LC-MS/MS Analysis of Cyanotoxins in Bivalve Mollusks—Method Development, Validation and First Evidence of Occurrence of Nodularin in Mussels (Mytilus edulis) and Oysters (Magallana gigas) from the West Coast of Sweden

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Method Development
2.2. Method Validation
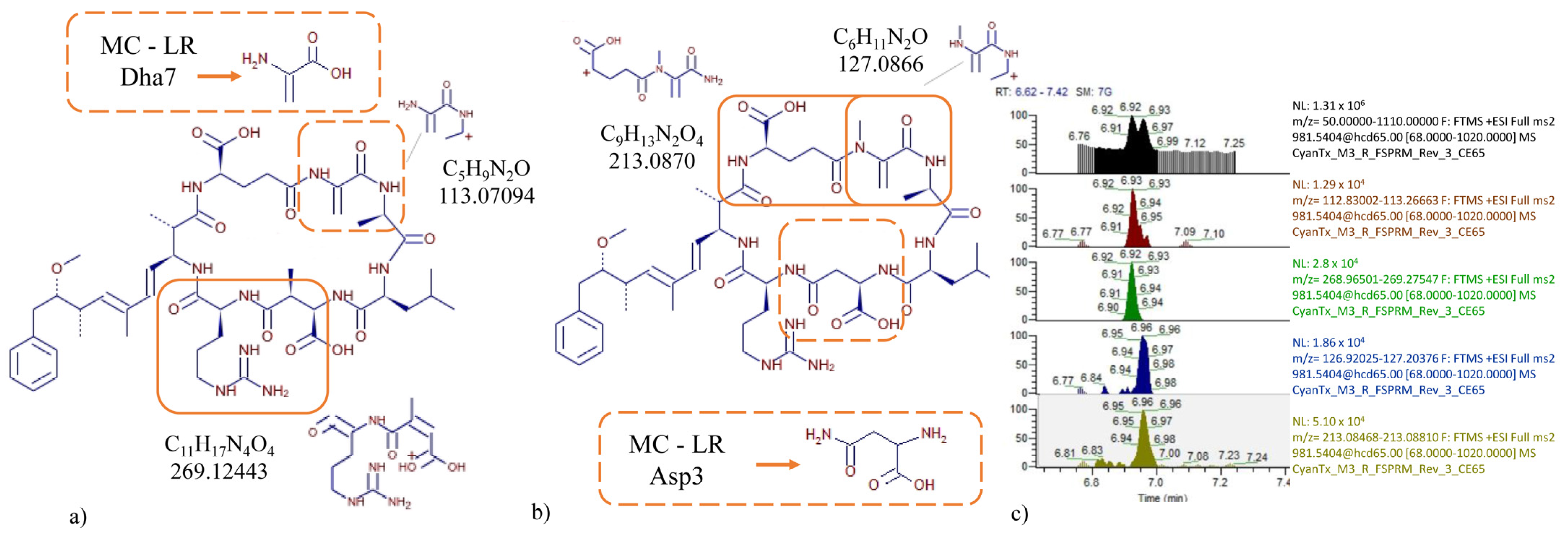
2.2.1. Specificity
2.2.2. Calibration, Linearity and Matrix Effects in ESI-MS
2.2.3. Limit of Quantification
2.2.4. Recovery
2.2.5. Results from Qualitative and Quantitative LC-MS/MS Analysis of Cyanotoxins in Field Samples
Analysis of Phytoplankton
Analysis of Cyanotoxins in Authentic Bivalve Samples
3. Conclusions
4. Material and Methods
4.1. Chemicals, Consumables and Standards
4.2. Samples and Sample Preparation
4.2.1. Bivalve Samples
4.2.2. Phytoplankton Samples
4.3. LC-MS/MS (QQQ)
4.4. LC-HRMS/MS (Hybrid Q-Orbitrap)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huisman, J.; Codd, G.A.; Paerl, H.W.; Ibelings, B.W.; Verspagen, J.M.H.; Visser, P.M. Cyanobacterial blooms. Nat. Rev. Microbiol. 2018, 16, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, W.W.; Azevedo, S.M.; An, J.S.; Molica, R.J.; Jochimsen, E.M.; Lau, S.; Rinehart, K.L.; Shaw, G.R.; Eaglesham, G.K. Human fatalities from cyanobacteria: Chemical and biological evidence for cyanotoxins. Environ. Health Perspect. 2001, 109, 663–668. [Google Scholar] [CrossRef] [PubMed]
- van der Merwe, D. Chapter 31—Cyanobacterial (Blue-Green Algae) Toxins. In Handbook of Toxicology of Chemical Warfare Agents, 2nd ed.; Gupta, R.C., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 421–429. [Google Scholar]
- Dixit, A.; Dhaked, R.K.; Alam, S.I.; Singh, L. MILITARY POTENTIAL OF BIOLOGICAL NEUROTOXINS. Toxin Rev. 2005, 24, 175–207. [Google Scholar] [CrossRef]
- Walter, J.M.; Coutinho, F.H.; Dutilh, B.E.; Swings, J.; Thompson, F.L.; Thompson, C.C. Ecogenomics and Taxonomy of Cyanobacteria Phylum. Front. Microbiol. 2017, 8, 2132. [Google Scholar] [CrossRef] [PubMed]
- van Apeldoorn, M.E.; van Egmond, H.P.; Speijers, G.J.; Bakker, G.J. Toxins of cyanobacteria. Mol. Nutr. Food Res. 2007, 51, 7–60. [Google Scholar] [CrossRef]
- Rastogi, R.P.; Madamwar, D.; Incharoensakdi, A. Bloom Dynamics of Cyanobacteria and Their Toxins: Environmental Health Impacts and Mitigation Strategies. Front. Microbiol. 2015, 6, 01254. [Google Scholar] [CrossRef]
- Rastogi, R.P.; Sinha, R.P.; Incharoensakdi, A. The cyanotoxin-microcystins: Current overview. Rev. Environ. Sci. Biotechnol. 2014, 13, 215–249. [Google Scholar] [CrossRef]
- Neilan, B.A.; Pearson, L.A.; Muenchhoff, J.; Moffitt, M.C.; Dittmann, E. Environmental conditions that influence toxin biosynthesis in cyanobacteria. Environ. Microbiol. 2013, 15, 1239–1253. [Google Scholar] [CrossRef]
- Häder, D.P.; Villafañe, V.E.; Helbling, E.W. Productivity of aquatic primary producers under global climate change. Photochem. Photobiol. Sci. 2014, 13, 1370–1392. [Google Scholar] [CrossRef]
