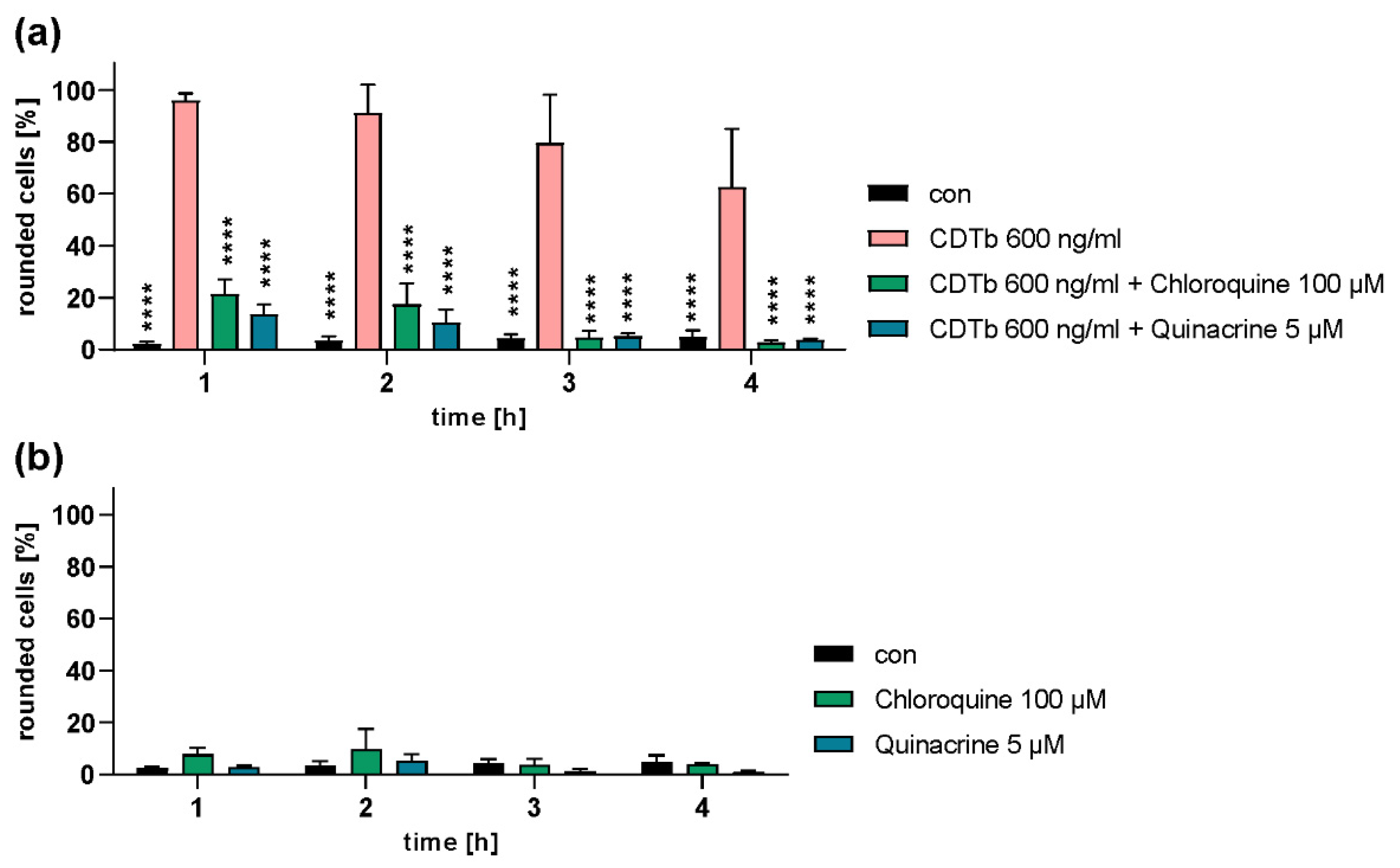
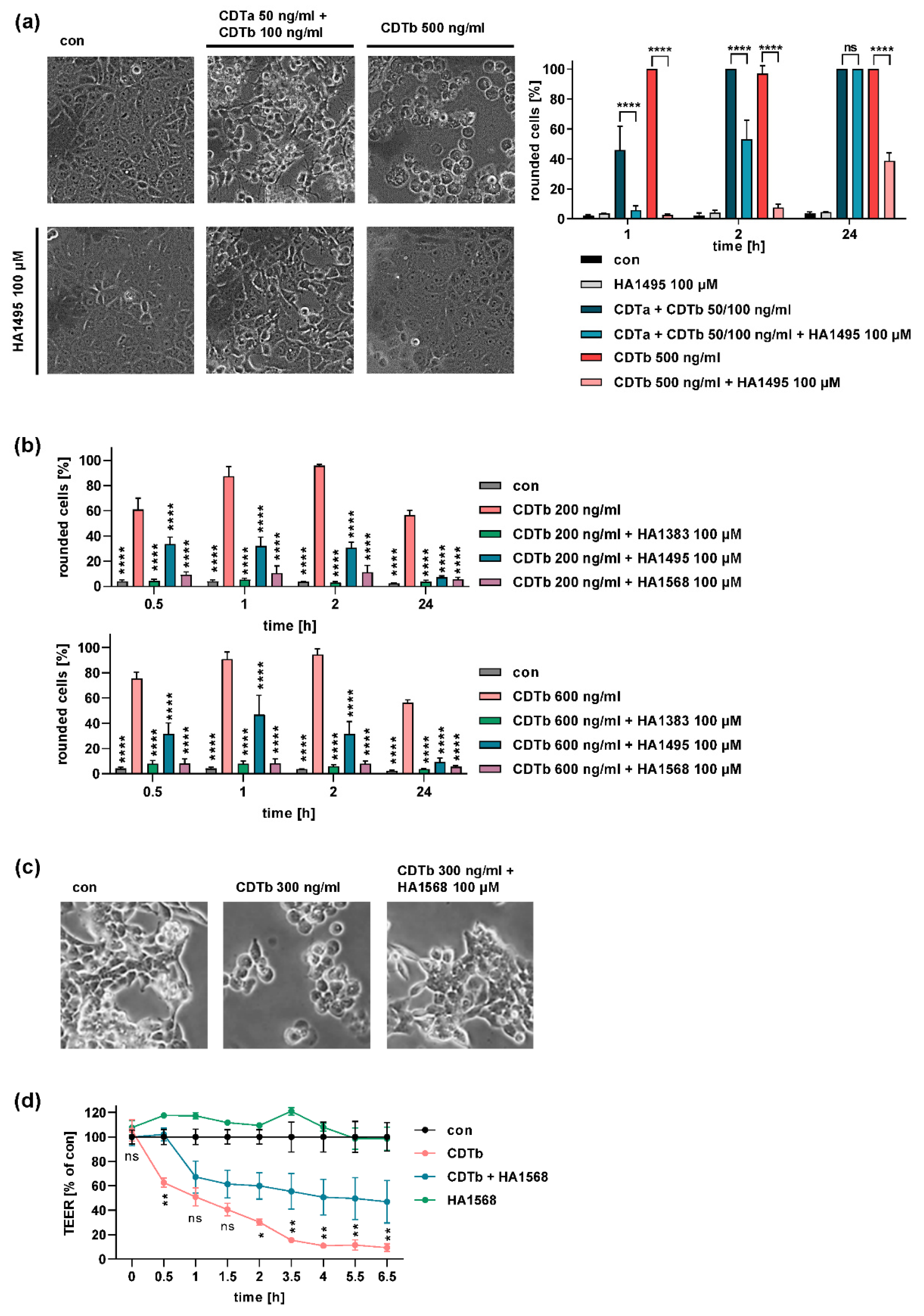
2.4. Chloroquine and Chloroquine Derivatives Protect Cells from CDTb Cytotoxicity
2.5. Chloroquine Blocks CDTb Pores In Vitro
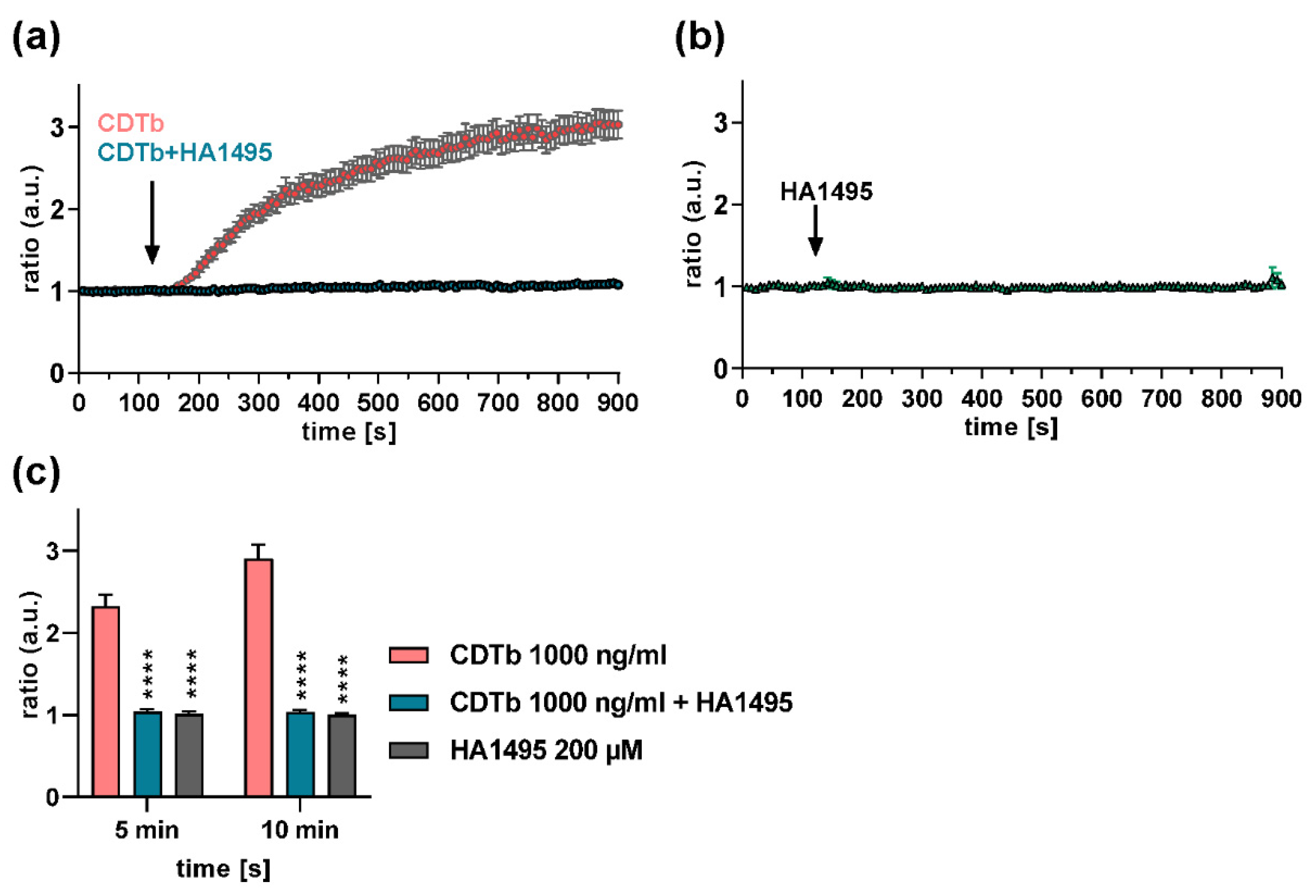
2.6. A Chloroquine Derivative Prevents Ca2+ Influx through CDTb Pores in Cells
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Cell Culture and Intoxication Experiments
4.3. Cell Viability
4.4. Fluorescence Microscopy
4.5. TEER Measurements
4.6. Lipid Bilayer Experiments
4.7. Calcium (Ca2+) Imaging
4.8. Reproducibility of Experiments and Statistics
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gerding, D.N.; Johnson, S.; Rupnik, M.; Aktories, K. Clostridium Difficile Binary Toxin CDT: Mechanism, Epidemiology, and Potential Clinical Importance. Gut Microbes 2014, 5, 15–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, S.; Gerding, D.N. Current and Future Trends in Clostridioides (Clostridium) Difficile Infection Management. Anaerobe 2019, 58, 95–102. [Google Scholar] [CrossRef]
- Kampouri, E.; Croxatto, A.; Prod’hom, G.; Guery, B. Clostridioides Difficile Infection, Still a Long Way to Go. J. Clin. Med. 2021, 10, 389. [Google Scholar] [CrossRef]
- Aktories, K.; Schwan, C.; Jank, T. Clostridium Difficile Toxin Biology. Annu. Rev. Microbiol. 2017, 71, 281–307. [Google Scholar] [CrossRef] [PubMed]
- Perelle, S.; Gibert, M.; Bourlioux, P.; Corthier, G.; Popoff, M.R. Production of a Complete Binary Toxin (Actin-Specific ADP-Ribosyltransferase) by Clostridium Difficile CD196. Infect. Immun. 1997, 65, 1402–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popoff, M.R.; Rubin, E.J.; Gill, D.M.; Boquet, P. Actin-Specific ADP-Ribosyltransferase Produced by a Clostridium Difficile Strain. Infect. Immun. 1988, 56, 2299–2306. [Google Scholar] [CrossRef] [Green Version]
- Barth, H. Uptake of Binary Actin ADP-Ribosylating Toxins. Rev. Physiol. Biochem. Pharmacol. 