Proteotransciptomics of the Most Popular Host Sea Anemone Entacmaea quadricolor Reveals Not All Toxin Genes Expressed by Tentacles Are Recruited into Its Venom Arsenal

 , , ,
, , ,
Abstract
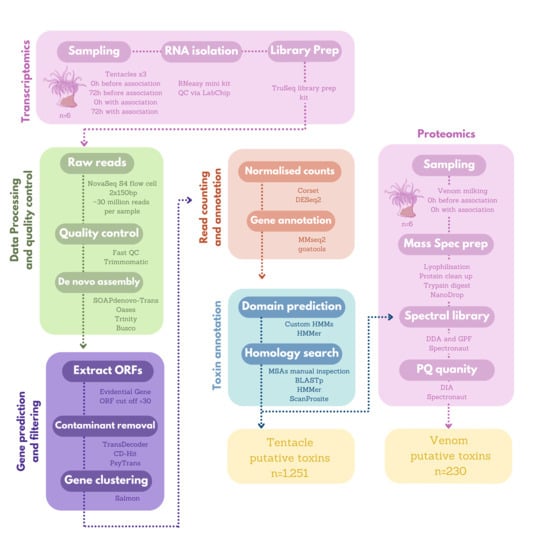
:
1. Introduction
2. Results
2.1. De Novo Tentacle Transcriptome Assembly
2.2. Venom Proteome

2.3. Putative Toxins
2.4. Toxin Tentacle Gene Clusters Detected in Venom
2.5. Exploration of the Dominant E. quadricolor Venom Phenotype
2.5.1. Toxins Assigned to the Unknown Venom Function Category
2.5.2. Haemostatic and Haemorrhagic Venom Category
2.5.3. Protease Inhibitor Venom Category
2.5.4. Mixed Function Enzymes Venom Category
2.5.5. Neurotoxin Venom Category
2.5.6. Pore-Forming Venom Category
3. Discussion
3.1. Putative Toxins
3.2. Venom Proteome
3.3. Where Are the Toxin Genes That Are Expressed Going?
3.4. Dominant Venom Hypothesis
4. Conclusions
5. Materials and Methods
5.1. Study Species and Experimental Set-Up
5.2. Sea Anemone Venom and Tentacle Collection
5.3. Transcriptomics
5.3.1. RNA Isolation and Library Preparation
5.3.2. RNA-Seq Read Quality Control
5.3.3. De Novo Transcriptome Assembly
5.3.4. Contaminant Removal
5.3.5. Clustering and Read Counting
5.3.6. Gene Annotation and Functional Enrichment Analysis
5.3.7. Toxin Annotation Pipeline
5.3.8. Signal P
5.3.9. Normalised Abundance of Transcripts
5.4. Proteomics
5.4.1. Venom Protein Extraction for Mass Spectrometry
5.4.2. Spectral Library Creation via DDA and GPF
5.4.3. Normalised Abundance of Protein Expression Utilising DIA
5.5. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Madio, B.; King, G.F.; Undheim, E.A.B. Sea Anemone Toxins: A Structural Overview. Mar. Drugs 2019, 17, 325. [Google Scholar] [CrossRef] [PubMed]
- Zancolli, G.; Reijnders, M.; Waterhouse, R.M.; Robinson-Rechavi, M. Convergent evolution of venom gland transcriptomes across Metazoa. Proc. Natl. Acad. Sci. USA 2022, 119, e2111392119. [Google Scholar] [CrossRef]
- Prentis, P.J.; Pavasovic, A.; Norton, R.S. Sea Anemones: Quiet achievers in the field of peptide toxins. Toxins 2018, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Surm, J.M.; Stewart, Z.K.; Papanicolaou, A.; Pavasovic, A.; Prentis, P.J. The draft genome of Actinia tenebrosa reveals insights into toxin evolution. Ecol. Evol. 2019, 9, 11314–11328. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, L.M.; Elnahriry, K.A.; Stewart, Z.K.; Shafee, T.; Naseem, M.U.; Szanto, T.G.; van der Burg, C.A.; Smith, H.L.; Surm, J.M.; Undheim, E.A.B.; et al. Genomic, functional and structural analyses elucidate evolutionary innovation within the sea anemone 8 toxin family. BMC Biol. 2023, 21, 121. [Google Scholar] [CrossRef]
- Sachkova, M.Y.; Landau, M.; Surm, J.M.; Macrander, J.; Singer, S.A.; Reitzel, A.M.; Moran, Y. Toxin-like neuropeptides in the sea anemone Nematostella unravel recruitment from the nervous system to venom. Proc. Natl. Acad. Sci. USA 2020, 117, 27481–27492. [Google Scholar] [CrossRef]
- Surm, J.M.; Moran, Y. Insights into how development and life-history dynamics shape the evolution of venom. Evodevo 2021, 12, 1. [Google Scholar] [CrossRef]
- Sunagar, K.; Moran, Y. The rise andfall of an evolutionary innovation: Contrasting strategies of venom evolution in ancient and young animals. PLoS Genet. 2015, 11, e1005596. [Google Scholar] [CrossRef]
- Rodríguez, E.; Fautin, D.G.; Daly, M. World List of Actiniaria. World Register of Marine Species. 2022. Available online: https://www.marinespecies.org/aphia.php?p=taxdetails&id=1360 (accessed on 23 August 2022).