- El-Shehawy, R.; Gorokhova, E.; Fernández-Piñas, F.; del Campo, F.F. Global warming and hepatotoxin production by cyanobacteria: What can we learn from experiments? Water Res. 2012, 46, 1420–1429. [Google Scholar] [CrossRef]
- Häder, D.-P.; Gao, K. Interactions of anthropogenic stress factors on marine phytoplankton. Front. Environ. Sci. 2015, 3, 14. [Google Scholar] [CrossRef]
- EPA-810F11001; United States Environmental Protection Agency. Cyanobacteria and Cyanotoxins: Information for Drinking Water Systems. EPA: Washington, DC, USA, 2014.
- Massey, I.Y.; Wu, P.; Wei, J.; Luo, J.; Ding, P.; Wei, H.; Yang, F. A Mini-Review on Detection Methods of Microcystins. Toxins 2020, 12, 641. [Google Scholar] [CrossRef] [PubMed]
- Bouaïcha, N.; Miles, C.O.; Beach, D.G.; Labidi, Z.; Djabri, A.; Benayache, N.Y.; Nguyen-Quang, T. Structural Diversity, Characterization and Toxicology of Microcystins. Toxins 2019, 11, 714. [Google Scholar] [CrossRef] [PubMed]
- Sivonen, K.; Kononen, K.; Carmichael, W.W.; Dahlem, A.M.; Rinehart, K.L.; Kiviranta, J.; Niemela, S.I. Occurrence of the hepatotoxic cyanobacterium Nodularia spumigena in the Baltic Sea and structure of the toxin. Appl. Environ. Microbiol. 1989, 55, 1990–1995. [Google Scholar] [CrossRef] [PubMed]
- Jokela, J.; Heinilä, L.M.P.; Shishido, T.K.; Wahlsten, M.; Fewer, D.P.; Fiore, M.F.; Wang, H.; Haapaniemi, E.; Permi, P.; Sivonen, K. Production of High Amounts of Hepatotoxin Nodularin and New Protease Inhibitors Pseudospumigins by the Brazilian Benthic Nostoc sp. CENA543. Front. Microbiol. 2017, 8, 1963. [Google Scholar] [CrossRef]
- Turner, A.D.; Waack, J.; Lewis, A.; Edwards, C.; Lawton, L. Development and single-laboratory validation of a UHPLC-MS/MS method for quantitation of microcystins and nodularin in natural water, cyanobacteria, shellfish and algal supplement tablet powders. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018, 1074–1075, 111–123. [Google Scholar] [CrossRef]
- Imanishi, S.; Kato, H.; Mizuno, M.; Tsuji, K.; Harada, K. Bacterial degradation of microcystins and nodularin. Chem. Res. Toxicol. 2005, 18, 591–598. [Google Scholar] [CrossRef]
- Dittmann, E.; Wiegand, C. Cyanobacterial toxins-occurrence, biosynthesis and impact on human affairs. Mol. Nutr. Food Res. 2006, 50, 7–17. [Google Scholar] [CrossRef]
- Blahova, L.; Sehnal, L.; Lepsova-Skacelova, O.; Szmucova, V.; Babica, P.; Hilscherova, K.; Teikari, J.; Sivonen, K.; Blaha, L. Occurrence of cylindrospermopsin, anatoxin-a and their homologs in the southern Czech Republic—Taxonomical, analytical, and molecular approaches. Harmful Algae 2021, 108, 102101. [Google Scholar] [CrossRef]
- Chorus, I.; Welker, M. Toxic Cyanobacteria in Water, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2021; on behalf of the World Health Organization, Geneva, Switzerland. [Google Scholar]
- Pekar, H.; Westerberg, E.; Bruno, O.; Lääne, A.; Persson, K.M.; Sundström, L.F.; Thim, A.M. Fast, rugged and sensitive ultra high pressure liquid chromatography tandem mass spectrometry method for analysis of cyanotoxins in raw water and drinking water-First findings of anatoxins, cylindrospermopsins and microcystin variants in Swedish source waters and infiltration ponds. J. Chromatogr. A 2016, 1429, 265–276. [Google Scholar] [CrossRef]
