Transcriptome Analysis Reveals the Algicidal Mechanism of Brevibacillus laterosporus against Microcystis aeruginosa through Multiple Metabolic Pathways

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
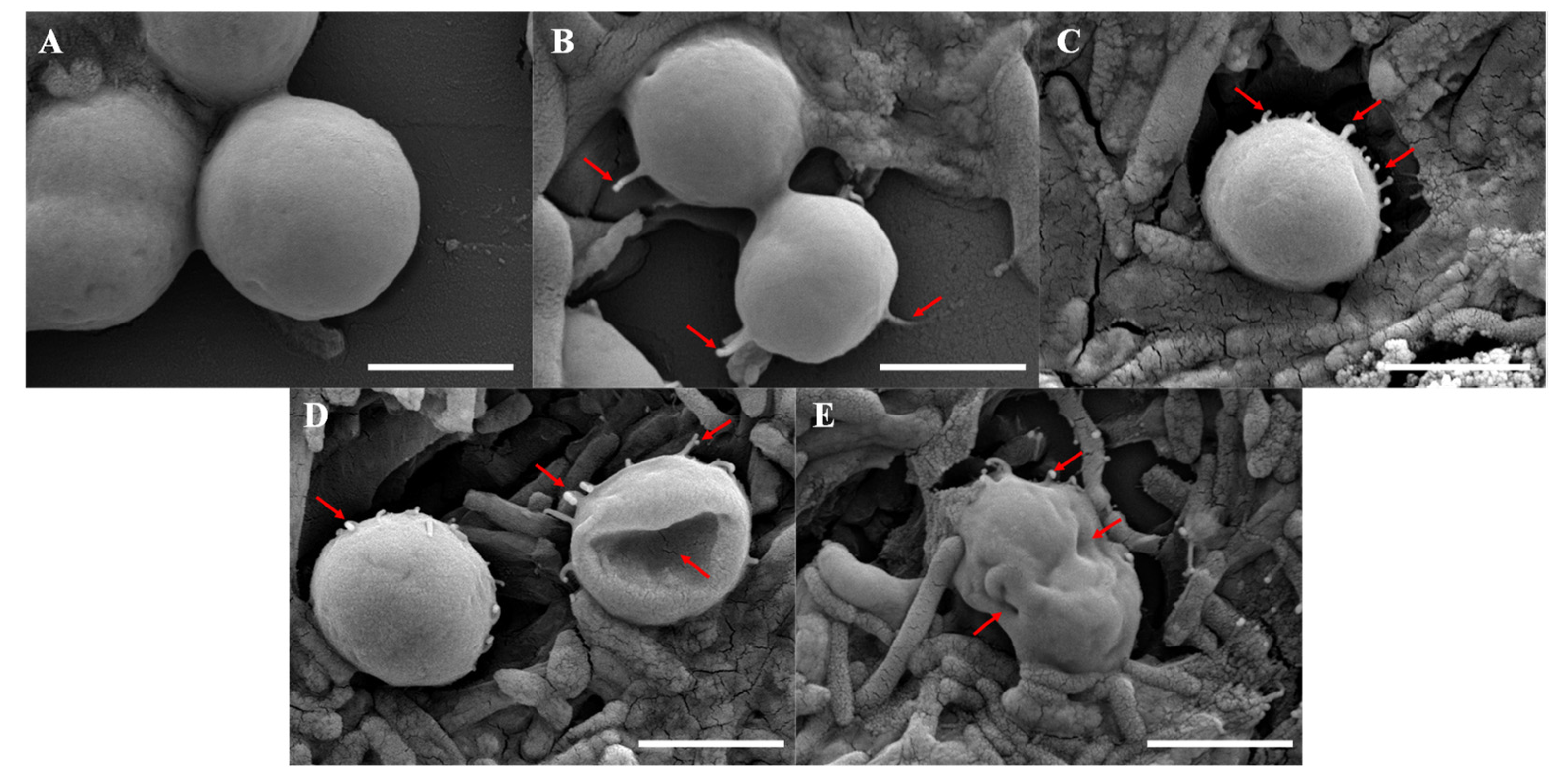
2.1. Effect of B. laterosporus on the Cell Morphology of M. aeruginosa
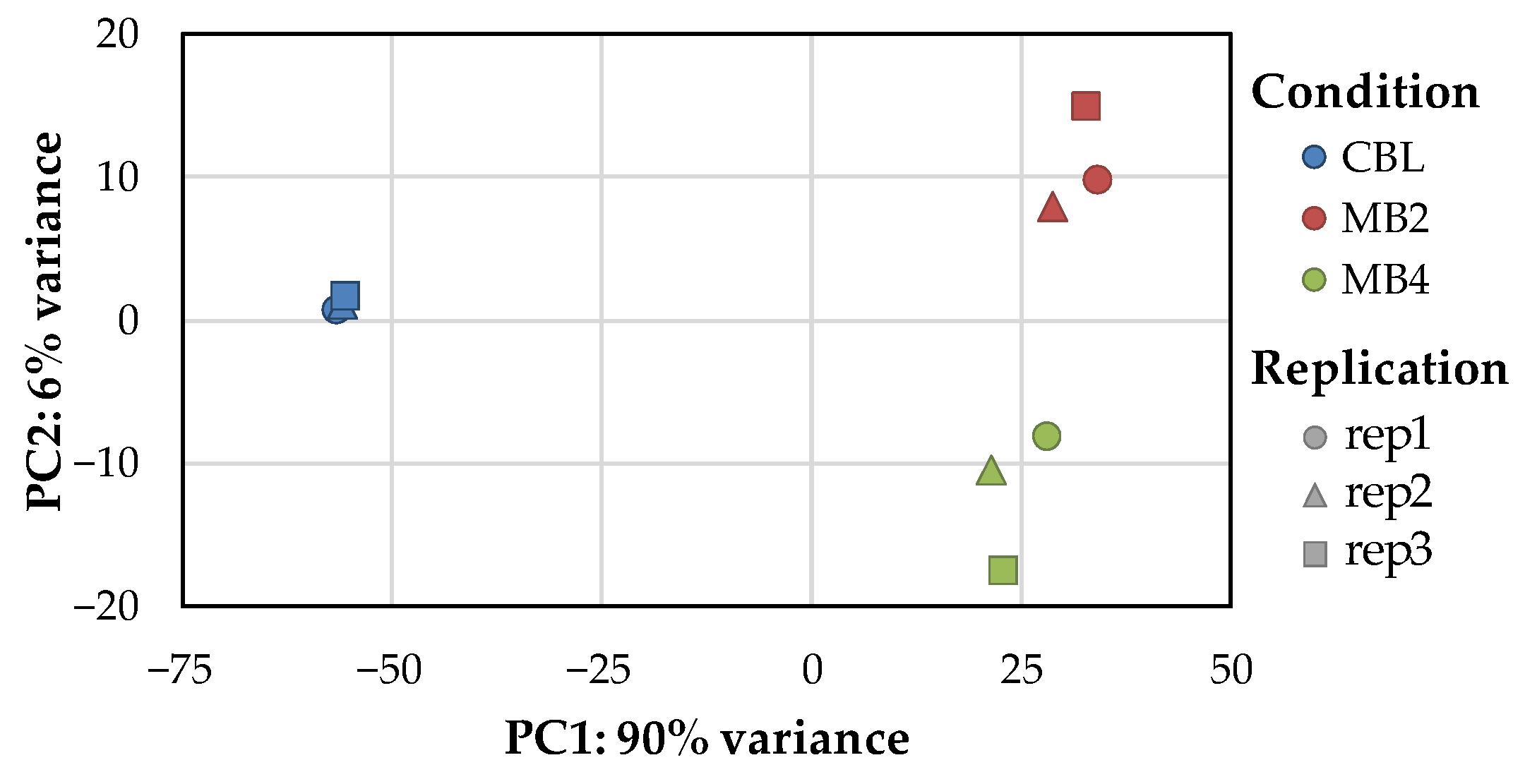
2.2. Transcriptome Sequencing, Assembly and Data Quality Analysis
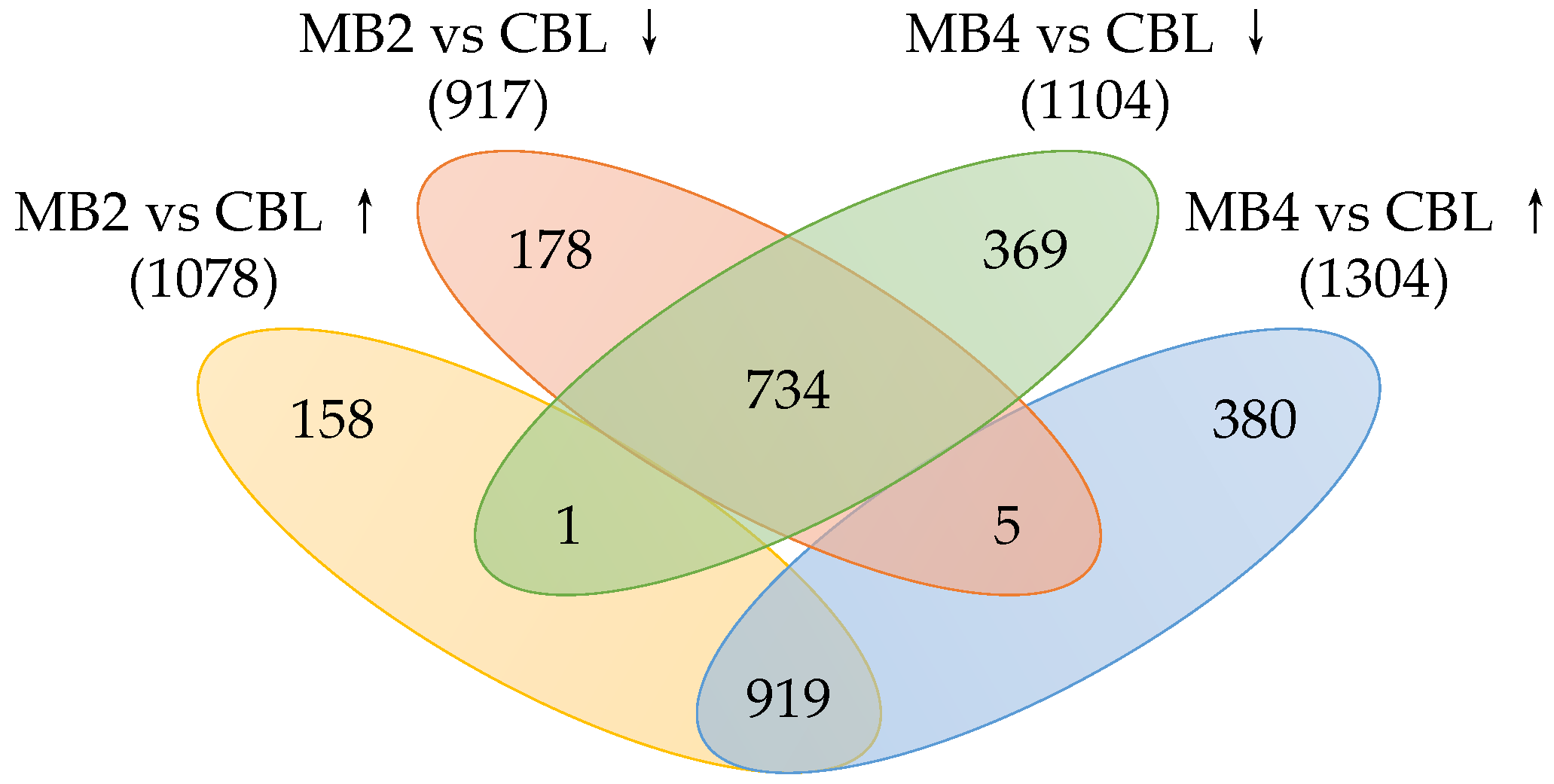
2.3. Identification of Differentially Expressed Genes
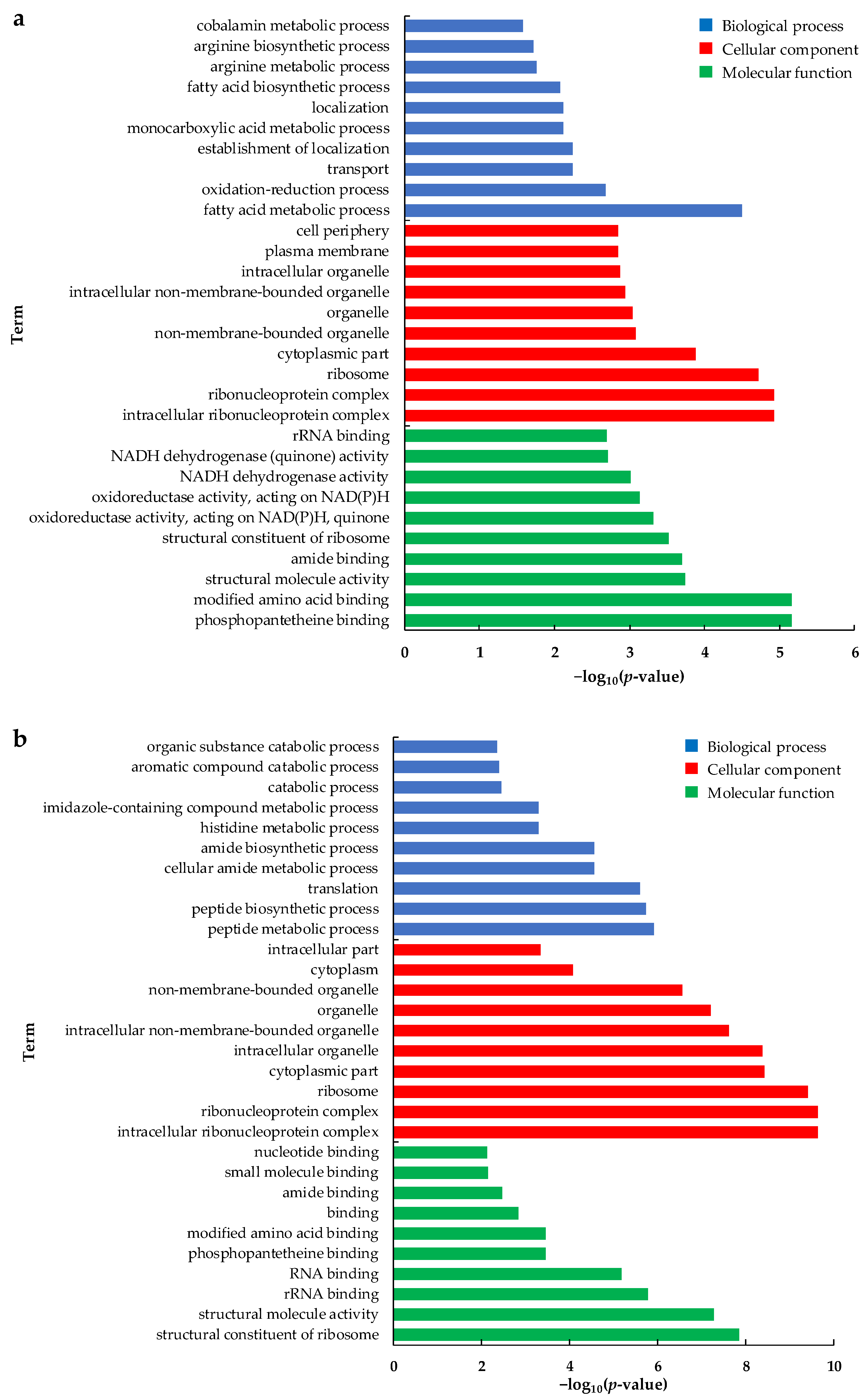
2.4. Enrichment Analysis of Differentially Expressed Gene Function
2.4.1. Gene Ontology (GO) Pathway Analysis
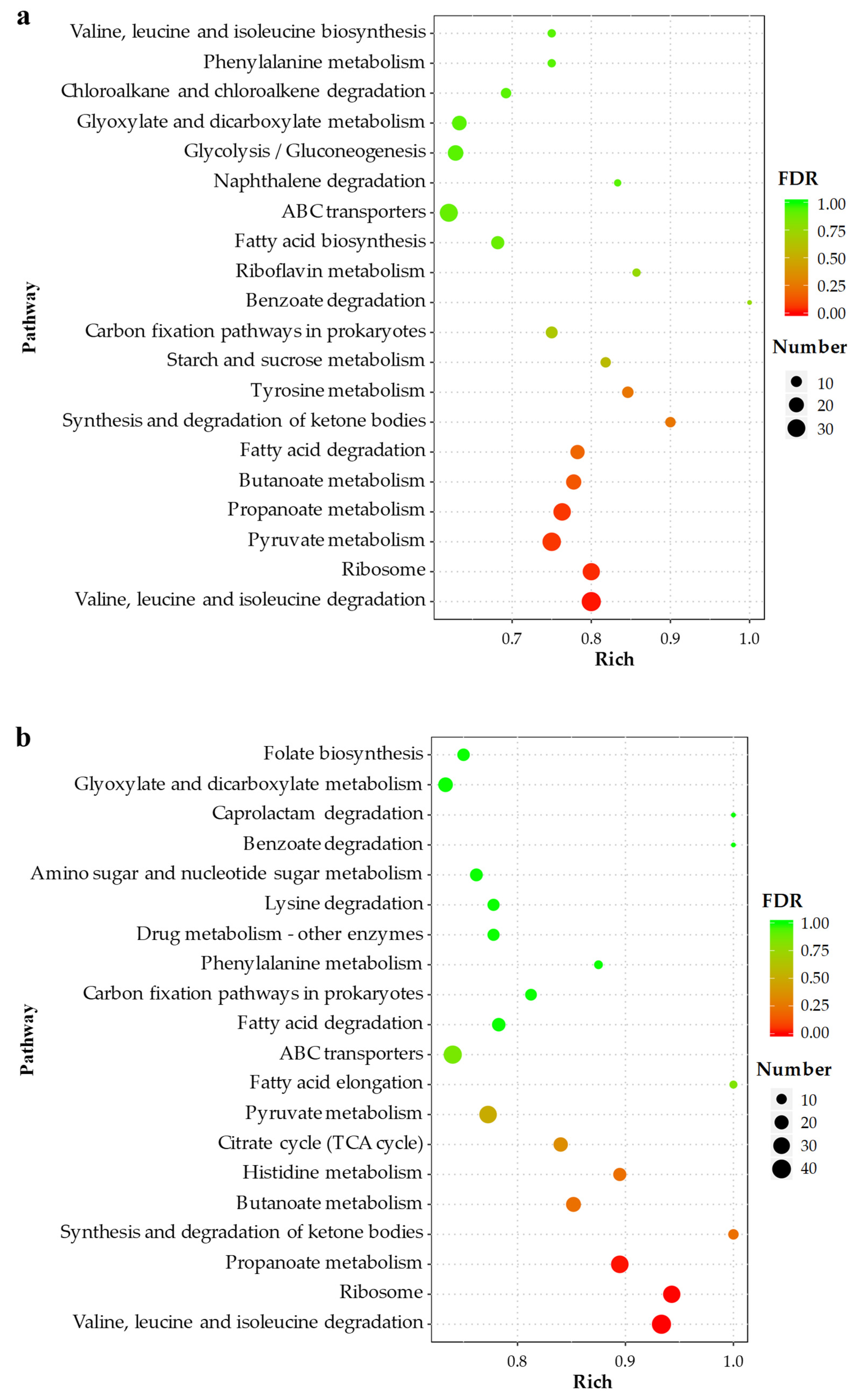
2.4.2. KEGG Pathway Analysis
2.5. Analysis of Differentially Expressed Genes
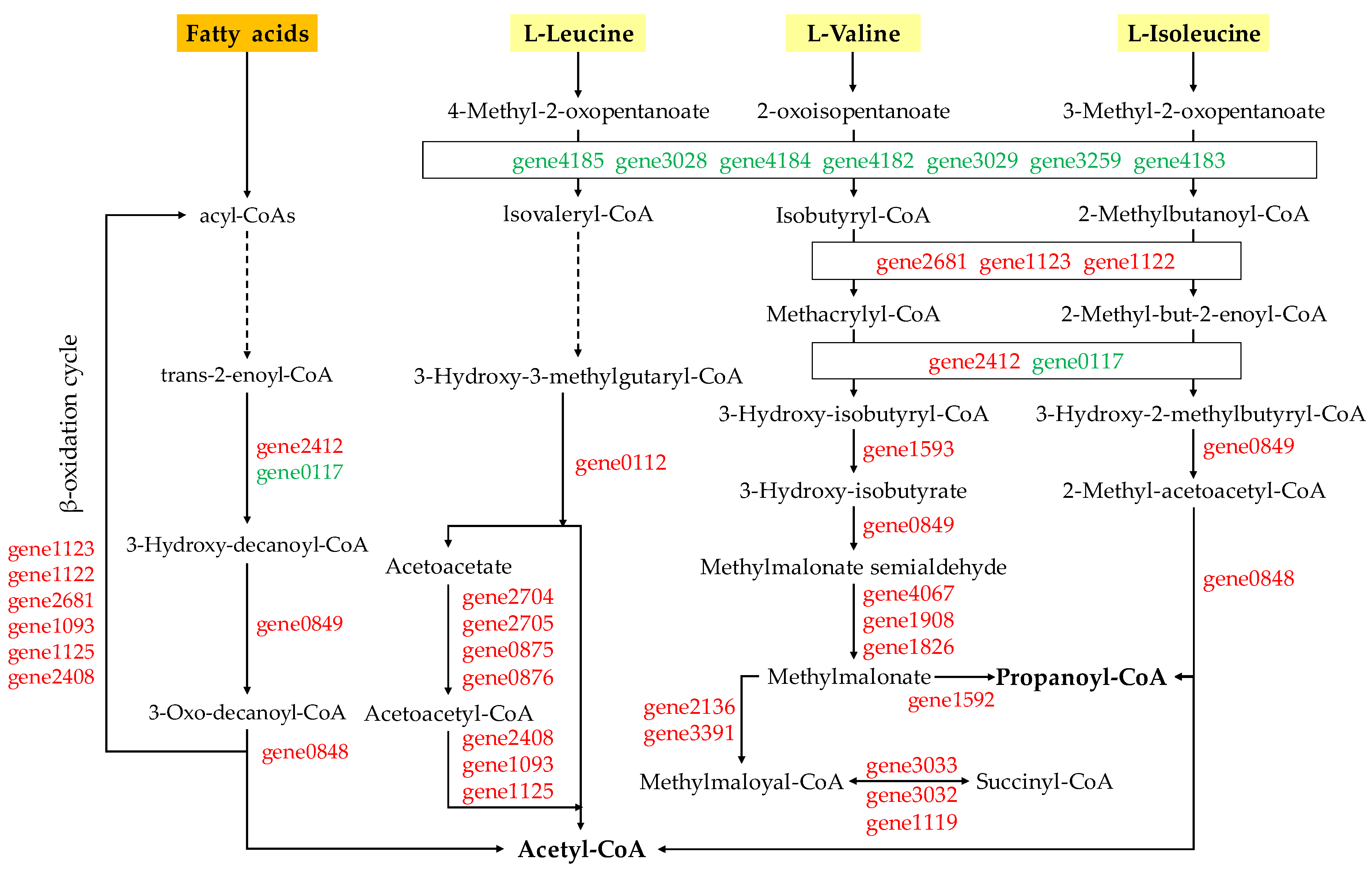
2.5.1. Degradation of Valine, Leucine, and Isoleucine and Fatty Acids
2.5.2. Transporter-Related Differential Genes
2.5.3. Hydrolase and Protease Related Differential Genes
2.5.4. Biosynthesis-Related Differential Genes of Other Secondary Metabolites
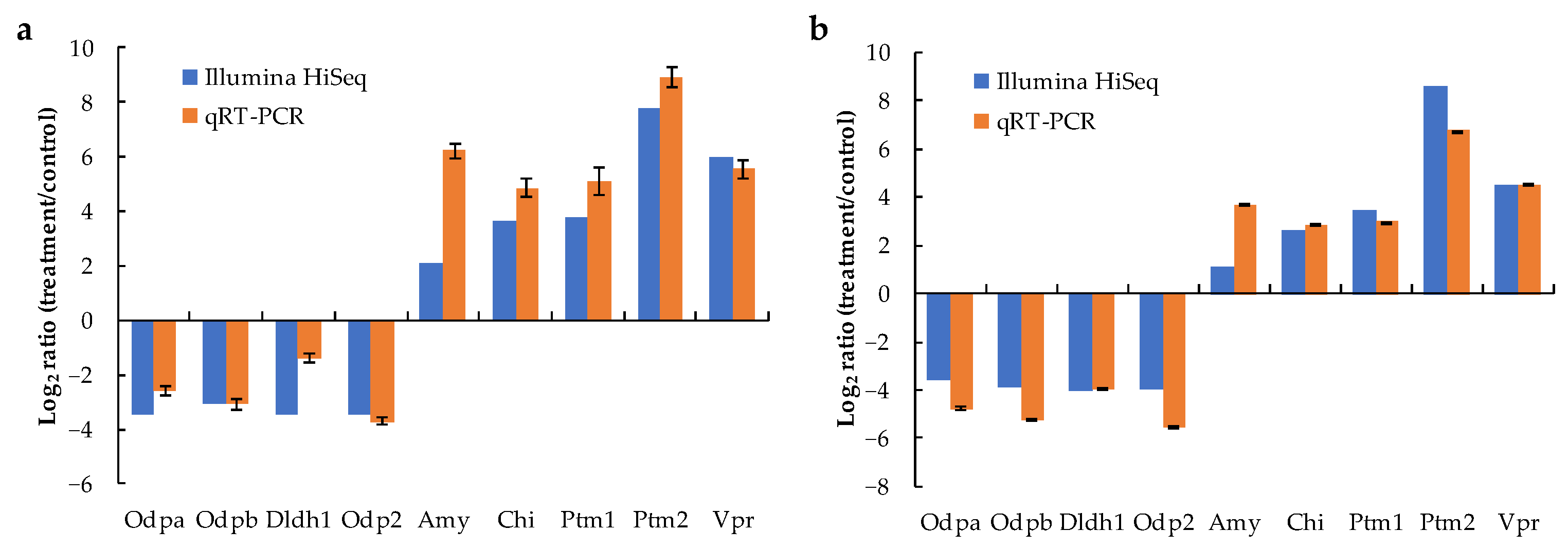
2.6. Results of qRT-PCR
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Co-Cultivation of B. laterosporus and M. aeruginosa
5.2. Specimen Preparation for Scanning Electron Microscopy
5.3. Transcriptome Sample Processing and Sequencing
5.4. Analysis of Differential Expressed Genes
5.5. Quantitative PCR Verification
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sidabutar, T.; Srimariana, E.S.; Wouthuyzen, S. Phytoplankton species potentially harmful algal blooms (HABs) in Jakarta Bay. IOP Conf. Ser. Earth Environ. Sci. 2021, 744, 012077. [Google Scholar] [CrossRef]
- Melaram, R.; Newton, A.R.; Chafin, J. Microcystin contamination and toxicity: Implications for agriculture and public health. Toxins 2022, 14, 350. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Sun, P.; Zhang, J.; Esquivel-Elizondo, S.; Wu, Y. Microorganisms-based methods for harmful algal blooms control: A review. Bioresour. Technol. 2018, 248, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Zhang, P.; Liu, C.; Xue, Y.; Lian, B. The potential use of bacterium strain R219 for controlling of the bloom-forming cyanobacteria in freshwater lake. World J. Microbiol. Biotechnol. 2009, 26, 465–472. [Google Scholar] [CrossRef]
- Yang, C.; Hou, X.; Wu, D.; Chang, W.; Zhang, X.; Dai, X.; Du, H.; Zhang, X.; Igarashi, Y.; Luo, F. The characteristics and algicidal mechanisms of cyanobactericidal bacteria, a review. World J. Microbiol. Biotechnol. 2020, 36, 188. [Google Scholar] [CrossRef]
- Shao, J.; Li, R.; Lepo, J.E.; GU, J. Potential for control of harmful cyanobacterial blooms using biologically derived substances: Problems and prospects. J. Environ. Manag. 2013, 125, 149–155. [Google Scholar] [CrossRef]