- Titus, B.M.; Benedict, C.; Laroche, R.; Gusmao, L.C.; Van Deusen, V.; Chiodo, T.; Meyer, C.P.; Berumen, M.L.; Bartholomew, A.; Yanagi, K.; et al. Phylogenetic relationships among the clownfish-hosting sea anemones. Mol. Phylogenet. Evol. 2019, 139, 106526. [Google Scholar] [CrossRef]
- Burke da Silva, K.; Nedosyko, A. Sea Anemones and Anemonefish: A match made in heaven. In The Cnidaria, Past, Present and Future; Springer: Cham, Switzerland, 2016; pp. 425–438. [Google Scholar]
- Fautin, D.G. The Anemonefish Symbiosis: What is know and what is not. Symbiosis 1991, 10, 23–46. [Google Scholar]
- Tang, K.L.; Stiassny, M.L.J.; Mayden, R.L.; DeSalle, R. Systematics of damselfishes. Ichthyol. Herpetol. 2021, 109, 258–318. [Google Scholar] [CrossRef]
- Smith, E.G.; Surm, J.M.; Macrander, J.; Simhi, A.; Amir, G.; Sachkova, M.Y.; Lewandowska, M.; Reitzel, A.M.; Moran, Y. Micro and macroevolution of sea anemone venom phenotype. Nat. Commun. 2023, 14, 249. [Google Scholar] [CrossRef]
- Dutertre, S.; Jin, A.-H.; Vetter, I.; Hamilton, B.; Sunagar, K.; Lavergne, V.; Dutertre, V.; Fry, B.G.; Antunes, A.; Venter, D.J.; et al. Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails. Nat. Commun. 2014, 5, 3521. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, L.M.; Mitchell, M.L.; Madio, B.; Hurwood, D.A.; King, G.F.; Undheim, E.A.B.; Norton, R.S.; Prentis, P.J. Tentacle morphological variation coincides with differential expression of toxins in sea anemones. Toxins 2021, 13, 452. [Google Scholar] [CrossRef] [PubMed]
- Macrander, J.; Broe, M.; Daly, M. Tissue-Specific Venom Composition and Differential Gene Expression in Sea Anemones. Genome Biol. Evol. 2016, 8, 2358–2375. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, S.J.; Schmitt, R.J. Growth, reproduction and survival of a tropical sea anemone (Actiniaria): Benefits of hosting anemonefish. Coral Reefs 2004, 24, 67–73. [Google Scholar] [CrossRef]
- Frisch, A.J.; Rizzari, J.R.; Munkres, K.P.; Hobbs, J.-P.A. Anemonefish depletion reduces survival, growth, reproduction and fishery productivity of mutualistic anemone–anemonefish colonies. Coral Reefs 2016, 35, 375–386. [Google Scholar] [CrossRef]
- Szczebak, J.T.; Henry, R.P.; Al-Horani, F.A.; Chadwick, N.E. Anemonefish oxygenate their anemone hosts at night. J. Exp. Biol. 2013, 216, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Schligler, J.; Blandin, A.; Beldade, R.; Mills, S.C. Aggression of an orange-fin anemonefish to a blacktip reef shark: A potential example of fish mobbing? Mar. Biodivers. 2022, 52, 17. [Google Scholar] [CrossRef]
- Jungo, F.; Bougueleret, L.; Xenarios, I.; Poux, S. The UniProtKB/Swiss-Prot Tox-Prot program: A central hub of integrated venom protein data. Toxicon 2012, 60, 551–557. [Google Scholar] [CrossRef]
- Nedosyko, A.M.; Young, J.E.; Edwards, J.W.; Burke da Silva, K. Searching for a toxic key to unlock the mystery of anemonefish and anemone symbiosis. PLoS ONE 2014, 9, e98449. [Google Scholar] [CrossRef]