- WHO. World Health Organisation Report, 2003. Guidelines for Safe Recreational Water Environments. Volume 1, Coastal and Fresh Waters; 9241545801; World Health Organization: Geneva, Switzerland, 2003. [Google Scholar]
- Abdallah, M.F.; Van Hassel, W.H.R.; Andjelkovic, M.; Wilmotte, A.; Rajkovic, A. Cyanotoxins and Food Contamination in Developing Countries: Review of Their Types, Toxicity, Analysis, Occurrence and Mitigation Strategies. Toxins 2021, 13, 786. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, S.; Jiang, X. Cyanobacterial Toxins in Freshwater and Food: Important Sources of Exposure to Humans. Annu. Rev. Food Sci. Technol. 2017, 8, 281–304. [Google Scholar] [CrossRef]
- Turner, A.D.; Turner, F.R.I.; White, M.; Hartnell, D.; Crompton, C.G.; Bates, N.; Egginton, J.; Branscombe, L.; Lewis, A.M.; Maskrey, B.H. Confirmation Using Triple Quadrupole and High-Resolution Mass Spectrometry of a Fatal Canine Neurotoxicosis following Exposure to Anatoxins at an Inland Reservoir. Toxins 2022, 14, 804. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Foss, A.; Miller, M.A.; Gibson, Q. Detection of cyanotoxins (microcystins/nodularins) in livers from estuarine and coastal bottlenose dolphins (Tursiops truncatus) from Northeast Florida. Harmful Algae 2018, 76, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, N.; Nikitin, O. Cyanotoxins as a possible cause of fish and waterfowl death in the Kazanka River (Russia), In Proceedings of the International Multidisciplinary Scientific GeoConference-SGEM, Albena, Bulgaria, 2–8 July 2018. [CrossRef]
- Benayache, N.Y.; Afri-Mehennaoui, F.Z.; Kherief-Nacereddine, S.; Vo-Quoc, B.; Hushchyna, K.; Nguyen-Quang, T.; Bouaïcha, N. Massive fish death associated with the toxic cyanobacterial Planktothrix sp. bloom in the Béni-Haroun Reservoir (Algeria). Environ. Sci. Pollut. Res. Int. 2022, 29, 80849–80859. [Google Scholar] [CrossRef]
- Biré, R.; Bertin, T.; Dom, I.; Hort, V.; Schmitt, C.; Diogène, J.; Lemée, R.; De Haro, L.; Nicolas, M. First Evidence of the Presence of Anatoxin-A in Sea Figs Associated with Human Food Poisonings in France. Mar. Drugs 2020, 18, 285. [Google Scholar] [CrossRef] [PubMed]
- Wood, R. Acute animal and human poisonings from cyanotoxin exposure—A review of the literature. Environ. Int. 2016, 91, 276–282. [Google Scholar] [CrossRef]
- De Pace, R.; Vita, V.; Bucci, M.S.; Gallo, P.; Bruno, M. Microcystin Contamination in Sea Mussel Farms from the Italian Southern Adriatic Coast following Cyanobacterial Blooms in an Artificial Reservoir. J. Ecosyst. 2014, 2014, 374027. [Google Scholar] [CrossRef]
- Aparicio-Muriana, M.D.M.; Lara, F.J.; Olmo-Iruela, M.D.; García-Campaña, A.M. Determination of Multiclass Cyanotoxins in Blue-Green Algae (BGA) Dietary Supplements Using Hydrophilic Interaction Liquid Chromatography-Tandem Mass Spectrometry. Toxins 2023, 15, 127. [Google Scholar] [CrossRef]