2004, 152, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, P.; Barth, H.; Minton, N.; Aktories, K. Cellular Uptake and Mode-of-Action of Clostridium Difficile Toxins. Adv. Exp. Med. Biol. 2018, 1050, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K.; Papatheodorou, P.; Schwan, C. Binary Clostridium Difficile Toxin (CDT)—A Virulence Factor Disturbing the Cytoskeleton. Anaerobe 2018, 53, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Ernst, K. Chaperones and ADP-Ribosylating Bacterial Toxins. In Microbial Toxins; Gopalakrishnakone, P., Stiles, B., Alape-Girón, A., Dubreuil, J.D., Mandal, M., Eds.; Toxinology; Springer: Dordrecht, The Netherlands, 2016; pp. 1–22. ISBN 978-94-007-6725-6. [Google Scholar]
- Ernst, K.; Schnell, L.; Barth, H. Host Cell Chaperones Hsp70/Hsp90 and Peptidyl-Prolyl Cis/Trans Isomerases Are Required for the Membrane Translocation of Bacterial ADP-Ribosylating Toxins. Curr. Top. Microbiol. Immunol. 2016, 406, 163–198. [Google Scholar] [CrossRef]
- Geric, B.; Carman, R.J.; Rupnik, M.; Genheimer, C.W.; Sambol, S.P.; Lyerly, D.M.; Gerding, D.N.; Johnson, S. Binary Toxin-Producing, Large Clostridial Toxin-Negative Clostridium Difficile Strains Are Enterotoxic but Do Not Cause Disease in Hamsters. J. Infect. Dis. 2006, 193, 1143–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehne, S.A.; Collery, M.M.; Kelly, M.L.; Cartman, S.T.; Cockayne, A.; Minton, N.P. Importance of Toxin A, Toxin B, and CDT in Virulence of an Epidemic Clostridium Difficile Strain. J. Infect. Dis 2014, 209, 83–86. [Google Scholar] [CrossRef]
- Wigelsworth, D.J.; Ruthel, G.; Schnell, L.; Herrlich, P.; Blonder, J.; Veenstra, T.D.; Carman, R.J.; Wilkins, T.D.; Van Nhieu, G.T.; Pauillac, S.; et al. CD44 Promotes Intoxication by the Clostridial Iota-Family Toxins. PLoS ONE 2012, 7, e51356. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, P.; Carette, J.E.; Bell, G.W.; Schwan, C.; Guttenberg, G.; Brummelkamp, T.R.; Aktories, K. Lipolysis-Stimulated Lipoprotein Receptor (LSR) Is the Host Receptor for the Binary Toxin Clostridium Difficile Transferase (CDT). Proc. Natl. Acad. Sci. USA 2011, 108, 16422–16427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papatheodorou, P.; Hornuss, D.; Nölke, T.; Hemmasi, S.; Castonguay, J.; Picchianti, M.; Aktories, K. Clostridium Difficile Binary Toxin CDT Induces Clustering of the Lipolysis-Stimulated Lipoprotein Receptor into Lipid Rafts. MBio 2013, 4, e00244-13. [Google Scholar] [CrossRef] [Green Version]