- Madio, B.; Undheim, E.A.B.; King, G.F. Revisiting venom of the sea anemone Stichodactyla haddoni: Omics techniques reveal the complete toxin arsenal of a well-studied sea anemone genus. J. Proteom. 2017, 166, 83–92. [Google Scholar] [CrossRef]
- Ramirez-Carreto, S.; Vera-Estrella, R.; Portillo-Bobadilla, T.; Licea-Navarro, A.; Bernaldez-Sarabia, J.; Rudino-Pinera, E.; Verleyen, J.J.; Rodriguez, E.; Rodriguez-Almazan, C. Transcriptomic and proteomic analysis of the tentacles and mucus of Anthopleura dowii Verrill, 1869. Mar. Drugs 2019, 17, 436. [Google Scholar] [CrossRef]
- Liao, Q.; Gong, G.; Poon, T.C.W.; Ang, I.L.; Lei, K.M.K.; Siu, S.W.I.; Wong, C.T.T.; Radis-Baptista, G.; Lee, S.M. Combined transcriptomic and proteomic analysis reveals a diversity of venom-related and toxin-like peptides expressed in the mat anemone Zoanthus natalensis (Cnidaria, Hexacorallia). Arch. Toxicol. 2019, 93, 1745–1767. [Google Scholar] [CrossRef]
- Levin, M.; Butter, F. Proteotranscriptomics—A facilitator in omics research. Comput. Struct. Biotechnol. J. 2022, 20, 3667–3675. [Google Scholar] [CrossRef] [PubMed]
- Moran, Y.; Praher, D.; Schlesinger, A.; Ayalon, A.; Tal, Y.; Technau, U. Analysis of soluble protein contents from the nematocysts of a model sea anemone sheds light on venom evolution. Mar. Biotechnol. 2013, 15, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Delgado, A.; Benedict, C.; Macrander, J.; Daly, M. Never, Ever Make an Enemy… out of an anemone: Transcriptomic comparison of clownfish hosting sea anemone venoms. Mar. Drugs 2022, 20, 730. [Google Scholar] [CrossRef] [PubMed]
- Kashimoto, R.; Tanimoto, M.; Miura, S.; Satoh, N.; Laudet, V.; Khalturin, K. Transcriptomes of giant sea anemones from Okinawa as a tool for understanding their phylogeny and symbiotic relationships with anemonefish. Zool. Sci. 2022, 39, 374–387, 314. [Google Scholar] [CrossRef] [PubMed]
- Barua, A.; Kashimoto, R.; Khalturin, K.; Satoh, N.; Laudet, V. The genetic basis for adaptation in giant sea anemones to their symbiosis with anemonefish and Symbiodiniaceae. bioRxiv 2022. [Google Scholar] [CrossRef]
- Marcionetti, A.; Rossier, V.; Roux, N.; Salis, P.; Laudet, V.; Salamin, N. Insights into the genomics of clownfish adaptive radiation: Genetic basis of the mutualism with sea anemones. Genome Biol. Evol. 2019, 11, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Hoepner, C.; Fobert, E.; Abbott, C.; Burke da Silva, K. No Place Like Home: Can omics uncover the secret behind the sea anemone and anemonefish symbiotic relationship? In Evolution, Development and Ecology of Anemonefishes; Laudet, V., Ravasi, T., Eds.; CRC Press: Boca Raton, FL, USA, 2022; pp. 197–208. [Google Scholar]
- Forêt, S.; Ong, J. Psytrans, Original Work Published 2014; Python, 2017. Available online: https://github.com/jueshengong/psytrans (accessed on 1 November 2022).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Stewart, Z.K.; Undheim, E.A.B.; Prentis, P.J. Gene families conserved and found in the venom of several sea anemone species 2020. Unpublished work.