- Lee, S.O.; Kato, J.; Takiguchi, N.; Kuroda, A.; Ikeda, T.; Mitsutani, A.; Ohtake, H. Involvement of an extracellular protease in algicidal activity of the marine bacterium Pseudoalteromonas sp. strain A28. Appl. Environ. Microbiol. 2000, 66, 4334–4339. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.H.; Sang, M.; Hwang, S.J.; Han, M.S. In situ bacterial mitigation of the toxic cyanobacterium Microcystis aeruginosa: Implications for biological bloom control. Limnol. Oceanogr. Methods 2008, 6, 513–522. [Google Scholar] [CrossRef]
- Yu, Y.; Zeng, Y.; Li, J.; Yang, C.; Zhang, X.; Luo, F.; Dai, X. An algicidal Streptomyces amritsarensis strain against Microcystis aeruginosa strongly inhibits microcystin synthesis simultaneously. Sci. Total Environ. 2018, 650, 34–43. [Google Scholar] [CrossRef]
- Han, G.; Ma, H.; Ren, S.; Gao, X.; He, X.; Zhu, S.; Deng, R.; Zhang, S. Insights into the mechanism of cyanobacteria removal by the algicidal fungi Bjerkandera adusta and Trametes versicolor. MicrobiologyOpen 2020, 9, e1042. [Google Scholar] [CrossRef]
- Dai, W.; Chen, X.; Wang, X.; Xu, Z.; Gao, X.; Jiang, C.; Deng, R.; Han, G. The algicidal fungus Trametes versicolor F21a eliminating blue algae via genes encoding degradation enzymes and metabolic pathways revealed by transcriptomic analysis. Front. Microbiol. 2018, 9, 826. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, C.; Dai, W.; Ren, S.; Tao, F.; He, X.; Han, G.; Wang, W. Proteomic analysis reveals large amounts of decomposition enzymes and major metabolic pathways involved in algicidal process of Trametes versicolor F21a. Sci. Rep. 2017, 7, 3907. [Google Scholar] [CrossRef] [PubMed]
- Krachkovskii, S.A.; Sobol’, A.G.; Ovchinnikova, T.V.; Tagaev, A.A.; Yakimenko, Z.A.; Azizbekyan, R.R.; Kuznetsova, N.I.; Shamshina, T.N.; Arseniev, A.S. Isolation, biological properties, and spatial structure of antibiotic loloatin A. Russ. J. Bioorg. Chem. 2002, 28, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Huang, X.; Li, C. A preliminary study of the algicidal mechanism of bioactive metabolites of Brevibacillus laterosporus on Oscillatoria in prawn ponds. Sci. World J. 2014, 2041, 869149. [Google Scholar]
- Zhang, Y.; Chen, D.; Cai, J.; Zhang, N.; Li, F.; Li, C.; Huang, X. Complete genome sequence analysis of Brevibacillus laterosporus Bl-zj reflects its potential algicidal response. Curr. Microbiol. 2021, 78, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, D.; Zhang, N.; Li, F.; Luo, X.; Li, Q.; Li, C.; Huang, X. Transcriptional analysis of Microcystis aeruginosa co-cultured with algicidal bacteria Brevibacillus laterosporus. Int. J. Environ. Res. Public Health 2021, 18, 8615. [Google Scholar] [CrossRef]
- Bruins, M.R.; Kapil, S.; Oehme, F.W. Microbial resistance to metals in the environment. Ecotoxicol. Environ. Saf. 2000, 45, 198–207. [Google Scholar] [CrossRef]
