1. Introduction
Pathogenic bacteria continuously produce bacterial toxins, which exert various toxic effects in human beings. Some of these toxins are complex multi-component structures such as the class of AB
5 toxins. These toxins are characterized by their structural composition, and as the name implies, such toxins consist of an A and B subunit. The A subunit, which is the catalytic subunit, and thus induces the toxic effect, can be split into two parts. The A1 subunit and the A2 subunit can be separated by a furin cleavage site, but the fragments subsequently remain linked via a disulfide bridge [
1]. The A1 fragment contains the catalytic domain whilst the A2 fragment acts as an anchor to the B subunit. The B subunit forms a stable pentameric ring, which acts as the receptor-binding domain and attaches to the cell surface. When the A subunit is cleaved at the serine–protease cleavage site, the alpha helical A2 fragment anchors the A1 fragment to the core of the pentameric B ring. The A1 fragment is subsequently internalized into the cell [
2,
3,
4].
Two of the best-characterized AB
5 toxins are the cholera toxin (Ctx) and the heat-labile enterotoxin (LT). Infections with bacterial strains secreting these toxins into the intestinal lumen can cause mild-to-severe disease progressions. Watery stool or even severe diarrhea, dehydration and vomiting are typical symptoms of such an infection [
4,
5,
6]. The B subunit of Ctx and LT generally target the monosialoganglioside GM1 receptor, which is expressed in jejunal epithelial cells [
7]. Further studies have later shown that the B subunit is cross-reactive and that glycoproteins on the cell surface are responsible for the binding which leads to toxicity in the absence of GM1 [
8,
9]. After binding to the cell surface, Ctx as well as LT catalyze the ADP-ribosylation of the heterotrimeric G protein, specifically the Gs alpha subunit. This eventually leads to the continuous activation of adenylate cyclase and production of cyclic adenosine monophosphate (cAMP) [
4,
10]. Thus, the infected intestinal cell secretes chloride and water, leading to typical symptoms of diarrhea. In vitro assays confirmed the activation of the adenylate cyclase and showed that Chinese hamster ovary (CHO) target cells were elongated after intoxication with Ctx and LT, suggesting morphological changes attributed to the increased cAMP level [
10,
11].
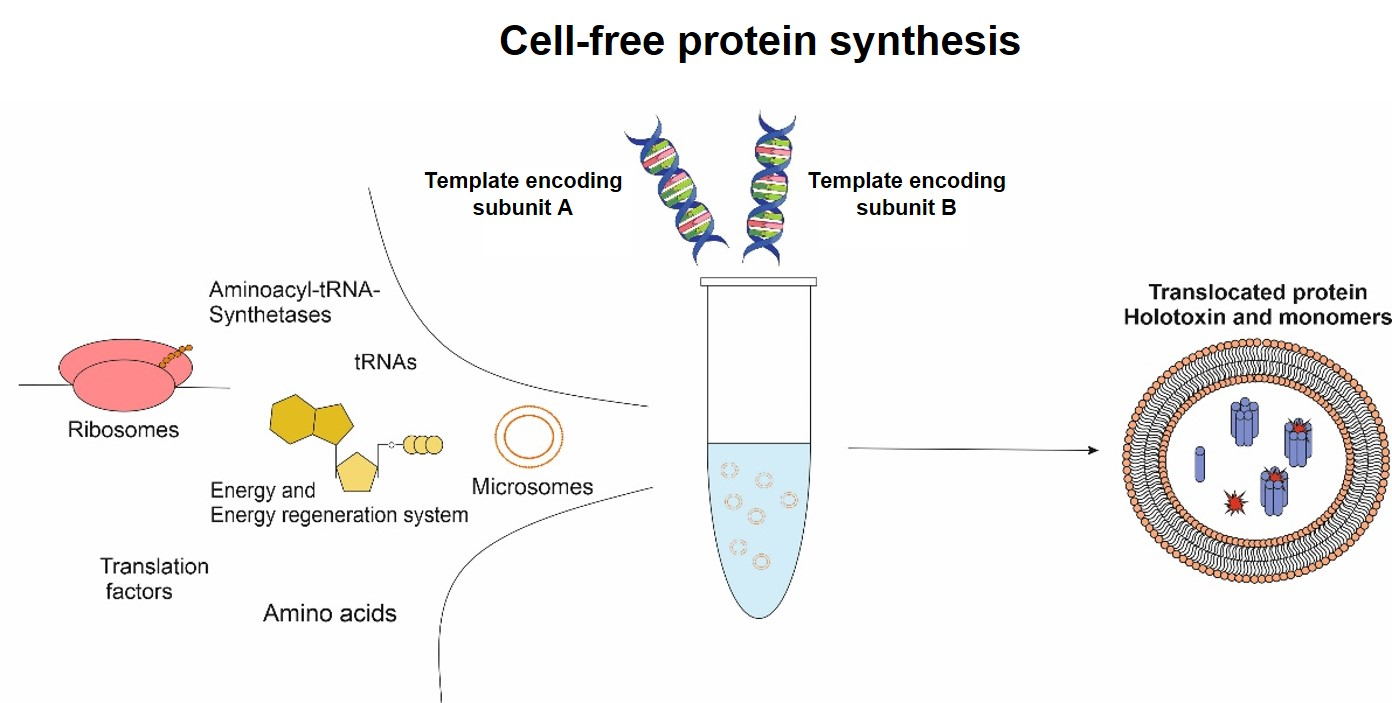
Toxins, especially multi-component toxins, are “difficult-to-express” proteins, but studying and understanding the mechanism of action is mandatory to counteract toxic effects. An alternative to conventional in vivo expression systems is cell-free protein synthesis (CFPS). As a cell lysate is used, the synthesis of a toxin typically does not harm the expression system itself [
12,
13]. This is important when using eukaryotic systems for synthesis, as many bacterial toxins target eukaryotic cells and their signaling systems [
4,
14]. Further, using a eukaryotic cell-free system allows for the direct application of the protein of interest for functionality assessments avoiding prior purification steps, as the eukaryotic cell-free system enables the endotoxin-free production of the protein of interest [