- Sigrist, J.A.C.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucleic Acids Res. 2012, 41, D344–D347. [Google Scholar] [CrossRef]
- Shiomi, K.; Qian, W.-H.; Lin, X.-Y.; Shimakura, K.; Nagashima, Y.; Ishida, M. Novel polypeptide toxins with crab lethality from the sea anemone Anemonia erythraea. Biochim. Biophys. Acta (BBA)—Gen. Subj. 1997, 1335, 191–198. [Google Scholar] [CrossRef]
- Bos, M.H.; Boltz, M.; St Pierre, L.; Masci, P.P.; de Jersey, J.; Lavin, M.F.; Camire, R.M. Venom factor V from the common brown snake escapes hemostatic regulation through procoagulant adaptations. Blood 2009, 114, 686–692. [Google Scholar] [CrossRef]
- Capasso, C.; Rizzi, M.; Menegatti, E.; Ascenzi, P.; Bolognesi, M. Crystal structure of the bovine α-chymotrypsin:kunitz inhibitor complex. An example of multiple protein:protein recognition sites. J. Mol. Recognit. 1997, 10, 26–35. [Google Scholar] [CrossRef]
- An, D.; Pinheiro-Junior, E.L.; Béress, L.; Gladkikh, I.; Leychenko, E.; Undheim, E.A.B.; Peigneur, S.; Tytgat, J. AsKC11, a kunitz peptide from Anemonia sulcata, is a novel activator of G protein-coupled inward-rectifier potassium channels. Mar. Drugs 2022, 20, 140. [Google Scholar] [CrossRef] [PubMed]
- Foray, M.-F.; Lancelin, J.-M.; Hollecker, M.; Marion, D. Sequence-specific 1H-NMR assignment and secondary structure of black mamba dendrotoxin I, a highly selective blocker of voltage-gated potassium channels. Eur. J. Biochem. 1993, 211, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Razpotnik, A.; Krizaj, I.; Sribar, J.; Kordis, D.; Macek, P.; Frangez, R.; Kem, W.R.; Turk, T. A new phospholipase A2 isolated from the sea anemone Urticina crassicornis—Its primary structure and phylogenetic classification. FEBS J. 2010, 277, 2641–2653. [Google Scholar] [CrossRef]
- Xiao, H.; Pan, H.; Liao, K.; Yang, M.; Huang, C. Snake venom PLA(2), a promising target for broad-spectrum antivenom drug development. Biomed. Res. Int. 2017, 2017, 6592820. [Google Scholar] [CrossRef] [PubMed]
- Joubert, F.J.; Strydom, D.J. Snake Venoms. Eur. J. Biochem. 1978, 87, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Rouault, M.; Rash, L.D.; Escoubas, P.; Boilard, E.; Bollinger, J.; Lomonte, B.; Maurin, T.; Guillaume, C.; Canaan, S.; Deregnaucourt, C.; et al. Neurotoxicity and other pharmacological activities of the snake venom phospholipase A2 OS2: The N-terminal region is more important than enzymatic activity. Biochemistry 2006, 45, 5800–5816. [Google Scholar] [CrossRef] [PubMed]
- Castro-Amorim, J.; Novo de Oliveira, A.; Da Silva, S.L.; Soares, A.M.; Mukherjee, A.K.; Ramos, M.J.; Fernandes, P.A. Catalytically active snake venom PLA2 enzymes: An overview of its elusive mechanisms of reaction. J. Med. Chem. 2023, 66, 5364–5376. [Google Scholar] [CrossRef] [PubMed]
- Mebs, D. Anemonefish symbiosis: Vulnerability and resistance of fish to the toxin of the sea anemone. Toxicon 1994, 32, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.A.; Robinson, S.D.; Hamilton, B.F.; Undheim, E.A.B.; King, G.F. Deadly Proteomes: A practical guide to proteotranscriptomics of animal venoms. Proteomics 2020, 20, 1900324. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Lan, X.; Li, T.; Xiang, Y.; Zhao, F.; Zhang, Y.; Lee, W.-H. Proteotranscriptomic analysis and discovery of the profile and diversity of toxin-like proteins in Centipede. Mol. Cell. Proteom. 2018, 17, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, L.M.; Undheim, E.A.B.; Madio, B.; Hamilton, B.R.; Daly, M.; Hurwood, D.A.; King, G.F.; Prentis, P.J. Venoms for all occasions: The functional toxin profiles of different anatomical regions in sea anemones are related to their ecological function. Mol. Ecol. 2022, 31, 866–883. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Perez, D.; Campos, A.; Alexei Rodriguez, A.; Turkina, M.V.; Ribeiro, T.; Osorio, H.; Vasconcelos, V.; Antunes, A. Proteomic analyses of the unexplored sea anemone Bunodactis verrucosa. Mar. Drugs 2018, 16, 42. [Google Scholar] [CrossRef]