- Sheedlo, M.J.; Anderson, D.M.; Thomas, A.K.; Lacy, D.B. Structural Elucidation of the Clostridioides Difficile Transferase Toxin Reveals a Single-Site Binding Mode for the Enzyme. Proc. Natl. Acad. Sci. USA 2020, 117, 6139–6144. [Google Scholar] [CrossRef]
- Kaiser, E.; Kroll, C.; Ernst, K.; Schwan, C.; Popoff, M.; Fischer, G.; Buchner, J.; Aktories, K.; Barth, H. Membrane Translocation of Binary Actin-ADP-Ribosylating Toxins from Clostridium Difficile and Clostridium Perfringens Is Facilitated by Cyclophilin A and Hsp90. Infect. Immun. 2011, 79, 3913–3921. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, E.; Böhm, N.; Ernst, K.; Langer, S.; Schwan, C.; Aktories, K.; Popoff, M.; Fischer, G.; Barth, H. FK506-Binding Protein 51 Interacts with Clostridium Botulinum C2 Toxin and FK506 Inhibits Membrane Translocation of the Toxin in Mammalian Cells. Cell. Microbiol. 2012, 14, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Ernst, K.; Langer, S.; Kaiser, E.; Osseforth, C.; Michaelis, J.; Popoff, M.R.; Schwan, C.; Aktories, K.; Kahlert, V.; Malesevic, M.; et al. Cyclophilin-Facilitated Membrane Translocation as Pharmacological Target to Prevent Intoxication of Mammalian Cells by Binary Clostridial Actin ADP-Ribosylated Toxins. J. Mol. Biol. 2015, 427, 1224–1238. [Google Scholar] [CrossRef]
- Ernst, K.; Schmid, J.; Beck, M.; Hägele, M.; Hohwieler, M.; Hauff, P.; Ückert, A.K.; Anastasia, A.; Fauler, M.; Jank, T.; et al. Hsp70 Facilitates Trans-Membrane Transport of Bacterial ADP-Ribosylating Toxins into the Cytosol of Mammalian Cells. Sci. Rep. 2017, 7, 2724. [Google Scholar] [CrossRef]
- Gülke, I.; Pfeifer, G.; Liese, J.; Fritz, M.; Hofmann, F.; Aktories, K.; Barth, H. Characterization of the Enzymatic Component of the ADP-Ribosyltransferase Toxin CDTa from Clostridium Difficile. Infect. Immun. 2001, 69, 6004–6011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aktories, K.; Schwan, C.; Papatheodorou, P.; Lang, A.E. Bidirectional Attack on the Actin Cytoskeleton. Bacterial Protein Toxins Causing Polymerization or Depolymerization of Actin. Toxicon 2012, 60, 572–581. [Google Scholar] [CrossRef]