15]. The open system of CFPS enables the addition of factors affecting the functionality of the individual protein. Labeled amino acids can be added to the reaction mixture for quantitative and qualitative analysis. As the modification of toxins is of significance for studying the toxin’s characteristics, orthogonal translation systems can be applied. Such a system consists of an orthogonal synthetase, tRNA and a non-canonical amino acid (ncAA). The presence of an amber stop codon in the protein-encoding template and the subsequent implementation of the orthogonal systems allow for the fast and efficient modification of proteins via the incorporation of the individual ncAA which can further be labeled with fluorescent dyes [
16,
17]. Hence, cell-free protein synthesis is a qualified option to synthesize and characterize a wide variety of toxic proteins such as pore-forming and apoptosis inducing proteins [
18,
19,
20]. The synthesis of AB
5 toxins has generally been performed in prokaryotic cell-based systems such as via overexpression in
Escherichia coli (
E. coli) [
1,
10,
21]. In this study, eukaryotic cell-free systems and orthogonal labeling systems were used to produce, modify and characterize the two AB
5 toxins Ctx and LT. Two well established cell-free systems, namely the mammalian CHO-derived and the insect-
Sf21-derived lysates, were used. It was further shown that CFPS could be used to develop a Trojan horse application as the LTB subunit was fused to Streptavidin (Strep) in silico, and after the synthesis of the fusion protein it was labeled with a biotin-conjugated fluorescent dye. Finally, the CtxA subunit was modified with an amber stop codon and ncAAs were incorporated using orthogonal systems. Subsequently, the subunit was fluorescently labeled using copper catalyzed click chemistry and a Staudinger ligation. Such mutational analysis studies and the combination with amber suppression and fluorescent labeling qualifies CFPS for the development of targeted toxins and for studying the intracellular trafficking of toxins, thereby identifying their mechanism of action. This is the first study using eukaryotic cell-free systems for studying and modifying Ctx and LT, therefore facilitating CFPS as a platform technology for analyzing the pathways of toxins such as AB
5 toxins.
3. Discussion
A major group of toxins expressed by pathogenic bacteria is the group of AB
5 toxins. This group of toxins is rather diverse and was therefore subcategorized into different families. After the detection of the novel AB
5 toxin subtilase, a new family was announced in the early 2000s [
25], showing that there is an increasing demand for stable and easy-to-handle production systems for the synthesis and characterization of active AB
5 toxins. The two best-characterized and most-well-known representatives of this group are Ctx and LT. Our study aimed to synthesize, characterize and modify Ctx and LT in eukaryotic cell-free systems. In prior studies, Ctx and LT were isolated from prokaryotic cell-based systems [
1,
10,