- Moran, Y.; Genikhovich, G.; Gordon, D.; Wienkoop, S.; Zenkert, C.; Ozbek, S.; Technau, U.; Gurevitz, M. Neurotoxin localization to ectodermal gland cells uncovers an alternative mechanism of venom delivery in sea anemones. Proc. Biol. Sci. 2012, 279, 1351–1358. [Google Scholar] [CrossRef]
- Pintor, A.F.; Krockenberger, A.K.; Seymour, J.E. Costs of venom production in the common death adder (Acanthophis antarcticus). Toxicon 2010, 56, 1035–1042. [Google Scholar] [CrossRef]
- Balamurugan, J.; Kumar, T.T.A.; Kannan, R.; Pradeep, H.D. Acclimation behaviour and bio-chemical changes during anemonefish (Amphiprion sebae) and sea anemone (Stichodactyla haddoni) symbiosis. Symbiosis 2015, 64, 127–138. [Google Scholar] [CrossRef]
- Moran, Y.; Cohen, L.; Kahn, R.; Karbat, I.; Gordon, D.; Gurevitz, M. Expression and mutagenesis of the sea anemone toxin Av2 reveals key amino acid residues important for activity on voltage-gated sodium channels. Biochemistry 2006, 45, 8864–8873. [Google Scholar] [CrossRef]
- Halaby, D.M.; Mornon, J.P.E. The immunoglobulin superfamily: An insight on its tissular, species, and functional diversity. J. Mol. Evol. 1998, 46, 389–400. [Google Scholar] [CrossRef]
- Holding, M.L.; Drabeck, D.H.; Jansa, S.A.; Gibbs, H.L. Venom resistance as a model for understanding the molecular basis of complex coevolutionary adaptations. Integr. Comp. Biol. 2016, 56, 1032–1043. [Google Scholar] [CrossRef]
- Perales, J.; Neves-Ferreira, A.G.C.; Valente, R.H.; Domont, G.B. Natural inhibitors of snake venom hemorrhagic metalloproteinases. Toxicon 2005, 45, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Bastos, V.A.; Gomes-Neto, F.; Perales, J.; Neves-Ferreira, A.G.; Valente, R.H. Natural inhibitors of snake venom metalloendopeptidases: History and current challenges. Toxins 2016, 8, 250. [Google Scholar] [CrossRef]
- Elliot, J.K.; Mariscal, R.N.; Roux, K.H. Do anemonefishes use molecular mimicry to avoid being stung by host anemones? J. Exp. Mar. Biol. Ecol. 1994, 79, 99–113. [Google Scholar] [CrossRef]
- Pennington, M.W.; Chang, S.C.; Chauhan, S.; Huq, R.; Tajhya, R.B.; Chhabra, S.; Norton, R.S.; Beeton, C. Development of highly selective Kv1.3-blocking peptides based on the sea anemone peptide ShK. Mar. Drugs 2015, 13, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Klompen, A.M.L.; Macrander, J.; Reitzel, A.M.; Stampar, S.N. Transcriptomic Analysis of Four Cerianthid (Cnidaria, Ceriantharia) Venoms. Mar. Drugs 2020, 18, 413. [Google Scholar] [CrossRef] [PubMed]
- Pérez, A.V.; Rucavado, A.; Sanz, L.; Calvete, J.J.; Gutiérrez, J.M. Isolation and characterization of a serine proteinase with thrombin-like activity from the venom of the snake Bothrops asper. Braz. J. Med. Biol. Res. 2008, 41, 12–17. [Google Scholar] [CrossRef]