- Oh, E.J.; Bae, Y.H.; Kim, K.H.; Park, Y.C.; Seo, J.H. Effects of overexpression of acetaldehyde dehydrogenase 6 and acetyl-CoA synthetase 1 on xylitol production in recombinant Saccharomyces cerevisiae. Biocatal. Agric. Biotechnol. 2012, 1, 15–19. [Google Scholar] [CrossRef]
- Liu, X. ABC family transporters. Adv. Exp. Med. Biol. 2019, 1141, 13–100. [Google Scholar]
- Zhang, F.; Fan, Y.; Zhang, D.; Chen, S.; Bai, X.; Ma, X.; Xie, Z.; Xu, H. Effect and mechanism of the algicidal bacterium Sulfitobacter porphyrae ZFX1 on the mitigation of harmful algal blooms caused by Prorocentrum donghaiense. Environ. Pollut. 2020, 263 Pt A, 114475. [Google Scholar] [CrossRef]
- Hu, Q.; Yin, X.; Li, H.; Wang, X.; Jiang, Z.; Li, L.; Ni, H.; Li, Q.; Zhu, Y. Characterisation of a novel laminarinase from Microbulbifer sp. ALW1 and the antioxidant activity of its hydrolysates. Int. J. Food Sci. Technol. 2021, 56, 4129–4138. [Google Scholar] [CrossRef]
- Redhead, K.; Wright, S.J. Isolation and properties of fungi that lyse blue-green algae. Appl. Environ. Microbiol. 1978, 35, 962–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genes, C.; Baquero, E.; Echeverri, F.; Maya, J.D.; Triana, O. Mitochondrial dysfunction in Trypanosoma cruzi: The role of Serratia marcescens prodigiosin in the alternative treatment of chagas disease. Parasite Vectors 2011, 4, 66. [Google Scholar] [CrossRef] [Green Version]
- Clements, T.; Ndlovu, T.; Khan, W. Broad-spectrum antimicrobial activity of secondary metabolites produced by Serratia marcescens strains. Microbiol. Res. 2019, 229, 126329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yang, Y.; Xie, W.; He, W.; Xie, J.; Liu, W. Identifying algicides of Enterobacter hormaechei F2 for control of the harmful alga Microcystis aeruginosa. Int. J. Environ. Res. Public Health 2022, 19, 7556. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Wei, Z.; Hui, W. Physiological response and morphological changes of Heterosigma akashiwo to an algicidal compound prodigiosin. J. Hazard. Mater. 2020, 385, 121530. [Google Scholar]
- Zhang, H.; Peng, Y.; Zhang, S.; Cai, G.; Li, Y.; Yang, X.; Yang, K.; Chen, Z.; Zhang, J.; Wang, H.; et al. Algicidal effects of prodigiosin on the harmful alga Phaeocystis globosa. Front. Microbiol. 2016, 7, 602. [Google Scholar]
- Yang, K.; Chen, Q.; Zhang, D.; Zhang, H.; Lei, X.; Chen, Z.; Li, Y.; Hong, Y.; Ma, X.; Zheng, W.; et al. The algicidal mechanism of prodigiosin from Hahella sp. KA22 against Microcystis aeruginosa. Sci. Rep. 2017, 7, 7750. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Wei, H.; Li, X.; Li, Y.; Li, X.; Yin, L.; Pu, Y. Isolation and characterization of an algicidal bacterium indigenous to Lake Taihu with a red pigment able to lyse Microcystis aeruginosa. Biomed. Environ. Sci. 2013, 26, 148–154. [Google Scholar]
- Qian, H.; Li, J.; Pan, X.; Sun, Z.; Ye, C.; Jin, G.; Fu, Z. Effects of streptomycin on growth of algae Chlorella vulgaris and Microcystis aeruginosa. Environ. Toxicol. 2012, 27, 229–237. [Google Scholar] [CrossRef]