21]. Prokaryotic cell-free systems generally lack the ability to form PTMs such as disulfide bridges. The addition of supplements, such as a redox system, is necessary to functionally synthesize active proteins containing selected PTMs. As bacterial strains can further encode endotoxins, proteins expressed in prokaryotic systems must be purified for further analyses. A eukaryotic cell-free system circumvents these drawbacks [
13].
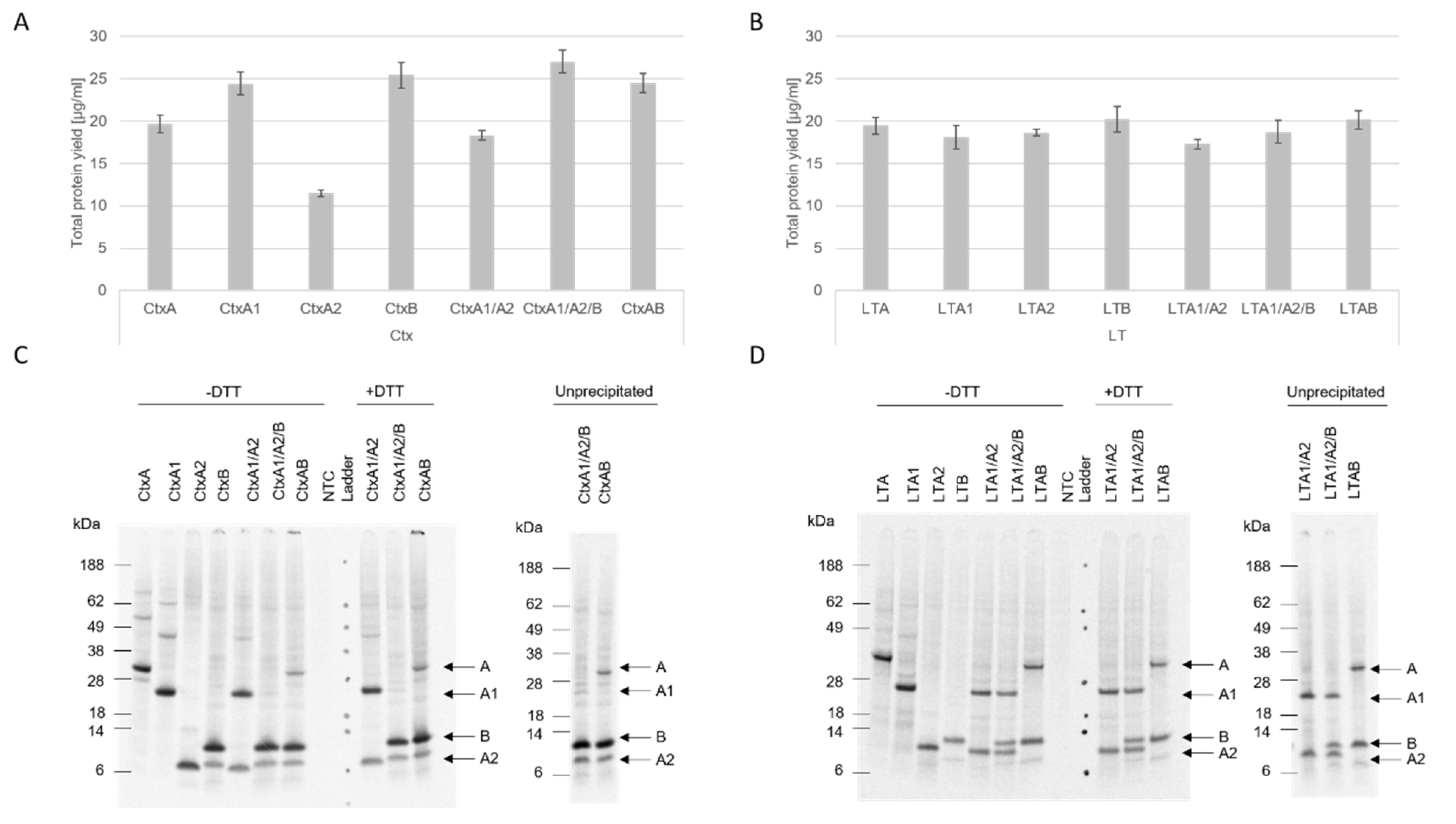
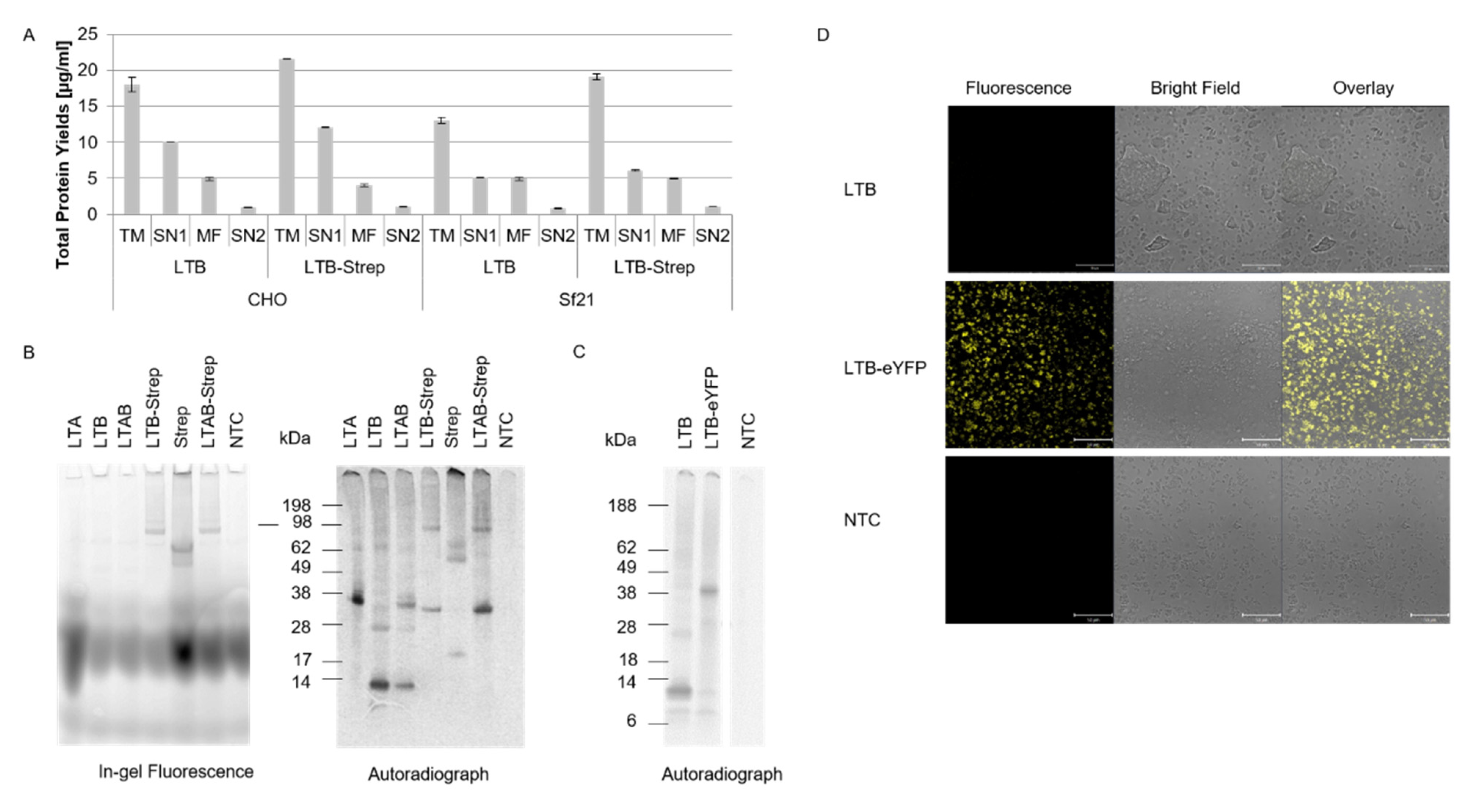
Our data showed that both eukaryotic systems, CHO and
Sf21, resulted in reproducible total protein yields and functional activity. Nonetheless, higher protein yields were detected in the CHO system. In prior work, we demonstrated that the CHO system can be used as a high-yield system [
12], thus our data add to prior findings that different toxin subunits with diverse molecular weights can be efficiently synthesized in eukaryotic systems, especially the CHO system. Strikingly, the CtxA2 subunit resulted in the lowest protein yields in both CHO and
Sf21, and the LTA2 subunit showed similarly low yields in the
Sf21 system. These data indicate that the A2 subunit is difficult to express in comparison to the other subunits, which might be caused by its small size. Other studies generally expressed A2 chimeras with other toxin compounds and investigated the holotoxin formation [
23,
26], thus data on the synthesis of A2 itself are rather limited and future studies should investigate the single synthesis of A2 fragments.
The structure of AB
5 toxins is characteristic for this class of toxins. The A subunit is cleaved into the A1 and A2 fragment which are subsequently linked together via a disulfide bridge [
1]. Further, the A2 fragment non-covalently links the A subunit to the stable pentameric ring [
4]. The data presented here indicate no holotoxin formation after an SDS-PAGE. Few multimerizations of the single subunits were detected. Jobling et al. showed a clear holotoxin formation of the AB
5 multimer [
27]. As these proteins were purified from
E. coli strains, a higher total protein yield might have been loaded onto the gel. Prior studies have also shown that CtxB and LTB form two types of multimers with either a native or a non-native conformation. The non-native multimers appear to occur at acidic pH values and form different secondary conformations as compared to native multimers. While both types can bind to the receptor, the native subunit multimers are SDS-resistant, but SDS degrades the non-native multimers [
22,
28]. Additionally, Zrimi and colleagues identified that 35 µM of CtxB only showed a pentameric state, while a concentration of 8.6 µM led to monomeric states [
22]. As shown in this study, slight multimeric structures in the CtxB subunit were detected, suggesting that a higher protein yield might be needed to visualize the pentameric state. In the case of LTB, no multimerizations were detectable in the autoradiograph. Either LTB only formed non-native multimers, which cleaved to the monomeric state in the SDS-PAGE, or the concentration of the formed multimers was not yet sufficient to be detected by autoradiography.