- Da Silva, J.A.A.; Oliveira, K.C.; Camillo, M.A.P. Gyroxin increases blood-brain barrier permeability to Evans blue dye in mice. Toxicon 2011, 57, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Boldrini-França, J.; Pinheiro-Junior, E.L.; Peigneur, S.; Pucca, M.B.; Cerni, F.A.; Borges, R.J.; Costa, T.R.; Carone, S.E.I.; de Matos Fontes, M.R.; Sampaio, S.V.; et al. Beyond hemostasis: A snake venom serine protease with potassium channel blocking and potential antitumor activities. Sci. Rep. 2020, 10, 4476. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.S.; Caldeira, C.A.S.; Diniz-Sousa, R.; Romero, D.L.; Marcussi, S.; Moura, L.A.; Fuly, A.L.; de Carvalho, C.; Cavalcante, W.L.G.; Gallacci, M.; et al. Pharmacological characterization of cnidarian extracts from the Caribbean Sea: Evaluation of anti-snake venom and antitumor properties. J. Venom. Anim. Toxins 2018, 24, 22. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, H. Snake Venom Protease Inhibitors: Enhanced Identification, Expanding Biological Function, and Promising Future. In Snake Venoms; Gopalakrishnakone, P., Inagaki, H., Mukherjee, A.K., Rahmy, T.R., Vogel, C.-W., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 1–26. [Google Scholar]
- Manjunatha Kini, R. Excitement ahead: Structure, function and mechanism of snake venom phospholipase A2 enzymes. Toxicon 2003, 42, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.C.; Huq, R.; Chhabra, S.; Beeton, C.; Pennington, M.W.; Smith, B.J.; Norton, R.S. N-terminally extended analogues of the K+ channel toxin from Stichodactyla helianthus as potent and selective blockers of the voltage-gated potassium channel Kv1.3. FEBS J. 2015, 282, 2247–2259. [Google Scholar] [CrossRef] [PubMed]
- Sencic, L.; Macek, P. New method for isolation of venom from the sea anemone Actinia cari purification and characterization of cytolytic toxins. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1990, 97, 687–693. [Google Scholar] [CrossRef]
- Hoepner, C.M.; Abbott, C.A.; da Silva, K.B. The Ecological Importance of Toxicity: Sea Anemones Maintain Toxic Defence When Bleached. Toxins 2019, 11, 266. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC, Java. Original Work Published 2015. 2022. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 November 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef]
- Gilbert, D. Gene-omes built from mRNA seq not genome DNA. F1000Research 2016, 5, 1695. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Arriola, M.B.; Velmurugan, N.; Zhang, Y.; Plunkett, M.H.; Hondzo, H.; Barney, B.M. Genome sequences of Chlorella sorokiniana UTEX 1602 and Micractinium conductrix SAG 241.80: Implications to maltose excretion by a green alga. Plant J. 2018, 93, 566–586. [Google Scholar] [CrossRef] [PubMed]
- Camp, E.F.; Kahlke, T.; Signal, B.; Oakley, C.A.; Lutz, A.; Davy, S.K.; Suggett, D.J.; Leggat, W.P. Proteome metabolome and transcriptome data for three Symbiodiniaceae under ambient and heat stress conditions. Sci. Data 2022, 9, 153. [Google Scholar] [CrossRef]
- González-Pech, R.A.; Stephens, T.G.; Chen, Y.; Mohamed, A.R.; Cheng, Y.; Shah, S.; Dougan, K.E.; Fortuin, M.D.A.; Lagorce, R.; Burt, D.W.; et al. Comparison of 15 dinoflagellate genomes reveals extensive sequence and structural divergence in family Symbiodiniaceae and genus Symbiodinium. BMC Biol. 2021, 19, 73. [Google Scholar] [CrossRef] [PubMed]
- Levin, R.A.; Beltran, V.H.; Hill, R.; Kjelleberg, S.; McDougald, D.; Steinberg, P.D.; van Oppen, M.J.H. Sex, Scavengers, and Chaperones: Transcriptome secrets of divergent symbiodinium thermal tolerances. Mol. Biol. Evol. 2016, 33, 2201–2215. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A. TransDecoder, Original Work Published 2015. 2022. Available online: https://github.com/TransDecoder/TransDecoder (accessed on 1 November 2022).