- Stiles, B.G.; Pradhan, K.; Fleming, J.M.; Samy, R.P.; Barth, H.; Popoff, M.R. Clostridium and Bacillus Binary Enterotoxins: Bad for the Bowels, and Eukaryotic Being. Toxins 2014, 6, 2626–2656. [Google Scholar] [CrossRef] [Green Version]
- Landenberger, M.; Nieland, J.; Roeder, M.; Nørgaard, K.; Papatheodorou, P.; Ernst, K.; Barth, H. The Cytotoxic Effect of Clostridioides Difficile Pore-Forming Toxin CDTb. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183603. [Google Scholar] [CrossRef]
- Fischer, S.; Ückert, A.-K.; Landenberger, M.; Papatheodorou, P.; Hoffmann-Richter, C.; Mittler, A.-K.; Ziener, U.; Hägele, M.; Schwan, C.; Müller, M.; et al. Human Peptide α-Defensin-1 Interferes with Clostridioides Difficile Toxins TcdA, TcdB, and CDT. FASEB J. 2020, 34, 6244–6261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbmacher, M.; Fischer, S.; Landenberger, M.; Papatheodorou, P.; Aktories, K.; Barth, H. Human α-Defensin-5 Efficiently Neutralizes Clostridioides Difficile Toxins TcdA, TcdB, and CDT. Front. Pharmacol. 2020, 11, 1204. [Google Scholar] [CrossRef] [PubMed]
- Kronhardt, A.; Schwan, C.; Maier, E.; Popoff, M.R.; Benz, R. Clostridium Difficile CDT Toxin Forms Two Different Types of Channels in Lipid Bilayer Membranes. BAOJ Chem. 2018, 3, 35. [Google Scholar]
- Anderson, D.M.; Sheedlo, M.J.; Jensen, J.L.; Lacy, D.B. Structural Insights into the Transition of Clostridioides Difficile Binary Toxin from Prepore to Pore. Nat. Microbiol. 2020, 5, 102–107. [Google Scholar] [CrossRef]
- Schwan, C.; Stecher, B.; Tzivelekidis, T.; van Ham, M.; Rohde, M.; Hardt, W.-D.; Wehland, J.; Aktories, K. Clostridium Difficile Toxin CDT Induces Formation of Microtubule-Based Protrusions and Increases Adherence of Bacteria. PLoS Pathog. 2009, 5, e1000626. [Google Scholar] [CrossRef] [Green Version]
- Schwan, C.; Aktories, K. Formation of Nanotube-Like Protrusions, Regulation of Septin Organization and Re-Guidance of Vesicle Traffic by Depolymerization of the Actin Cytoskeleton Induced by Binary Bacterial Protein Toxins. Curr. Top. Microbiol. Immunol. 2017, 399, 35–51. [Google Scholar] [CrossRef]