- WHO. World Health Organization Repport, 2017. Guidelines for Drinking-Water Quality, 4th ed.; Incorporating the First Addendum; World Health Organization: Geneva, Switzerland, 2017; ISBN 978-92-4-154995-0. [Google Scholar]
- Ibelings, B.W.; Chorus, I. Accumulation of cyanobacterial toxins in freshwater “seafood” and its consequences for public health: A review. Environ. Pollut. 2007, 150, 177–192. [Google Scholar] [CrossRef]
- Bernes, C. Biologisk Mångfald i Sverige; Naturvårdsverket: Stockholm, Sweden, 2011; Volume Monitor 22. [Google Scholar]
- Bertos-Fortis, M.; Farnelid, H.M.; Lindh, M.V.; Casini, M.; Andersson, A.; Pinhassi, J.; Legrand, C. Unscrambling Cyanobacteria Community Dynamics Related to Environmental Factors. Front. Microbiol. 2016, 7, 625. [Google Scholar] [CrossRef] [PubMed]
- Willén, E. Phytoplankton and water quality characterization: Experiences from the Swedish large lakes Mälaren, Hjälmaren, Vättern and Vänern. Ambio 2001, 30, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.; Ahlgren, G.; Willén, E. Bioaccumulation of microcystins in the food web: A field study of four Swedish lakes. Inland Waters 2014, 4, 91–104. [Google Scholar] [CrossRef]
- Munkes, B.; Löptien, U.; Dietze, H. Cyanobacteria blooms in the Baltic Sea: A review of models and facts. Biogeosciences 2021, 18, 2347–2378. [Google Scholar] [CrossRef]
- Karlson, B.; Andersen, P.; Arneborg, L.; Cembella, A.; Eikrem, W.; John, U.; West, J.J.; Klemm, K.; Kobos, J.; Lehtinen, S.; et al. Harmful algal blooms and their effects in coastal seas of Northern Europe. Harmful Algae 2021, 102, 101989. [Google Scholar] [CrossRef] [PubMed]
- EPA-815B16011; Method 546—Determination of Total Microcystins and Nodularins in Drinking Water and Ambient Water by Adda Enzyme-Linked Immunosorbent Assay. Standards and Risk Management Division. Technical Support Center, Office of Ground Water and Drinking Water: Cincinnati, OH, USA, 2016.
- Lawton, L.A.; Edwards, C.; Codd, G.A. Extraction and high-performance liquid chromatographic method for the determination of microcystins in raw and treated waters. Analyst 1994, 119, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Meriluoto, J. Chromatography of microcystins. Anal. Chim. Acta 1997, 352, 277–298. [Google Scholar] [CrossRef]
- Metcalf, J.S.; Beattie, K.A.; Pflugmacher, S.; Codd, G.A. Immuno-crossreactivity and toxicity assessment of conjugation products of the cyanobacterial toxin, microcystin-LR. FEMS Microbiol. Lett. 2000, 189, 155–158. [Google Scholar] [CrossRef]
- Karlsson, K.M.; Spoof, L.E.; Meriluoto, J.A. Quantitative LC-ESI-MS analyses of microcystins and nodularin-R in animal tissue--matrix effects and method validation. Environ. Toxicol. 2005, 20, 381–389. [Google Scholar] [CrossRef]
- Klijnstra, M.D.; Faassen, E.J.; Gerssen, A. A Generic LC-HRMS Screening Method for Marine and Freshwater Phycotoxins in Fish, Shellfish, Water, and Supplements. Toxins 2021, 13, 823. [Google Scholar] [CrossRef]