- Wu, L.; Chen, X.; Huang, L.; Tian, J.; Ke, F.; Xu, J.; Chen, Y.; Zheng, M. A novobiocin derivative, XN4, inhibits the proliferation of chronic myeloid leukemia cells by inducing oxidative DNA damage. PLoS ONE 2015, 10, e0123314. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, N.; Guo, S. Structure and function analysis of protein HD73_0859 produced by Bacillus thuringiensis. Bio-Med. Mater. Eng. 2014, 24, 3891–3896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaskar, N.; Sudeepa, E.S.; Rashmi, H.N.; Selvi, A.T. Partial purification and characterization of protease of Bacillus proteolyticus CFR3001 isolated from fish processing waste and its antibacterial activities. Bioresour. Technol. 2007, 98, 2758–2764. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Pu, G.; Shao, C.; Cheng, S.; Cai, J.; Zhou, L.; Jia, Y.; Tian, X. Potential of extracellular enzymes from Trametes versicolor F21a in Microcystis spp. degradation. Mater. Sci. Eng. C 2015, 48, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Wang, P.; Wang, Y. Algicidal efficiency and mechanism of Phanerochaete chrysosporium against harmful algal bloom species. Algal Res. 2015, 12, 182–190. [Google Scholar] [CrossRef]
- Barbieri, G.; Voigt, B.; Albrecht, D.; Hecker, M.; Albertini, A.M.; Sonenshein, A.L.; Ferrari, E.; Belitsky, B.R. CodY regulates expression of the Bacillus subtilis extracellular proteases Vpr and Mpr. J. Bacteriol. 2015, 197, 1423–1432. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Chen, S. Microorganisms in sediment microbial fuel cells: Ecological niche, microbial response, and environmental function. Sci. Total Environ. 2020, 756, 144145. [Google Scholar] [CrossRef]
- Xiao, F.H.; Yu, Q.; Deng, Z.L.; Yang, K.; Ye, Y.; Ge, M.X.; Yan, D.; Wang, H.T.; Chen, X.Q.; Yang, L.Q.; et al. ETS1 acts as a regulator of human healthy aging via decreasing ribosomal activity. Sci. Adv. 2022, 8, eabf2017. [Google Scholar] [CrossRef]
- Rippka, R.; Deruelles, J.; Waterbury, J.B.; Herdman, M.; Stanier, R.Y. Generic assignments, strain histories and properties of pure cultures of cyanobacteria. J. Gen. Microbiol. 1979, 111, 1–61. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Data (bp) | Raw Reads No. | Clean Data (bp) | Clean Reads No. | Mapped Reads No. | Q20 (%) | Q30 (%) |
|---|---|---|---|---|---|---|---|
| CBL1 | 8,378,192,100 | 55,854,614 | 7,357,141,200 | 49,047,608 | 48,325,154 | 98.14 | 94.54 |
| CBL2 | 8,746,394,400 | 58,309,296 | 7,617,902,100 | 50,786,014 | 49,954,121 | 98.07 | 94.42 |
| CBL3 | 7,970,677,500 | 53,137,850 | 7,125,755,100 | 47,505,034 | 46,792,179 | 98.05 | 94.30 |
| MB2_1 | 11,260,710,900 | 75,071,406 | 9,110,216,700 | 60,734,778 | 15,744,400 | 97.73 | 94.22 |
| MB2_2 | 9,961,599,300 | 66,410,662 | 7,954,650,900 | 53,031,006 | 12,159,967 | 97.62 | 93.88 |