- Beitzinger, C.; Bronnhuber, A.; Duscha, K.; Riedl, Z.; Huber-Lang, M.; Benz, R.; Hajós, G.; Barth, H. Designed Azolopyridinium Salts Block Protective Antigen Pores In Vitro and Protect Cells from Anthrax Toxin. PLoS ONE 2013, 8, e66099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronnhuber, A.; Maier, E.; Riedl, Z.; Hajós, G.; Benz, R.; Barth, H. Inhibitions of the Translocation Pore of Clostridium Botulinum C2 Toxin by Tailored Azolopyridinium Salts Protects Human Cells from Intoxication. Toxicology 2014, 316, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Kreidler, A.-M.; Benz, R.; Barth, H. Chloroquine Derivatives Block the Translocation Pores and Inhibit Cellular Entry of Clostridium Botulinum C2 Toxin and Bacillus Anthracis Lethal Toxin. Arch. Toxicol. 2017, 91, 1431–1445. [Google Scholar] [CrossRef] [PubMed]
- Kronhardt, A.; Beitzinger, C.; Barth, H.; Benz, R. Chloroquine Analog Interaction with C2- and Iota-Toxin in Vitro and in Living Cells. Toxins 2016, 8, 237. [Google Scholar] [CrossRef] [Green Version]
- Bachmeyer, C.; Benz, R.; Barth, H.; Aktories, K.; Gilbert, M.; Popoff, M.R. Interaction of Clostridium Botulinum C2 Toxin with Lipid Bilayer Membranes and Vero Cells: Inhibition of Channel Function by Chloroquine and Related Compounds in Vitro and Intoxification in Vivo. FASEB J. 2001, 15, 1658–1660. [Google Scholar] [CrossRef]
- Bachmeyer, C.; Orlik, F.; Barth, H.; Aktories, K.; Benz, R. Mechanism of C2-Toxin Inhibition by Fluphenazine and Related Compounds: Investigation of Their Binding Kinetics to the C2II-Channel Using the Current Noise Analysis. J. Mol. Biol. 2003, 333, 527–540. [Google Scholar] [CrossRef]
- Schmid, A.; Benz, R.; Just, I.; Aktories, K. Interaction of Clostridium Botulinum C2 Toxin with Lipid Bilayer Membranes. Formation of Cation-Selective Channels and Inhibition of Channel Function by Chloroquine. J. Biol. Chem. 1994, 269, 16706–16711. [Google Scholar] [CrossRef]
- Blöcker, D.; Bachmeyer, C.; Benz, R.; Aktories, K.; Barth, H. Channel Formation by the Binding Component of Clostridium Botulinum C2 Toxin: Glutamate 307 of C2II Affects Channel Properties In Vitro and PH-Dependent C2I Translocation In Vivo. Biochemistry 2003, 42, 5368–5377. [Google Scholar] [CrossRef] [PubMed]