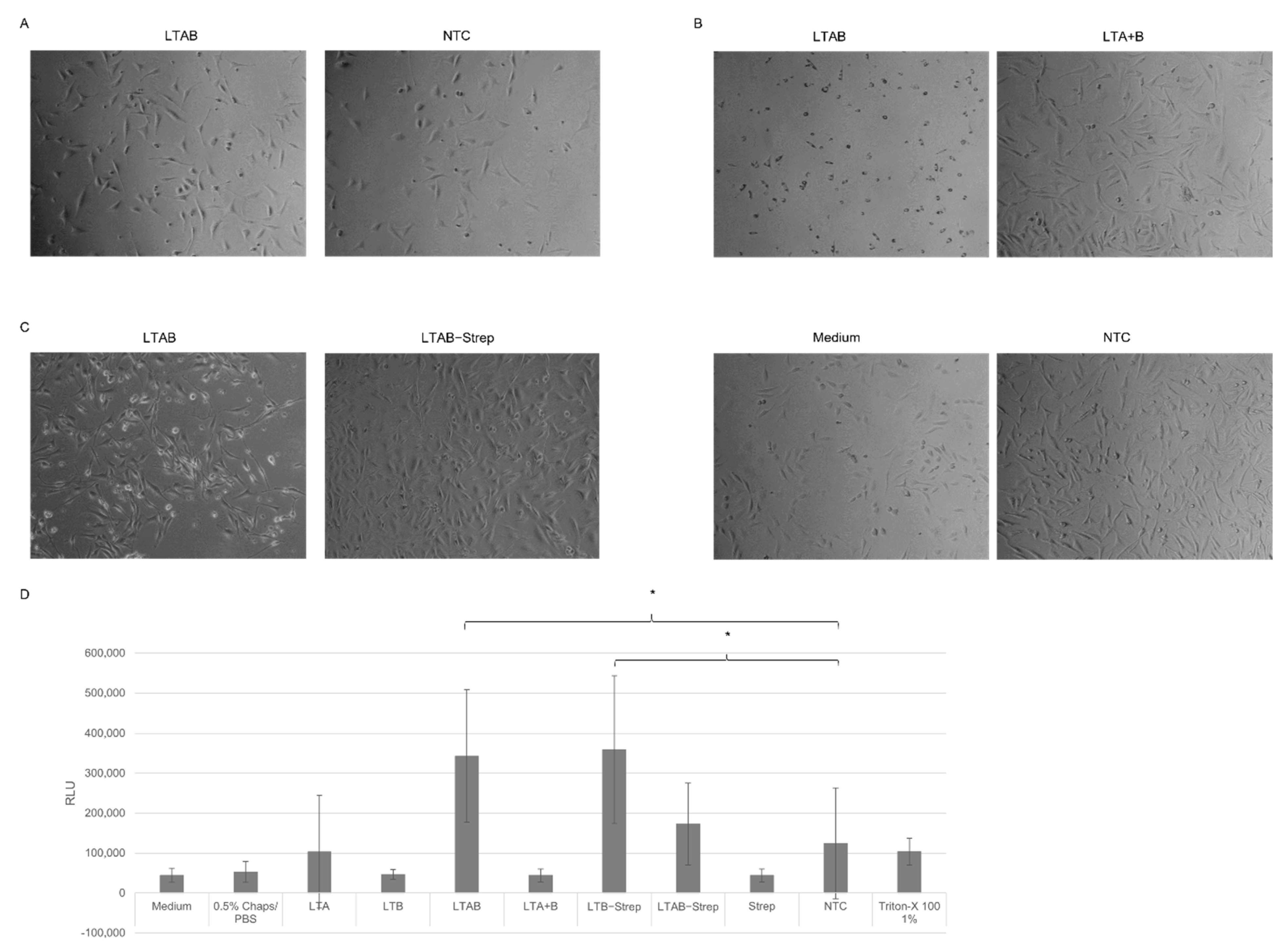
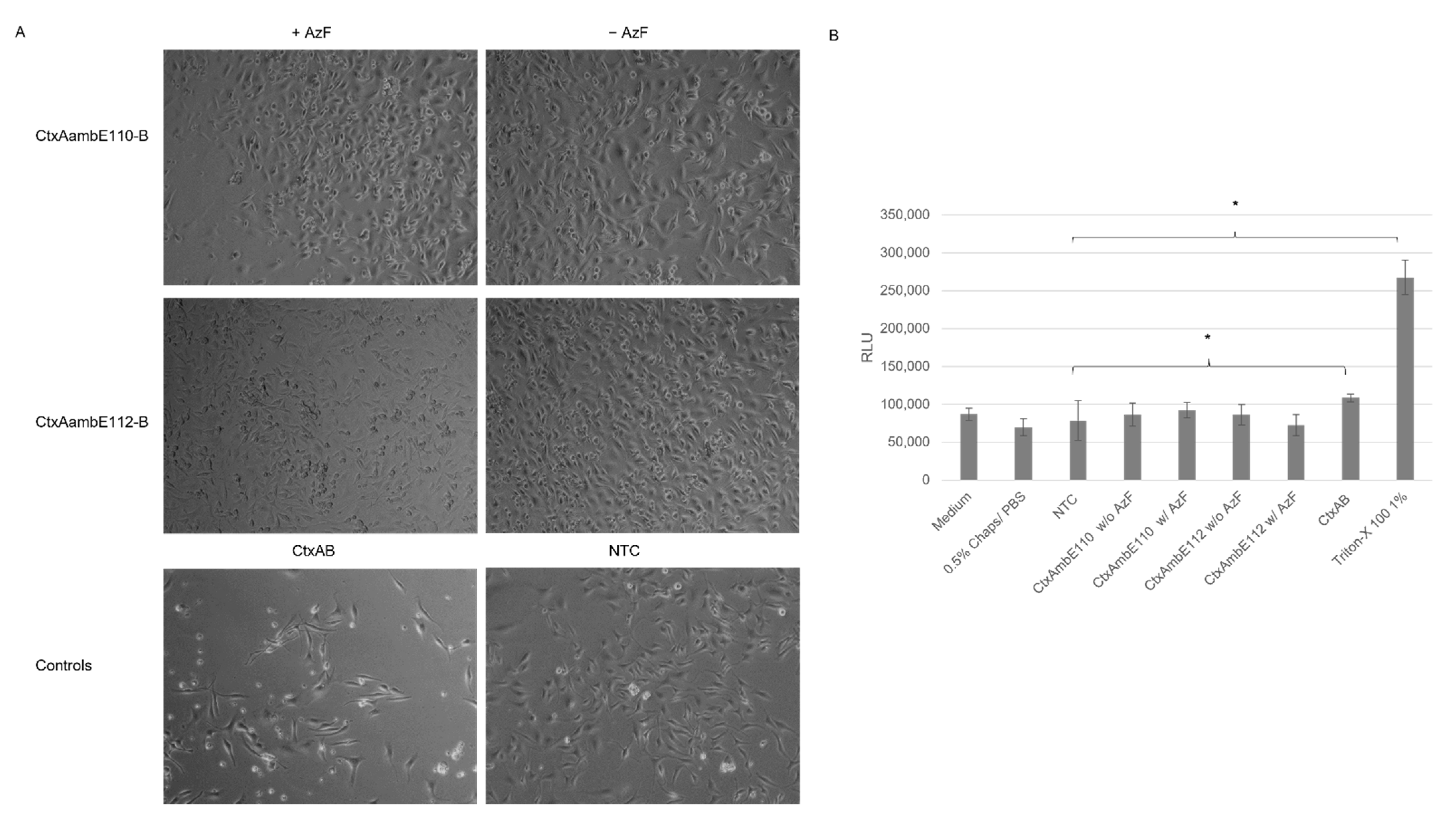
The cell-based assays showed that cell-free synthesized AB
5 toxins induced characteristic morphological elongation of cells at lower concentrations after 24 h, which is in line with previous findings, where elongated cells were detected after 4 h and 24 h of incubation [
10,
11]. After 48 h, Ctx and LT induced detached cells and morphological changes suggested non-viable cells. A cytotoxicity assay was performed in order to quantify these changes. Strikingly, LTB-Strep and the individual Ctx subunits demonstrated high background values. These background signals could derive from unspecific interactions with the lysate as well as mRNA present in the sample. Different background values for the individual lysates could be detected suggesting the possibility of purification steps for this particular cytotoxicity assay in order to specifically compare the individual experiments. Nonetheless, the cytotoxicity data and cell micrographs indicate that functional AB
5 complexes can be synthesized using cell-free protein synthesis. Complex toxin structures were active after cell-free co-expression of the subunits but not when mixing the subunits after their individual syntheses. This could indicate the importance of the formation of multimeric complexes by disulfide bridges and stable bonds for functionally active proteins. Prior work has shown that CHO and
Sf21 lysates can be used to synthesize proteins that include disulfide bridges [
29,
30]. Thus, the formation of disulfide bonds in the AB
5 complex of cell-free synthesized Ctx and LT could have led to functional proteins when co-expressing the subunits. When the individual subunits were mixed together following their individual syntheses, the complex formation seemed to be inefficient and did not lead to functional complexes. In addition to this, prior studies have also shown that free CtxA and CtxB subunits do not assemble to complete holotoxins [
31]. If we assume this is also true for LT, our data add new information to the assembly of LT and clearly align with the findings for Ctx. Ctx is supposed to be the more potent toxin when comparing Ctx and LT, as Ctx forms more stable holotoxins [
23,
24,
32]. Nonetheless, the pentameric B ring of LT was shown to be more resistant to external factors [
22]. Despite these findings, no differences in the activities of WT holotoxins could be observed from the cell-free, synthesized holotoxins generated in this study.
The modified toxins used in this study did not show the same activity as WT proteins. When LTB was fused to Strep, the activity was decreased, which suggests that the holotoxin formation was not as stable as with WT LTB. As LTA is already less potent than CtxA [
23,