- Wilding, C.S.; Fletcher, N.; Smith, E.K.; Prentis, P.; Weedall, G.D.; Stewart, Z. The genome of the sea anemone Actinia equina (L.): Meiotic toolkit genes and the question of sexual reproduction. Mar. Genom. 2020, 53, 100753. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novoassembled transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar] [CrossRef]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H.; Consortium, T.U. UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2014, 31, 926–932. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2020, 49, D325–D334. [Google Scholar] [CrossRef]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; Ramírez, F.; Warwick Vesztrocy, A.; Naldi, A.; Mungall, C.J.; Yunes, J.M.; Botvinnik, O.; Weigel, M.; et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 8, 10872. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- de Castro, E.; Sigrist, C.J.A.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Zhao, S.; Ye, Z.; Stanton, R. Misuse of RPKM or TPM normalization when comparing across samples and sequencing protocols. RNA 2020, 26, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, M.-C.; Konaté, M.M.; Chen, L.; Das, B.; Karlovich, C.; Williams, P.M.; Evrard, Y.A.; Doroshow, J.H.; McShane, L.M. TPM, FPKM, or Normalized Counts? A Comparative Study of Quantification Measures for the Analysis of RNA-seq Data from the NCI Patient-Derived Models Repository. J. Transl. Med. 2021, 19, 269. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.-W. PieDonut/webr, R. 2020. Available online: https://cardiomoon.github.io/webr/reference/PieDonut.html (accessed on 1 November 2023).
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Pennington, M.W.; Byrnes, M.E.; Zaydenberg, I.; Khaytin, I.; De Chastonay, J.; Krafte, D.S.; Hill, R.; Mahnir, V.M.; Volberg, W.A.; Gorczyca, W.; et al. Chemical synthesis and characterization of ShK toxin: A potent potassium channel inhibitor from a sea anemone. Int. J. Pept. Protein Res. 1995, 46, 354–358. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| RNA Gene Clusters | Protein Gene Clusters | |
|---|---|---|
| ORF sequences | 279,274 | 5375 |
| Annotated ORF sequences | 72,218 | 5213 |
| ORFs with GO terms | 46,288 | 3901 |
| Unique UniRef90 Hits | 18,469 | 3718 |
| ORFs with signal sequence | 11,807 | 1224 |
| Putative toxin ORFs | 1251 | 230 |
| Putative toxin ORFs with signal sequence | 515 | 149 |
| Putative toxin unique UniRef90 Hits | 296 | 124 |
| Venom Category | RNA Gene Clusters | TF | Protein Gene Clusters | TF | Percentage (%) 1 |
|---|---|---|---|---|---|
| Allergen and innate immunity | 9 | 3 | 4 | 2 | 44.4 |
| Auxiliary | 67 | 1 | 19 | 1 | 28.4 |
| Haemostatic and haemorrhagic | 409 | 5 | 55 | 5 | 13.4 |
| Mixed function enzymes | 24 | 1 | 1 | 1 | 4.2 |
| Neurotoxins | 122 | 9 | 21 | 5 | 17.2 |
| Pore forming | 45 | 5 | 16 | 4 | 35.6 |
| Protease inhibitors | 38 | 2 | 5 | 1 | 13.2 |
| Unknown | 537 | 16 | 109 | 11 | 20.3 |
| (a) Venom Category | Toxin Protein Family | RNA Gene Clusters | Rank of GC in Proteome |
|---|---|---|---|
| Haemostatic and haemorrhagic | Coagulation factor V-like | 315 | 2 |
| Unknown | IG-like | 300 | 1 |
| Unknown | Uncharacterised toxins | 104 | 3 |
| Neurotoxin | ShK-like | 85 | 5 |
| Auxiliary | Peptidase M12A | 67 | 4 |
| Haemostatic and haemorrhagic | Peptidase S1 | 37 | 3 |