- Van Hassel, W.H.R.; Masquelier, J.; Andjelkovic, M.; Rajkovic, A. Towards a Better Quantification of Cyanotoxins in Fruits and Vegetables: Validation and Application of an UHPLC-MS/MS-Based Method on Belgian Products. Separations 2022, 9, 319. [Google Scholar] [CrossRef]
- Van Hassel, W.H.R.; Ahn, A.-C.; Huybrechts, B.; Masquelier, J.; Wilmotte, A.; Andjelkovic, M. LC-MS/MS Validation and Quantification of Cyanotoxins in Algal Food Supplements from the Belgium Market and Their Molecular Origins. Toxins 2022, 14, 513. [Google Scholar] [CrossRef]
- Van Hassel, W.H.R.; Andjelkovic, M.; Durieu, B.; Marroquin, V.A.; Masquelier, J.; Huybrechts, B.; Wilmotte, A. A Summer of Cyanobacterial Blooms in Belgian Waterbodies: Microcystin Quantification and Molecular Characterizations. Toxins 2022, 14, 61. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, K.; Sipiä, V.; Kankaanpää, H.; Meriluoto, J. Mass spectrometric detection of nodularin and desmethylnodularin in mussels and flounders. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2003, 784, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Mazur-Marzec, H.; Tymińska, A.; Szafranek, J.; Pliński, M. Accumulation of nodularin in sediments, mussels, and fish from the Gulf of Gdańsk, southern Baltic Sea. Environ. Toxicol. 2007, 22, 101–111. [Google Scholar] [CrossRef]
- Sundaravadivelu, D.; Sanan, T.T.; Venkatapathy, R.; Mash, H.; Tettenhorst, D.; DAnglada, L.; Frey, S.; Tatters, A.O.; Lazorchak, J. Determination of Cyanotoxins and Prymnesins in Water, Fish Tissue, and Other Matrices: A Review. Toxins 2022, 14, 213. [Google Scholar] [CrossRef]
- Haddad, S.P.; Bobbitt, J.M.; Taylor, R.B.; Lovin, L.M.; Conkle, J.L.; Chambliss, C.K.; Brooks, B.W. Determination of microcystins, nodularin, anatoxin-a, cylindrospermopsin, and saxitoxin in water and fish tissue using isotope dilution liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2019, 1599, 66–74. [Google Scholar] [CrossRef] [PubMed]
- 2002/657/EC: Commission Decision of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results (Text with EEA Relevance) (Notified under Document Number C(2002) 3044). 2002, pp. 8–36. Available online: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX%3A32002D0657 (accessed on 17 March 2023).
- Lawrence, J.F.; Niedzwiadek, B.; Menard, C. Quantitative determination of paralytic shellfish poisoning toxins in shellfish using prechromatographic oxidation and liquid chromatography with fluorescence detection: Collaborative study. J. AOAC Int. 2005, 88, 1714–1732. [Google Scholar] [CrossRef]
- Patria, F.P.; Pekar, H.; Zuberovic-Muratovic, A. Multi-Toxin Quantitative Analysis of Paralytic Shellfish Toxins and Tetrodotoxins in Bivalve Mollusks with Ultra-Performance Hydrophilic Interaction LC-MS/MS-An In-House Validation Study. Toxins 2020, 12, 452. [Google Scholar] [CrossRef]