24,
32], the modified LTB subunit might have led to a further decrease in activity in comparison to the WT construct. The fusion of the Strep molecule to the C-terminus of the LTB might have led to a reduced multimerization of the B subunit. The mutated CtxA subunits showed no effect on CHO-K1 cells. As both mutations were situated within the active center of the fragment, this outcome was expected. Prior studies have already investigated this active core and mutated the same or similar positions. Position E112 has been widely investigated as a non-toxic mutant for adjuvant applications and a mutation at E112 was shown to lead to a reduced toxicity [
33,
34]. Aligning with these previous studies, our findings demonstrated that a single mutation within the active center disrupts the toxins functionality. Silencing a toxin with a point mutation in cell-free systems will be beneficial for future studies. This will allow for the defined analysis of the toxins pathway within the targeted cell.
In order to inhibit the toxic effect of a pathogen, the mechanism of action of the underlying toxin has to be fully understood. Tracking the toxin within the cell enables the clarification of the pathogenesis of diverse novel toxins. Few studies attempted to fluorescently label toxins. The labeling of proteins by generating a fusion protein with a fluorescent protein such as YFP leads to enlarged proteins, which might affect the activity of the protein. As shown in our study, the 12 kDa LTB is significantly smaller than eYFP. The LTB-eYFP fusion protein could still be translocated into the microsomal vesicles, indicating that larger fusion proteins might be a powerful analytical tool. Another study could even track the C-terminal fragment of tetanus toxin within the cell after it was fused to the green fluorescent protein [
35]. A further idea to label a toxin fragment is to fuse the protein to streptavidin and, subsequently, couple a biotin-conjugated fluorophore to the protein. Such couplings of streptavidin–biotin are very strong and do not require the addition of further chemicals. Studies have even shown that cell-staining and antigen-labeling is possible within such systems [
36]. All possible fluorophores and payloads that are linked to biotin can be “clicked” to the Strep modified construct, allowing for the diverse modifications of a single protein. The modified LTB-Strep construct in our study could be labeled with a biotin-conjugated fluorophore and was further detected by in-gel fluorescence. Thus, in future experiments, the fluorescent dye can easily be exchanged by a toxic moiety coupled to the B subunit. Under these conditions, the B subunit may further act as a Trojan horse, targeting the cells, and inducing a specific effect by the coupled moiety.