- Stiles, B.G. Clostridial Binary Toxins: Basic Understandings That Include Cell Surface Binding and an Internal “Coup de Grâce”. Curr. Top. Microbiol. Immunol. 2017, 406, 135–162. [Google Scholar] [CrossRef]
- Nagahama, M.; Umezaki, M.; Tashiro, R.; Oda, M.; Kobayashi, K.; Shibutani, M.; Takagishi, T.; Ishidoh, K.; Fukuda, M.; Sakurai, J. Intracellular Trafficking of Clostridium Perfringens Iota-Toxin b. Infect. Immun. 2012, 80, 3410–3416. [Google Scholar] [CrossRef] [Green Version]
- Nagahama, M.; Umezaki, M.; Oda, M.; Kobayashi, K.; Tone, S.; Suda, T.; Ishidoh, K.; Sakurai, J. Clostridium Perfringens Iota-Toxin b Induces Rapid Cell Necrosis. Infect. Immun. 2011, 79, 4353–4360. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.; Popoff, M.R.; Barth, H. Human Alpha-Defensin-1 Protects Cells from Intoxication with Clostridium Perfringens Iota Toxin. Pathog. Dis. 2018, 76. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Godoy-Ruiz, R.; Adipietro, K.A.; Peralta, C.; Ben-Hail, D.; Varney, K.M.; Cook, M.E.; Roth, B.M.; Wilder, P.T.; Cleveland, T.; et al. Structure of the Cell-Binding Component of the Clostridium Difficile Binary Toxin Reveals a Di-Heptamer Macromolecular Assembly. Proc. Natl. Acad. Sci. USA 2020, 117, 1049–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Cárdenas, S.; Torres-Martos, E.; Mora-Delgado, J.; Sánchez-Calvo, J.M.; Santos-Peña, M.; Zapata López, Á.; Dolores López-Prieto, M.; Pérez-Cortés, S.; Carlos Alados, J. The Prognostic Value of Toxin B and Binary Toxin in Clostridioides Difficile Infection. Gut Microbes 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Riedel, T.; Neumann-Schaal, M.; Wittmann, J.; Schober, I.; Hofmann, J.D.; Lu, C.-W.; Dannheim, A.; Zimmermann, O.; Lochner, M.; Groß, U.; et al. Characterization of Clostridioides Difficile DSM 101085 with A−B−CDT+ Phenotype from a Late Recurrent Colonization. Genome Biol. Evol. 2020, 12, 566–577. [Google Scholar] [CrossRef] [Green Version]
- Cowardin, C.A.; Buonomo, E.L.; Saleh, M.M.; Wilson, M.G.; Burgess, S.L.; Kuehne, S.A.; Schwan, C.; Eichhoff, A.M.; Koch-Nolte, F.; Lyras, D.; et al. The Binary Toxin CDT Enhances Clostridium Difficile Virulence by Suppressing Protective Colonic Eosinophilia. Nat. Microbiol. 2016, 1, 16108. [Google Scholar] [CrossRef] [Green Version]