- Lemoine, P.; Roy-Lachapelle, A.; Prévost, M.; Tremblay, P.; Solliec, M.; Sauvé, S. Ultra-fast analysis of anatoxin—A using laser diode thermal desorption-atmospheric pressure chemical ionization-tandem mass spectrometry: Validation and resolution from phenylalanine. Toxicon 2013, 61, 165–174. [Google Scholar] [CrossRef]
- Kankaanpää, H.; Leiniö, S.; Olin, M.; Sjövall, O.; Meriluoto, J.; Lehtonen, K.K. Accumulation and depuration of cyanobacterial toxin nodularin and biomarker responses in the mussel Mytilus edulis. Chemosphere 2007, 68, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Testai, E.; Buratti, F.M.; Funari, E.; Manganelli, M.; Vichi, S.; Arnich, N.; Biré, R.; Fessard, V.; Sialehaamoa, A. Review and analysis of occurrence, exposure and toxicity of cyanobacteria toxins in food. EFSA Support. Publ. 2016, 13, 998E. [Google Scholar] [CrossRef]
- Birbeck, J.A.; Peraino, N.J.; O’Neill, G.M.; Coady, J.; Westrick, J.A. Dhb Microcystins Discovered in USA Using an Online Concentration LC-MS/MS Platform. Toxins 2019, 11, 653. [Google Scholar] [CrossRef] [PubMed]
- Roy-Lachapelle, A.; Vo Duy, S.; Munoz, G.; Dinh, Q.T.; Bahl, E.; Simon, D.F.; Sauv, S. Analysis of multiclass cyanotoxins (microcystins, anabaenopeptins, cylindrospermopsin and anatoxins) in lake waters using on-line SPE liquid chromatography high-resolution Orbitrap mass spectrometry. Anal. Methods 2019, 11, 5289–5300. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Toxins | Cal. Range | Regression | %ME | ||
|---|---|---|---|---|---|
| (ng/mL) | Type | WLS * | r2 | Mussel | |
| Anatoxin A | 0.39–25 | Linear | 1/X2 | 0.998 | 78 |
| Homoanatoxin A | 0.39–25 | Linear | 1/X2 | 0.998 | 80 |
| Cylindrospermopsin | 0.78–25 | Quadratic | 1/Y | 0.988 | 16 |
| Microcystins RR Sum ** | 0.78–25 | Linear | 1/X | 0.993 | 115 |
| Microcystin RR | 0.39–25 | Linear | 1/X | 0.992 | 107 |
| Nodularin | 0.39–25 | Linear | 1/X | 0.998 | 104 |
| Microcystin LA | 0.39–25 | Linear | 1/Y | 0.977 | 125 |
| Microcystin LR [Dha7] | 0.78–25 | Linear | 1/X | 0.991 | 171 |
| Microcystin LR [Asp3] | 0.78–25 | Linear | 1/X | 0.992 | 162 |
| Microcystin LF | 0.39–25 | Linear | 1/X2 | 0.944 | 61 |
| Microcystin LR | 0.39–25 | Linear | 1/X | 0.992 | 167 |
| Microcystin LY | 0.39–25 | Linear | 1/Y | 0.989 | 118 |
| Microcystin HilR | 0.39–25 | Linear | 1/X | 0.987 | 175 |
| Microcystin LW | 0.78–25 | Linear | 1/X2 | 0.978 | 82 |
| Microcystin YR | 0.39–25 | Linear | 1/X | 0.992 | 208 |
| Microcystin HtyR | 0.39–25 | Linear | 1/X | 0.990 | 181 |
| Microcystin WR | 0.78–25 | Linear | 1/X | 0.984 | 179 |
| Toxins | Calculated (ng/mL) | LOD * (µg/kg) | LOQ * (µg/kg) | Recovery (%) | RSDr (%) | RSDR (%) |
|---|---|---|---|---|---|---|
| Anatoxin A | 0.94 | 2.51 | 7.52 | 60.3 | 1.6 | 6.6 |
| Homoanatoxin A | 0.81 | 2.17 | 6.50 | 52.1 | 1.6 | 6.9 |
| Cylindrospermopsin | 1.16 | 3.10 | 9.30 | 18.6 | 7.4 | 15.0 |
| Microcystins RR Sum ** | 1.41 | 3.75 | 11.26 | 90.3 | 1.5 | 5.6 |
| Microcystin RR | 1.48 | 3.95 | 11.85 | 94.9 | 1.3 | 7.6 |
| Nodularin | 0.98 | 2.61 | 7.83 | 62.7 | 2.4 | 7.1 |
| Microcystin LA | 1.09 | 2.92 | 8.76 | 70.2 | 2.4 | 8.6 |