A major disadvantage of these systems is that the label has to be fused to the protein of interest either at the N- or at the C-terminus. This might especially be a drawback in multicomponent proteins such as AB
5 toxins as the assembly of the complex might be hindered. Nonetheless, such fusion proteins were also tested for Ctx and LT, and it was shown that the labeled B subunits assembled and attacked the cells [
37,
38], suggesting that larger fusion proteins were possible. An alternative to such fusion proteins are site-specifically labeled proteins. In 2020, a study showed the incorporation of ncAAs into microcystins and the subsequent labeling of these constructs with copper-catalyzed click chemistry in living cells. Autofluorescence signals and unspecific labeling were detected but could be distinguished from specifically labeled peptides [
39].
These data show that bio-orthogonal systems can be used for studying intracellular trafficking. The data presented in our study add to these findings, as a site-specific fluorescent label could be added to the CtxA subunit by using an amber stop codon and an orthogonal system. As indicated in
Figure 5C, the full-length protein of the CtxA subunit harboring the incorporated ncAAs showed a reduced protein band in the autoradiograph as compared to the WT CtxA subunit. Nonetheless, the data acquired here show that small reaction volumes were sufficient for studying the toxicity of the different proteins. As CFPS is a scalable technology, small reaction volumes can be applied for parallel screenings, such as toxic mutants. The upscaling of cell-free reactions to mL or liter batches was shown in prior studies, thus facilitating CFPS for further down-stream processes [
40,
41].
Overall, the data presented in this study show that CFPS represents a versatile platform for the synthesis, characterization and modification of not only AB5 toxins but also proteinaceous toxins in general. Mutational studies can be performed in a high-throughput manner using cell-free systems as an amber stop codon can be inserted at any desired position within the gene of interest, thus enabling the screening of toxic domains.
CFPS will facilitate the investigation of the mechanism of action of novel toxins by diverse labeling methods and subsequent intracellular trafficking in the future. The modification and conjugation of individual toxin subunits will also improve the development of targeted toxins and Trojan horses for therapeutic use. Overall, this study demonstrated that cell-free systems enable the production of toxins for diagnostic and therapeutic applications using the example of the AB5 toxins, Ctx and LT.
4. Materials and Methods
4.1. DNA Template Design
Gene sequences encoding the cholera toxin A subunit, the partial A1 sequence, the partial A2 sequence (UniProt accession: P01555) and the B subunit (UniProt accession: P01556), as well as gene sequences encoding the A and B subunit (UniProt accession: P06717 and P32890, respectively) of the heat-labile enterotoxin, were modified for cell-free protein synthesis. Therefore, the DNA templates were modified according to Brödel et al. [
42] and the native signal sequences were replaced with the Melittin signal sequence. LTA was modified with a C-terminal double strep tag. These sequences were obtained by
de novo gene synthesis (Biocat GmbH, Heidelberg, Germany), cloned into the pUC57-1.8K vector backbone and plasmids were directly used for cell-free synthesis.
For further analyses, several additional templates were generated using an expression PCR (E-PCR) [
43]. All constructs were amplified using an E-PCR with the HiFidelity polymerase and its corresponding kit (Qiagen GmbH, Hilden, Germany). Standard HiFi PCR protocol (f.c. 0.2 ng/µL DNA, 1x HiFi Buffer including dNTPs, 0.05 U/µL HiFi polymerase). After each individual PCR, the templates were analyzed by agarose gel electrophoresis.
4.2. LTA1 and LTA2 Templates
At first, LTA1 and LTA2 were additionally generated. For LTA1, the forward primer N0 (5′-ATGATATCTCGAGCGGCCGCTAGCTAATACGACTCACTATAGGGAGAC CACAACGGTTTCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATAAACAATG-3′) and the gene-specific reverse primer, X-LTA1-oe-C0-R (5′-CTTGGTTAGTTAGTTATTAGATTGTTCTTGATGAATT-3′), which included an overhang to the regulatory sequences, were required for CFPS. In a second step, the generated PCR template was amplified with N0 and reverse C0 (5′-ATGATATCACCGGTGAATTCGGATCCAAAAAACCCCTCAAGACCCGTTTAGAGGCCCCAAGGGGTACAGATCTTGGTTAGTTAGTTATTA-3′). For the LTA2 template, a gene-specific primer with an overhang to the regulatory sequences for CFPS X-NCM-oe-LTA2-F (5′-TACATTTCTTACATCTATGCGGACACAGGTGATACTTGTAAT-3′) and C0 were used in the first step. Next, the template was fused to the NCM-F primer that included the CrPV IRES site and the Mel signal peptide
(5′-ATGATATCTCGAGCGGCCGCTAGCTAATACGACTCACTATAGGGAGACCACAACGGTTTCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATAAACAAAAGCAAAAATGTGATCTTGCTTGTAAATACAATTTTGAGAGGTTAATAAATTACAAGTAGTGCTATTTTTGTATTTAGGTTAGCTATTTAGCTTTACGTTCCAGGATGCCTAGTGGCAGCCCCACAATATCCAGGAAGCCCTCTCTGCGGTTTTTCAGATTAGGTAGTCGAAAAACCTAAGAAATTTACCTGCTAAATTCTTAGTCAACGTTGCCCTTGTTTTTATGGTCGTATACATTTCTTACATCTATGCGGAC-3′).
The templates from the first PCR step and the NCM-F primer were applied in an equal molar template ratio of 12.5 nM in a standard HiFi PCR protocol.
4.3. Modification of Wild-Type Constructs
In the next step, the LTB gene was fused to the streptavidin (Strep) gene (UniProt accession: P22629) in silico. Strep was fused to the C-terminus of the LTB gene. Two mutated CtxA subunits were designed by exchanging the glutamic acid codon at position 110 or 112 (amino acid number counting without the signal peptide) to an amber stop codon (TAG). The modified LTB-Strep construct, as well as the mutated CtxA constructs (CtxAambE110 and CtxAambE112), were also obtained by de novo gene synthesis (Biocat GmbH) as a lyophilizate. The standard HiFi protocol was performed while applying the N0 and C0 primers.