| MB2_3 | 9,498,113,100 | 63,320,754 | 7,660,962,000 | 51,073,080 | 12,936,077 | 97.65 | 93.90 |
| MB4_1 | 10,501,108,500 | 70,007,390 | 8,108,475,900 | 54,056,506 | 15,140,888 | 97.72 | 94.15 |
| MB4_2 | 9,475,501,500 | 63,170,010 | 7,402,896,600 | 49,352,644 | 42,864,396 | 97.29 | 93.41 |
| MB4_3 | 9,642,770,100 | 64,285,134 | 7,021,052,700 | 46,807,018 | 13,102,733 | 96.94 | 92.90 |
| Average | 9,492,785,267 | 63,285,235 | 7,706,561,467 | 51,377,076 | 28,557,768 | 97.69 | 93.97 |
| Level 2 | MB2 vs. CBL | MB4 vs. CBL | ||||
|---|---|---|---|---|---|---|
| Up | Down | Total | Up | Down | Total | |
| Amino acid metabolism | 122 | 47 | 169 | 131 | 76 | 207 |
| Carbohydrate metabolism | 93 | 90 | 183 | 93 | 116 | 209 |
| Lipid metabolism | 51 | 15 | 66 | 52 | 22 | 74 |
| Metabolism of cofactors and vitamins | 28 | 36 | 64 | 26 | 41 | 67 |
| Energy metabolism | 34 | 16 | 40 | 32 | 21 | 53 |
| Nucleotide metabolism | 6 | 32 | 38 | 7 | 43 | 50 |
| Translation | 0 | 33 | 33 | 1 | 47 | 48 |
| Xenobiotics biodegradation and metabolism | 24 | 14 | 38 | 24 | 21 | 45 |
| Membrane transport | 13 | 21 | 34 | 13 | 28 | 41 |
| Metabolism of other amino acids | 18 | 9 | 27 | 13 | 16 | 29 |
| Replication and repair | 1 | 9 | 10 | 7 | 14 | 21 |
| Folding, sorting and degradation | 4 | 10 | 14 | 6 | 13 | 19 |
| Biosynthesis of other secondary metabolites | 11 | 5 | 16 | 10 | 7 | 17 |
| Metabolism of terpenoids and polyketides | 11 | 4 | 15 | 11 | 6 | 17 |
| Cellular community—prokaryotes | 4 | 5 | 9 | 6 | 6 | 12 |
| Infectious diseases: Bacterial | 1 | 3 | 4 | 2 | 2 | 4 |
| Signal transduction | 3 | 0 | 3 | 3 | 1 | 4 |
| Immune system | 1 | 1 | 2 | 1 | 1 | 2 |
| Transcription | 0 | 1 | 1 | 0 | 2 | 2 |
| Environmental adaptation | 1 | 0 | 1 | 1 | 0 | 1 |
| Cell growth and death | 0 | 0 | 0 | 0 | 3 | 3 |
| Glycan biosynthesis and metabolism | 1 | 0 | 1 | 0 | 0 | 0 |
| Gene ID | MB2 vs. CBL | MB4 vs. CBL | Description | ||
|---|---|---|---|---|---|
| log2 (Fold Change) | p-Value | log2 (Fold Change) | p-Value | ||
| gene2217 | 7.04 | 1.04 × 10−17 | 2.29 | 1.80 × 10−11 | ABC transporter |
| gene3791 | 3.08 | 1.12 × 10−3 | 2.09 | 1.31 × 10−3 | ABC transporter family protein |
| gene2788 | 3.56 | 2.60 × 10−5 | 1.68 | 1.13 × 10−8 | ABC transporter family protein |
| gene4041 | 7.42 | 1.34 × 10−8 | 1.96 | 1.33 × 10−2 | ABC transporter permease protein |
| gene2787 | 3.40 | 7.31 × 10−6 | 1.63 | 2.79 × 10−2 | ABC transporter permease protein |
| gene0214 | 3.53 | 1.37 × 10−5 | 5.08 | 6.01 × 10−14 | ABC transporter permease protein |
| gene3837 | 4.19 | 2.00 × 10−15 | 1.48 | 1.64 × 10−3 | ABC transporter ATP-binding/permease protein TycE |
| gene3838 | 4.42 | 1.73 × 10−7 | 2.33 | 2.00 × 10−3 | ABC transporter ATP binding/permease protein TycD |
| gene3493 | 2.68 | 7.63 × 10−6 | 6.01 | 2.16 × 10−4 | ABC transporter substrate-binding protein |