| Microcystin LR [Dha7] | 1.35 | 3.59 | 10.78 | 86.4 | 3.0 | 9.4 |
| Microcystin LR [Asp3] | 1.16 | 3.10 | 9.29 | 74.4 | 3.0 | 11.9 |
| Microcystin LF | 0.79 | 2.10 | 6.29 | 50.4 | 5.0 | 18.0 |
| Microcystin LR | 1.22 | 3.25 | 9.74 | 78.1 | 1.7 | 6.6 |
| Microcystin LY | 1.19 | 3.17 | 9.50 | 76.1 | 1.9 | 7.8 |
| Microcystin HilR | 1.32 | 3.52 | 10.55 | 84.5 | 1.6 | 6.3 |
| Microcystin LW | 0.96 | 2.57 | 7.70 | 61.7 | 3.4 | 12.3 |
| Microcystin YR | 1.32 | 3.53 | 10.58 | 84.7 | 5.5 | 16.9 |
| Microcystin HtyR | 1.15 | 3.07 | 9.21 | 73.8 | 1.6 | 6.4 |
| Microcystin WR | 1.51 | 4.04 | 12.11 | 97.0 | 1.6 | 6.4 |
| Toxins | Low-Level Spike (50 µg/kg) | Mid-Level Spike (100 µg/kg) | High-Level Spike (200 µg/kg) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Rec. (%) | RSDr (%) | RSDR (%) | Rec. (%) | RSDr (%) | RSDR (%) | Rec. (%) | RSDr (%) | RSDR (%) | |
| Anatoxin A | 60.6 | 1.6 | 4.1 | 57.9 | 1.8 | 6.0 | 58.9 | 1.2 | 12.4 |
| Homoanatoxin A | 52.2 | 1.5 | 4.5 | 48.6 | 1.7 | 5.2 | 54.0 | 2.1 | 8.4 |
| Cylindrospermopsin | 18.6 | 7.4 | 15.0 | 19.1 | 4.7 | 11.3 | 30.7 | 4.5 | 13.5 |
| Microcystins RR Sum ** | 72.4 | 2.8 | 6.0 | 66.1 | 1.7 | 5.4 | 70.5 | 2.0 | 6.7 |
| Microcystin RR | 81.6 | 2.8 | 13.6 | 71.5 | 1.7 | 7.1 | 79.8 | 2.1 | 5.1 |
| Nodularin | 63.5 | 2.2 | 4.4 | 62.3 | 1.6 | 6.1 | 74.2 | 1.7 | 6.7 |
| Microcystin LA | 69.5 | 3.1 | 12.3 | 65.6 | 1.4 | 7.5 | 75.3 | 3.0 | 27.7 |
| Microcystin LR [Dha7] | 78.7 | 3.0 | 8.9 | 74.6 | 1.7 | 5.1 | 88.7 | 2.8 | 9.3 |
| Microcystin LR [Asp3] | 70.3 | 2.8 | 8.4 | 67.3 | 2.3 | 6.7 | 82.5 | 2.5 | 9.9 |
| Microcystin LF | 51.4 | 2.7 | 7.5 | 49.2 | 4.3 | 10.7 | 83.0 | 7.3 | 25.5 |
| Microcystin LR | 75.2 | 3.4 | 7.8 | 72.4 | 2.7 | 6.6 | 86.2 | 2.0 | 7.9 |
| Microcystin LY | 68.9 | 1.6 | 4.4 | 65.5 | 1.1 | 6.4 | 73.5 | 2.6 | 11.9 |
| Microcystin HilR | 77.9 | 2.9 | 6.7 | 76.0 | 2.3 | 6.5 | 87.4 | 1.7 | 7.3 |
| Microcystin LW | 53.2 | 1.5 | 7.2 | 50.6 | 1.9 | 7.8 | 72.2 | 5.2 | 17.9 |
| Microcystin YR | 77.6 | 3.4 | 10.9 | 71.6 | 1.8 | 5.5 | 88.8 | 2.9 | 9.9 |
| Microcystin HtyR | 71.5 | 3.8 | 8.8 | 69.5 | 2.0 | 6.7 | 82.9 | 2.9 | 8.6 |
| Microcystin WR | 81.7 | 3.3 | 7.6 | 76.8 | 1.4 | 5.2 | 87.2 | 2.0 | 6.3 |
| Sample Identity | Sampling Station | Date | Time | Filtered Volume (mL) | Nodularin * (µg/L) |
|---|---|---|---|---|---|
| SLV-01, Släggö, HOSE, 20210713 | Släggö | 2021-07-13 | 07:00 | 500 | <LOQ |
| SLV-02, Å17, HOSE, 20210713 | Å17 | 2021-07-13 | 15:45 | 500 | <LOQ |
| SLV-03, N14, HOSE, 20210714 | N14 | 2021-07-14 | 05:05 | 500 | 8.4 |
| SLV-04, Anholt, HOSE, 20210714 | Anholt | 2021-07-14 | 08:00 | 500 | 6.1 |
| SLV-05, BY2, HOSE, 20210715 | BY2 | 2021-07-15 | 05:15 | 500 | 10.8 |
| SLV-06, BY5, HOSE, 20210715, | BY5 | 2021-07-15 | 14:45 | 400 | 5.7 |
| SLV-07, BY15, HOSE, 20210716 | BY15 | 2021-07-16 | 18:00 | 400 | 12.4 |
| SLV-08, BY38, HOSE, 20210717 | BY38 | 2021-07-17 | 11:30 | 500 | 16.2 |
| SLV-09, RefM1V1, HOSE, 20210717 | RefM1V1 | 2021-07-17 | 23:20 | 400 | 26.1 |
| SLV-10, BY4, SURFACE, 20210715 | BY4 | 2021-07-15 | 11:20 | ND | ** |