A fusion construct of LTB with the fluorescent protein eYFP was generated to assess the co-translational translocation in the cell-free system. Therefore, the LTB template was modified by using the HiFi PCR scheme. In the first step, the N0 forward primer and the gene-specific reverse primer with an overhang to the eYFP X-LTB-oe-eYFP-R (5′-CTTGCTCACCTcTAGAcAGTTTTCCATACTGATTGCCGC-3′) were utilized. In a second step, the template was fused to an eYFP construct by mixing the two templates in a 1:1 molar ratio in a HiFi PCR and using N0 and C0 primers.
4.4. Strep Construct
In order to compare the synthesis of the modified LTB-Strep, not only to LTB but also to Strep itself, a 2-step E-PCR was performed. The Strep template was amplified from the LTB-Strep plasmid. Therefore, a standard HiFi PCR scheme, as described above, was applied using a gene-specific forward primer for Strep NCM-oe-Strep-F (5′-TACATTTCTTACATCTATGCGGACgacccctccaaggactcgaa-3′) and the C0 reverse primer. The primer NCM-oe-Strep-F contained an overlap of the regulatory sequences needed for CFPS, including the CrPV IRES site and the Mel signal peptide, in order to have a similar construct to LTB and LTB-Strep. Hence, a second PCR was performed in which the first PCR template was fused to the NCM-F primer.
The templates from the first PCR step and the NCM-F primer were applied in an equal molar template ratio of 12.5 nM in a standard HiFi PCR protocol.
4.5. DNA Template for Orthogonal tRNA
The template DNA for the orthogonal tRNA, which was used for the subsequent transcription, was generated using a Taq PCR reaction (0.01 ng/µL plasmid DNA, dNTPs 0.2 mM each (Qiagen), MgCl
2 2.5 mM (Thermo Fisher Scientific, Rockford, IL, USA) Taq-polymerase 0.025 U/µL (Thermo Fisher Scientific), 1× Taq-buffer (Thermo Fisher Scientific)). A specific O-methyl primer pair (0.5 µM each) was used as previously described [
44]. Finally, the PCR product was purified using Qiaquick PCR-Purification Kit (Qiagen) and analyzed by agarose gel electrophoresis.
4.6. Generation of Orthogonal Components
The orthogonal aminoacyl-tRNA synthetase [
44] (eAzPheRS), which is specific for p-propargyloxyphenylalanine (pPa) and p-azido-L-phenylalanine (AzF), was synthesized in the “RTS500 ProteoMaster
E. coli HY Kit” (Biotechrabbit GmbH, Berlin, Germany). Synthesis, purification and storage of eAzPheRS were handled as previously described [
17,
44].
Specific transcripts of suppressor tRNAs were transcribed in vitro overnight in a batch-formatted reaction at 37 °C, using a PCR product as a DNA template (f.c. 8 µg/mL) as described in Zemella et al., 2009 [
44]. The tRNA was purified using phenol-chloroform extraction with TRIzol-reagent (Life Technologies, Carlsbad, CA, USA). Subsequently, the purified tRNA was resuspended in ultrapure water and stored at −80 °C.
4.7. Cell-Free Protein Synthesis
Cell-free protein synthesis reactions were performed using translationally active lysates derived from cultured Chinese hamster ovary cells (CHO, DSMZ, Braunschweig, Germany) and cultured
Spodoptera frugiperda 21 cells (
Sf21, ECACC by Sigma Aldrich, Taufkirschen, Germany). Lysate preparation was performed as described previously [
45,
46,
47]. For individual subunit expression, the plasmid was added at a final concentration (f.c.) of 60 ng/µL. For co-expression syntheses, the templates of the subunits were added in a 1:1 (A1:A2), 1:5 (A:B) or 1:1:5 (A1:A2:B) molar DNA ratio. Whenever a template that was derived from a PCR was used, the PCR template concentration was estimated according to the DNA quantification standard (100 ng/µL, 1000 bp; Gensura, San Diego, CA, USA). Furthermore, a no-template control (NTC), consisting of a translation mixture without any DNA template was used as a background control.
4.8. Batch-Based Reactions
Cell-free protein synthesis was performed in a coupled transcription/translation mode at final volumes ranging from 20 to 85 µL. Reactions were conducted according to previously described protocols [
47,
48]. Briefly, the DNA template was added to a reaction mixture composed of 40% (
v/
v) lysate, 100 µM amino acids and energy components. PolyG primer (f.c. 10 µM, IBA, Göttingen, Germany) was additionally supplemented. For further analyses, such as autoradiography and liquid scintillation counting, cell-free protein synthesis reactions were supplemented with radioactive
14C-leucine (f.c. 50 μM, specific radioactivity 66.67 dpm/pmol, Perkin Elmer, Baesweiler, Germany). The reactions were incubated for 3 h at 30 °C for CHO lysate or 27 °C for
Sf21 lysate at an agitation of 500 rpm.
4.9. Repetitive Synthesis
The visualization of the co-translational translocation of the LTB-eYFP construct was undertaken after the repetitive synthesis scheme of the respective construct. Each cycle was performed in a 20 µL standard batch-based synthesis reaction in Sf21 lysate. Each synthesis took place for 3 h at 27 °C and 500 rpm. A total of three synthesis rounds were performed. After the first round, the microsomal vesicles that contained the protein of interest were separated from the soluble synthesis components by centrifugation (10 min, 16,000× g, 4 °C). This microsomal pellet was dissolved in a fresh reaction mixture as used for batch-based syntheses. The only exception was that the Sf21 lysate was depleted from microsomes by centrifugation. After the second synthesis, the microsomes were collected again and dissolved in a fresh reaction mixture without additional microsomes.
4.10. Orthogonal Translation
For site-specific incorporation of pPa and AzF, the respective ncAA (f.c. 2 mM, both from Iris Biotech GmbH, Marktredwitz, Germany), the pre-synthesized tRNA (f.c. 2.5 and 5 µM for Sf21 and CHO, respectively) were added to the standard coupled batch reaction as described above. A modified CHO lysate that integrated the eAzPheRS through stable transfection was used. Using this lysate, no eAzPheRS had to be separately supplemented.