| gene2004 | 3.42 | 2.74 × 10−7 | 3.01 | 1.19 × 10−20 | ABC transporter ATP-binding protein |
| gene1946 | 2.47 | 5.55 × 10−6 | 4.62 | 3.49 × 10−23 | ABC-2 type transporter family protein |
| gene1947 | 2.15 | 5.42 × 10−6 | 3.01 | 3.80 × 10−22 | Putative ABC transporter ATP-binding protein |
| gene0517 | 6.50 | 9.88 × 10−9 | 1.12 | 1.45 × 10−2 | Oligopeptide/dipeptide ABC transporter, ATP-binding, C-terminal domain protein |
| gene0714 | 2.15 | 1.09 × 10−2 | 1.45 | 1.66 × 10−2 | Peptide permease, major facilitator family transporter |
| gene1164 | 7.78 | 2.54 × 10−41 | 5.61 | 1.46 × 10−57 | Oligopeptide transporter, OPT family |
| gene2954 | 3.43 | 4.50 × 10−3 | 2.82 | 1.82 × 10−6 | Putative bacteriocin export ABC transporter, lactococcin group |
| gene3098 | 3.31 | 1.67 × 10−10 | 1.85 | 4.72 × 10−6 | Cyclic peptide transporter family protein |
| gene1210 | 1.83 | 3.92 × 10−3 | 4.71 | 5.95 × 10−43 | Uncharacterized MFS-type transporter YdgK |
| gene1223 | 3.71 | 3.11 × 10−8 | 3.23 | 1.02 × 10−24 | Glycerol-3-phosphate transporter |
| gene1160 | 1.33 | 5.63 × 10−3 | – | – | Putative multidrug resistance ABC transporter ATP-binding/permease protein YheH |
| gene1161 | 1.60 | 1.44 × 10−2 | – | – | Putative multidrug resistance ABC transporter ATP-binding/permease protein YheI |
| gene1174 | 1.12 | 4.87 × 10−2 | – | – | ABC transporter family protein |
| gene1714 | 2.70 | 4.84 × 10−2 | – | – | Phosphate ABC transporter, permease protein PstA |
| gene0216 | 3.44 | 2.08 × 10−2 | – | – | ABC transporter substrate binding protein |
| gene3053 | 1.05 | 2.63 × 10−2 | – | – | Oligopeptide ABC transporter permease protein |
| gene3055 | 1.74 | 2.24 × 10−2 | – | – | Oligopeptide ABC transporter ATP binding protein |
| gene3056 | 1.39 | 3.32 × 10−2 | – | – | Oligopeptide ABC transporter ATP binding protein |
| gene0492 | 1.89 | 1.72 × 10−3 | – | – | Citrate transporter family protein |
| gene1184 | – | – | 6.92 | 2.76 × 10−44 | Inner membrane transporter ycaM |
| gene2438 | – | – | 5.09 | 9.13 × 10−12 | Proton-coupled thiamine transporter ThiT |
| gene2761 | – | – | 1.85 | 4.33 × 10−10 | Formate/nitrite transporter |
| gene3336 | – | – | 5.41 | 1.44 × 10−5 | Efflux transporter, RND family, MFP subunit |
| gene4037 | – | – | 1.41 | 1.03 × 10−2 | ABC transporter family protein |
| Gene ID | MB2 vs. CBL | MB4 vs. CBL | Description | ||
|---|---|---|---|---|---|
| log2 (FoldChange) | p-Value | log2 (FoldChange) | p-Value | ||
| gene2281 | 1.47 | 4.43 × 10−3 | 1.54 | 1.24 × 10−6 | Cof-like hydrolase family protein |
| gene2308 | 10.39 | 1.83 × 10−9 | 8.98 | 6.29 × 10−27 | Cell wall hydrolase CwlJ |
| gene2656 | 2.72 | 7.69 × 10−5 | 3.27 | 2.63 × 10−13 | Dienelactone hydrolase family protein |
| gene2741 | 1.97 | 8.71 × 10−4 | 1.18 | 6.71 × 10−3 | Putative hydrolase |
| gene2775 | 1.30 | 2.05 × 10−2 | 1.87 | 3.04 × 10−7 | HAD-superfamily hydrolase YhcW |