- Eckert, C.; Emirian, A.; Le Monnier, A.; Cathala, L.; De Montclos, H.; Goret, J.; Berger, P.; Petit, A.; De Chevigny, A.; Jean-Pierre, H.; et al. Prevalence and Pathogenicity of Binary Toxin–Positive Clostridium Difficile Strains That Do Not Produce Toxins A and B. New Microbes New Infect. 2014, 3, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Abeyawardhane, D.L.; Godoy-Ruiz, R.; Adipietro, K.A.; Varney, K.M.; Rustandi, R.R.; Pozharski, E.; Weber, D.J. The Importance of Therapeutically Targeting the Binary Toxin from Clostridioides Difficile. Int. J. Mol. Sci. 2021, 22, 2926. [Google Scholar] [CrossRef]
- Papatheodorou, P.; Zamboglou, C.; Genisyuerek, S.; Guttenberg, G.; Aktories, K. Clostridial Glucosylating Toxins Enter Cells via Clathrin-Mediated Endocytosis. PLoS ONE 2010, 5, e10673. [Google Scholar] [CrossRef] [Green Version]
- Bahner, C.T.; Easley, W.K.; Pickens, M.D.; Lyons, H.D.; Norton, L.L.; Walden, B.G.; Biggerstaff, G.E. Quaternary Salts of Halogenated Pyridines and Quinolines1. J. Am. Chem. Soc. 1951, 73, 3499–3501. [Google Scholar] [CrossRef]
- Hajós, G.; Messmer, A. Ambident Reactivity of a Thiazolo[3,2-a]Pyridinium Salt with Nucleophiles. J. Heterocycl. Chem. 1984, 21, 809–811. [Google Scholar] [CrossRef]
- Timári, G.; Hajós, G.; Messmer, A. Synthesis, Alkylation and Ring Opening of Two Differently Fused Pyridoquinazolones. J. Heterocycl. Chem. 1990, 27, 2005–2009. [Google Scholar] [CrossRef]
- Palkó, R.; Riedl, Z.; Egyed, O.; Fábián, L.; Hajós, G. New Facile Tandem Route to Oxo- and Thioxo[1,2,4]Triazolo[1,5-a]Pyridinium Salts. J. Org. Chem. 2006, 71, 7805–7812. [Google Scholar] [CrossRef]
- Messmer, A.; Gelléri, A.; Hajós, G. Synthesis and Nitrogen Elimination of 3-Aryltetrazolo(1, 5-a)Pyridinium Salts and Its Angular Benzenologues: Formation of n-Arylamino-α-Pyridones. -Quinolones, -Isoquinolones, and Phenanthridones. Tetrahedron 1986, 42, 4827–4836. [Google Scholar] [CrossRef]