| SLV-11, BY5, SURFACE, 20210715 | BY5 | 2021-07-15 | 14:45 | ND | ** |
| SLV-12, BY10, SURFACE, 20210716 | BY10 | 2021-07-16 | 09:55 | ND | ** |
| SLV-13, BY39, SURFACE, 20210717 | BY39 | 2021-07-17 | 20:00 | ND | ** |
| SLV-14, Hanö, SURFACE, 20210718 | Hanö Bight | 2021-07-18 | 06:30 | ND | ** |
| Sample Name | Date | Sampled Species | Ion Ratio Duplicate * | Nodularin (µg/kg) | Sample Duplicate Diff. (%) | |
|---|---|---|---|---|---|---|
| Sample a | Sample b | |||||
| SLV-1, Cyano | w32, 02/8-20 | MG | 1.72 | 1.65 | 142.3 | 1.1 |
| SLV-2, Cyano | w33, 10/8-20 | ME | 1.66 | 1.57 | 28.8 | 0.1 |
| SLV-3, Cyano | w33, 10/8-20 | ME | 1.59 | 1.59 | 11.4 | 4.7 |
| SLV-4, Cyano | w33, 10/8-20 | ME | 1.64 | 1.59 | 27.3 | 0.5 |
| SLV-5, Cyano | w33, 09/8-20 | ME | 1.60 | 1.63 | 26.7 | 0.9 |
| SLV-6, Cyano | w34, 17/8-20 | ME | 1.63 | 1.57 | 31.2 | 5.1 |
| SLV-7, Cyano | w34, 19/8-20 | ME | 1.60 | 1.60 | 26.0 | 0.1 |
| SLV-8, Cyano | w34, 16/8-20 | ME | 1.58 | 1.60 | 33.1 | 3.4 |
| SLV-9, Cyano | w28, 13/7-21 | ME | 1.65 | 1.55 | 6.7 | 1.1 |
| SLV-10, Cyano | w28, 13/7-21 | ME | 1.58 | 1.55 | 7.6 | 4.0 |
| SLV-11, Cyano | w28, 18/7-21 | ME | 1.55 | 1.59 | 10.9 | 1.8 |
| SLV-12, Cyano | w28, 18/7-21 | MG | 1.53 | 1.61 | 24.1 | 0.6 |
| SLV-13, Cyano | w37, 18/9-22 | MG | 1.67 | 1.63 | 397.3 | 1.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
España Amórtegui, J.C.; Pekar, H.; Retrato, M.D.C.; Persson, M.; Karlson, B.; Bergquist, J.; Zuberovic-Muratovic, A. LC-MS/MS Analysis of Cyanotoxins in Bivalve Mollusks—Method Development, Validation and First Evidence of Occurrence of Nodularin in Mussels (Mytilus edulis) and Oysters (Magallana gigas) from the West Coast of Sweden. Toxins 2023, 15, 329. https://doi.org/10.3390/toxins15050329
España Amórtegui JC, Pekar H, Retrato MDC, Persson M, Karlson B, Bergquist J, Zuberovic-Muratovic A. LC-MS/MS Analysis of Cyanotoxins in Bivalve Mollusks—Method Development, Validation and First Evidence of Occurrence of Nodularin in Mussels (Mytilus edulis) and Oysters (Magallana gigas) from the West Coast of Sweden. Toxins. 2023; 15(5):329. https://doi.org/10.3390/toxins15050329
Chicago/Turabian StyleEspaña Amórtegui, Julio César, Heidi Pekar, Mark Dennis Chico Retrato, Malin Persson, Bengt Karlson, Jonas Bergquist, and Aida Zuberovic-Muratovic. 2023. "LC-MS/MS Analysis of Cyanotoxins in Bivalve Mollusks—Method Development, Validation and First Evidence of Occurrence of Nodularin in Mussels (Mytilus edulis) and Oysters (Magallana gigas) from the West Coast of Sweden" Toxins 15, no. 5: 329. https://doi.org/10.3390/toxins15050329
APA StyleEspaña Amórtegui, J. C., Pekar, H., Retrato, M. D. C., Persson, M., Karlson, B., Bergquist, J., & Zuberovic-Muratovic, A. (2023). LC-MS/MS Analysis of Cyanotoxins in Bivalve Mollusks—Method Development, Validation and First Evidence of Occurrence of Nodularin in Mussels (Mytilus edulis) and Oysters (Magallana gigas) from the West Coast of Sweden. Toxins, 15(5), 329. https://doi.org/10.3390/toxins15050329