| gene3428 | 5.31 | 5.85 × 10−5 | 4.89 | 7.79 × 10−8 | Glycoside hydrolase, family 18 |
| gene3564 | 2.65 | 4.09 × 10−4 | 4.51 | 1.49 × 10−34 | Putative polyketide biosynthesis zinc-dependent hydrolase PksB |
| gene3616 | 2.37 | 4.74 × 10−4 | 1.71 | 1.30 × 10−4 | Alpha/beta hydrolase fold family protein |
| gene3797 | 4.40 | 8.05 × 10−12 | 3.60 | 2.13 × 10−12 | Amidohydrolase family protein |
| gene0510 | 4.40 | 9.63 × 10−17 | 4.25 | 3.45 × 10−33 | Fumarylacetoacetate (FAA) hydrolase family protein |
| gene0800 | 4.04 | 1.44 × 10−2 | 6.09 | 2.09 × 10−4 | Glycosyl hydrolase |
| gene1702 | 2.73 | 9.47 × 10−6 | 4.61 | 2.82 × 10−25 | Metal dependent phosphohydrolase |
| gene1917 | 3.10 | 3.78 × 10−5 | 5.27 | 7.59 × 10−48 | Membrane-bound metal-dependent hydrolase |
| gene2086 | 1.21 | 3.56 × 10−2 | 1.25 | 3.42 × 10−4 | Alpha/beta hydrolase fold family protein |
| gene2140 | 2.10 | 3.17 × 10−2 | 1.09 | 2.07 × 10−3 | Amylopullulanase |
| gene3978 | 3.62 | 6.60 × 10−4 | 2.59 | 5.05 × 10−16 | Chitinase A1 |
| gene0479 | 3.74 | 1.88 × 10−8 | 3.48 | 1.75 × 10−5 | Peptidase M23 family protein |
| gene1871 | 7.77 | 1.15 × 10−17 | 8.58 | 5.53 × 10−18 | Peptidase M23 family protein |
| gene3430 | 5.06 | 9.12 × 10−7 | 3.75 | 1.63 × 10−7 | Peptidase M23/M37 family protein |
| gene0291 | 5.00 | 2.48 × 10−5 | 3.73 | 7.53 × 10−6 | Peptidase M20 family protein |
| gene1081 | 5.98 | 8.93 × 10−17 | 4.50 | 2.08 × 10−3 | Minor extracellular protease Vpr domain protein |
| gene2099 | 2.44 | 1.79 × 10−6 | 2.32 | 2.70 × 10−12 | Intracellular protease |
| gene0588 | 7.01 | 5.70 × 10−5 | 5.26 | 2.53 × 10−17 | Serine protease HtrA |
| gene2096 | 4.37 | 7.44 × 10−3 | – | – | Alpha/beta hydrolase fold family protein |
| gene1396 | 1.12 | 3.49 × 10−2 | – | – | Peptidyl-tRNA hydrolase |
| gene2293 | 2.27 | 3.88 × 10−3 | – | – | Oligoendopeptidase F |
| gene2996 | 1.52 | 1.58 × 10−2 | – | – | Peptidase SpoIVB |
| gene2763 | 1.60 | 7.96 × 10−4 | – | – | Metalloprotease YpwA |
| gene1264 | – | – | 4.10 | 1.38 × 10−2 | Acetyltransferases and hydrolases with the alpha/beta hydrolase fold |
| gene1950 | – | – | 1.70 | 9.00 × 10−5 | MazG nucleotide pyrophosphohydrolase domain protein |
| gene1974 | – | – | 1.63 | 9.93 × 10−4 | Glycoside hydrolase family 18 |
| Gene ID | MB2 vs. CBL | MB4 vs. CBL | Description | ||
|---|---|---|---|---|---|
| log2(Fold Change) | p-Value | log2(Fold Change) | p-Value | ||
| Novobiocin biosynthesis | |||||
| gene3280 | 2.01 | 1.82 × 10−2 | 1.18 | 1.39 × 10−3 | Histidinol-phosphate aminotransferase |
| gene3516 | −4.17 | 7.39 × 10−18 | −2.84 | 9.13 × 10−17 | Threonine-phosphate decarboxylase |
| Prodigiosin biosynthesis | |||||
| gene3924 | 4.36 | 6.65 × 10−7 | 3.83 | 1.70 × 10−6 | Short chain dehydrogenase family protein |
| gene1867 | 5.08 | 9.86 × 10−14 | 4.62 | 2.10 × 10−30 | 3-oxoacyl-[acyl-carrier-protein] reductase |