4.11. Protein Fractionation
As the cholera toxin subunits harbor a Melittin signal sequence, a translocation of the synthesized protein into the microsomal vesicles that are present in the CHO and
Sf21 lysate [
42] was expected. Therefore, microsomal vesicles were harvested. After the synthesis reaction, the crude translation mixture (TM) was centrifuged (16,000×
g, 10 min, 4 °C) resulting in the supernatant (SN1) containing the soluble subunits that were not translocated into the vesicles and the pelleted microsomes. The pellet was resuspended in phosphate-buffered saline (PBS, Merck, Darmstadt, Germany) containing 0.5% of the mild detergent Chaps (3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate, Amresco, Solon, OH, USA), which was termed the microsomal fraction (MF). MF was incubated at room temperature under rigorous agitation for 45 min. Finally, the fraction was centrifuged (16,000×
g, 10 min, 4 °C) resulting in the supernatant (SN2) containing the soluble subunits.
4.12. Fluorescent Labeling
Chemoselective labeling of pPa and AzF was performed. Shortly, a copper-catalyzed azide-alkyne cycloaddition was performed for constructs that incorporated pPa by adding 3[tris(3-hydroxypropyltriazolylmethyl)amine (ThPTA, f.c. 600 µM, Sigma Aldrich), sodium ascorbate (f.c. 5 mM, Sigma Aldrich), copper sulfate (f.c. 50 µM, Sigma Aldrich) and sulfo-Cy 5-azide (f.c. 5 µM, Lumiprobe, Hannover, Germany). The reaction took place in the dark at 25 °C for 90 min. A Staudinger ligation was used to label constructs that incorporated AzF. The sample was mixed with Dylight 632-Phosphine (f.c. 5 µM, Dyomics GmbH, Jena, Germany) and incubated in the dark for 90 min at 25 °C.
LTB-strep and Strep were labeled using a fluorescent dye that was fused to a biotin relying on the strep-biotin binding. Hence, the fluorescent dye Biotin-Atto488 (f.c. 1 µM, Sigma Aldrich) was mixed with the sample and incubated in the dark for 90 min at 25 °C.
4.13. Quantitative Protein Analysis
Total protein yield of synthesized proteins was determined by using hot trichloroacetic acid (TCA, Carl Roth GmbH, Karlsruhe, Germany) precipitation and liquid scintillation quantification as previously described [
20]. The total protein yield for co-expressed subunits was estimated using the sum of the molecular weight and the sum of the number of leucines of all expressed subunits according to the previously published protocol [
20].
4.14. Qualitative Protein Analysis
Proteins were either precipitated in acetone as described previously [
20] or the sample was directly mixed with LDS sample buffer (NUPAGE LDS sample buffer (Invitrogen, Thermo Fisher Scientific). If not stated otherwise, samples were heated to 70 °C for 10 min. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using precast gels (NuPAGE, 10% Bis-Tris, Life technologies) and autoradiography were performed according to previous protocols [
12,
17,
20].
In-gel fluorescence was performed for fluorescently labeled constructs directly after SDS-PAGE. The fluorescence scan was performed using the Amersham RGB Typhoon (GE Healthcare, Little Chalfont, Buckinghamshire, UK, DyLight632-Phosphine und Cy5-dyes: excitation 633 nm, emission 670 nm, Biotin-Atto488 dye, excitation 488 nm, emission 525 nm)
The SeeBlue PreStained Protein standard and SeeBlue Plus 2 PreStained Protein standard (Thermo Fisher Scientific) were used.
4.15. LTB-eYFP Fluorescence Analysis
An amount of 5 µL of the pre-synthesized protein was diluted in 20 µL PBS and added onto an µ-IBIDI-slide (Ibidi, Planegg, Germany). The translocation was assessed by confocal laser scanning microscopy (CLSM). The laser scanning microscope unit (LSM 510, Carl Zeiss Microscopy GmbH, Oberkochen, Germany) was used, and eYFP was exited with an argon laser at 488 nm. After passing a long-pass filter with a wavelength of 505 nm, the emitted light was captured using a photomultiplier.
4.16. Cell-Based Activity Assessment
For functional analysis, the effect of the AB5 complex and individual subunits was investigated in CHO-K1 cells (DSMZ, Braunschweig, Germany). In an initial experiment, the cell suspension (CHO-K1 cells in DMEM (Sigma Aldrich), 1% fetal bovine serum (Merck) and 1% Penicillium/Streptomycin (Merck)) with a concentration of 4000 cells/well were added to each well of a 96-well plate (Sarstedt, Nümbrecht, Germany). In the following experiments, 24-well plates (Sarstedt) with 25,000 cells/well were used. Cells were seeded shortly before adding the toxin and incubated at 37 °C and 5% CO2-flow. The toxins were synthesized in a batch-based reaction with either CHO or Sf21 lysate. All toxins were synthesized in two reactions, one reaction with additional 14C-labeled leucine, and the other reaction without 14C-leucine. The SN2 fraction of the labeled protein was quantitatively and qualitatively analyzed as described above. Toxin concentrations of the radiolabeled toxins were calculated and, consequently, assumed for non-labeled toxins, as both reactions were prepared simultaneously. In order to allow for the same detergent concentration of the final sample that was added to the cells, all samples were diluted and filled up to the same volume of 0.5% CHAPS/PBS and medium. The mixture of the toxin, CHAPS/PBS and medium was added to each well. A no-template control (NTC) was added by using a volume equivalent to the toxin fragment. Morphological changes were documented using a light microscope (Leica, Wetzlar, Germany) and phase contrast micrographs were captured with a CCD camera (Leica) after 48 h.
The cytotoxicity of the toxins was assessed by the CellTox Green Cytotoxicity Assay (Promega, Walldorft, Germany) according to the manufacturer’s protocol in a 96-well plate (Sarstedt). Cells were cultivated as described for morphological assays. CHO-K1 cells were incubated with toxins for 48 h.
A univariate analysis of variance (ANOVA) was used to assess the significant changes in the cytotoxicity assay. Significance was tested according to Bonferroni. Statistically significant changes were indicated in the Figures with *.

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