- Neumeyer, T.; Schiffler, B.; Maier, E.; Lang, A.E.; Aktories, K.; Benz, R. Clostridium Botulinum C2 Toxin. Identification of the Binding Site for Chloroquine and Related Compounds and Influence of the Binding Site on Properties of the C2II Channel. J. Biol. Chem. 2008, 283, 3904–3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlik, F.; Schiffler, B.; Benz, R. Anthrax Toxin Protective Antigen: Inhibition of Channel Function by Chloroquine and Related Compounds and Study of Binding Kinetics Using the Current Noise Analysis. Biophys. J. 2005, 88, 1715–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benz, R.; Schmid, A.; Vos-Scheperkeuter, G.H. Mechanism of Sugar Transport through the Sugar-Specific LamB Channel Of Escherichia Coli Outer Membrane. J. Membrain Biol. 1987, 100, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Kronhardt, A.; Rolando, M.; Beitzinger, C.; Stefani, C.; Leuber, M.; Flatau, G.; Popoff, M.R.; Benz, R.; Lemichez, E. Cross-Reactivity of Anthrax and C2 Toxin: Protective Antigen Promotes the Uptake of Botulinum C2I Toxin into Human Endothelial Cells. PLoS ONE 2011, 6, e23133. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ernst, K.; Landenberger, M.; Nieland, J.; Nørgaard, K.; Frick, M.; Fois, G.; Benz, R.; Barth, H. Characterization and Pharmacological Inhibition of the Pore-Forming Clostridioides difficile CDTb Toxin. Toxins 2021, 13, 390. https://doi.org/10.3390/toxins13060390
Ernst K, Landenberger M, Nieland J, Nørgaard K, Frick M, Fois G, Benz R, Barth H. Characterization and Pharmacological Inhibition of the Pore-Forming Clostridioides difficile CDTb Toxin. Toxins. 2021; 13(6):390. https://doi.org/10.3390/toxins13060390
Chicago/Turabian StyleErnst, Katharina, Marc Landenberger, Julian Nieland, Katharina Nørgaard, Manfred Frick, Giorgio Fois, Roland Benz, and Holger Barth. 2021. "Characterization and Pharmacological Inhibition of the Pore-Forming Clostridioides difficile CDTb Toxin" Toxins 13, no. 6: 390. https://doi.org/10.3390/toxins13060390
APA StyleErnst, K., Landenberger, M., Nieland, J., Nørgaard, K., Frick, M., Fois, G., Benz, R., & Barth, H. (2021). Characterization and Pharmacological Inhibition of the Pore-Forming Clostridioides difficile CDTb Toxin. Toxins, 13(6), 390. https://doi.org/10.3390/toxins13060390