| gene3734 | 6.01 | 4.80 × 10−11 | 4.52 | 7.65 × 10−20 | Malonyl CoA-acyl carrier protein transacylase |
| gene3394 | 6.55 | 2.29 × 10−33 | 5.96 | 6.87 × 10−66 | Malonyl CoA-acyl carrier protein transacylase |
| gene4412 | 1.37 | 6.10 × 10−3 | – | – | Putative 3-oxoacyl-[acyl-carrier protein] reductase |
| gene4621 | −2.01 | 8.40 × 10−6 | −3.57 | 8.84 × 10−25 | 3-oxoacyl-[acyl-carrier-protein] reductase |
| gene4622 | −2.30 | 3.87 × 10−7 | −3.55 | 5.31 × 10−25 | Malonyl CoA-acyl carrier protein transacylase |
| gene2031 | – | – | −2.14 | 2.97 × 10−10 | Enoyl-[acyl-carrier-protein] reductase |
| gene3922 | – | – | −1.85 | 4.54 × 10−5 | Malonyl CoA-acyl carrier protein transacylase |
| Acarbose and validamycin biosynthesis | |||||
| gene1620 | 3.30 | 8.16 × 10−3 | 2.25 | 4.18 × 10−4 | CDP-glucose 4,6-dehydratase |
| Streptomycin biosynthesis | |||||
| gene1620 | 3.30 | 8.16 × 10−3 | 2.25 | 4.18 × 10−4 | CDP-glucose 4,6-dehydratase |
| gene3643 | 2.42 | 4.89 × 10−5 | – | – | Myo-inositol-1-phosphate synthase family protein |
| gene0637 | – | – | 1.81 | 9.29 × 10−7 | Inositol monophosphatase family protein |
| Monobactam biosynthesis | |||||
| gene2420 | 1.97 | 1.33 × 10−3 | 1.75 | 1.95 × 10−5 | Aspartokinase 2 |
| gene2342 | 2.41 | 1.91 × 10−5 | – | – | Sulfate adenylyltransferase |
| gene3298 | −1.43 | 4.14 × 10−3 | – | – | Dihydrodipicolinate reductase |
| Carbapenem biosynthesis | |||||
| gene0853 | −1.04 | 3.28 × 10−2 | −1.09 | 7.42 × 10−3 | Glutamate-5-semialdehyde dehydrogenase |
| gene0853 | – | – | −1.75 | 1.93 × 10−7 | Glutamate-5-semialdehyde dehydrogenase |
| Phenazine biosynthesis | |||||
| gene3274 | – | – | 5.34 | 7.17 × 10−9 | Anthranilate synthase component |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Li, J.; Hu, Z.; Chen, D.; Li, F.; Huang, X.; Li, C. Transcriptome Analysis Reveals the Algicidal Mechanism of Brevibacillus laterosporus against Microcystis aeruginosa through Multiple Metabolic Pathways. Toxins 2022, 14, 492. https://doi.org/10.3390/toxins14070492
Zhang Y, Li J, Hu Z, Chen D, Li F, Huang X, Li C. Transcriptome Analysis Reveals the Algicidal Mechanism of Brevibacillus laterosporus against Microcystis aeruginosa through Multiple Metabolic Pathways. Toxins. 2022; 14(7):492. https://doi.org/10.3390/toxins14070492
Chicago/Turabian StyleZhang, Yulei, Jieyi Li, Zhangxi Hu, Dong Chen, Feng Li, Xianghu Huang, and Changling Li. 2022. "Transcriptome Analysis Reveals the Algicidal Mechanism of Brevibacillus laterosporus against Microcystis aeruginosa through Multiple Metabolic Pathways" Toxins 14, no. 7: 492. https://doi.org/10.3390/toxins14070492
APA StyleZhang, Y., Li, J., Hu, Z., Chen, D., Li, F., Huang, X., & Li, C. (2022). Transcriptome Analysis Reveals the Algicidal Mechanism of Brevibacillus laterosporus against Microcystis aeruginosa through Multiple Metabolic Pathways. Toxins, 14(7), 492. https://doi.org/10.3390/toxins14070492