| Haemostatic and haemorrhagic | Ficolin lectin family | 30 | 10 |
| Unknown | EGF-like | 25 | 11 |
| Mixed function enzymes | PLA2 | 24 | 13 |
| Pore Forming | Actinoporins | 23 | 7 |
| (b) Venom Category | Toxin Protein Family | Protein Gene Clusters | Rank of GC in Transcriptome |
| Unknown | IG-like | 56 | 2 |
| Haemostatic and haemorrhagic | Coagulation factor V-like | 22 | 1 |
| Haemostatic and haemorrhagic | Peptidase S1 | 21 | 6 |
| Unknown | Uncharacterised toxins | 21 | 3 |
| Auxiliary | Peptidase M12A | 19 | 5 |
| Neurotoxin | ShK-like | 14 | 4 |
| Unknown | U15 | 11 | 11 |
| Pore forming | Actinoporins | 7 | 10 |
| Haemostatic and haemorrhagic | Peptidase M12B | 6 | 12 |
| Unknown | Z3 | 6 | 13 |
| Unknown | U12 | 6 | 17 |
| Pore forming | DELTA-alicitoxin-Pse2b-like | 5 | 18 |
| Protease inhibitor | Venom Kunitz-type family | 5 | 15 |
| Haemostatic and haemorrhagic | Ficolin lectin family | 4 | 7 |
| (a) Venom Category | Toxin Protein Family | RNA Normalised Abundance 1 | Rank of TF in Proteome |
|---|---|---|---|
| Unknown | Z3 | 56,348 | 1 |
| Unknown | Sea Anemone 8 | 54,210 | Not present |
| Neurotoxin | ShK-like | 48,215 | 18 |
| Unknown | IG-like | 34,585 | 7 |
| Unknown | Uncharacterised toxins | 24,521 | 16 |
| Unknown | EGF-like | 19,030 | 5 |
| Mixed function enzyme | PLA2 | 11,305 | 4 |
| Auxiliary | Peptidase M12A | 8987 | 13 |
| Haemostatic and haemorrhagic | Factor V-like | 8767 | 19 |
| Unknown | CREC | 6308 | 15 |
| (b) Venom Category | Toxin Protein Family | Protein Normalised Abundance 2 | Rank of TF in Transcriptome |
| Unknown | Z3 | 16,933,968 | 1 |
| Haemostatic and haemorrhagic | Peptidase S1 | 9,294,230 | 11 |
| Protease inhibitor | Venom Kunitz | 6,409,895 | 13 |
| Mixed function enzyme | PLA2 | 4,873,099 | 7 |
| Unknown | EGF-like | 4,773,700 | 6 |
| Unknown | U15 | 4,298,257 | 27 |
| Unknown | IG-like | 3,884,603 | 4 |
| Unknown | U12 | 1,416,547 | 17 |
| Allergen and innate immunity | CAP | 1,367,399 | 14 |
| Neurotoxin | NEP 3 family | 911,820 | 16 |
| Time | Solvent B |
|---|---|
| 0 min | 2% |
| 5 min | 2% |
| 10 min | 8% |
| 60 min | 31.2% |
| 66 min | 50% |
| 69 min | 100% |
| 72 min | 100% |
| 75 min | 2% |
| Method | m/z |
|---|---|
| 1 | 350–1200 |
| 2 | 350–500 |
| 3 | 490–550 |
| 4 | 540–610 |
| 5 | 600–710 |
| 6 | 700–810 |
| 7 | 800–1200 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoepner, C.M.; Stewart, Z.K.; Qiao, R.; Fobert, E.K.; Prentis, P.J.; Colella, A.; Chataway, T.; Burke da Silva, K.; Abbott, C.A. Proteotransciptomics of the Most Popular Host Sea Anemone Entacmaea quadricolor Reveals Not All Toxin Genes Expressed by Tentacles Are Recruited into Its Venom Arsenal. Toxins 2024, 16, 85. https://doi.org/10.3390/toxins16020085
Hoepner CM, Stewart ZK, Qiao R, Fobert EK, Prentis PJ, Colella A, Chataway T, Burke da Silva K, Abbott CA. Proteotransciptomics of the Most Popular Host Sea Anemone Entacmaea quadricolor Reveals Not All Toxin Genes Expressed by Tentacles Are Recruited into Its Venom Arsenal. Toxins. 2024; 16(2):85. https://doi.org/10.3390/toxins16020085
Chicago/Turabian StyleHoepner, Cassie M., Zachary K. Stewart, Robert Qiao, Emily K. Fobert, Peter J. Prentis, Alex Colella, Tim Chataway, Karen Burke da Silva, and Catherine A. Abbott. 2024. "Proteotransciptomics of the Most Popular Host Sea Anemone Entacmaea quadricolor Reveals Not All Toxin Genes Expressed by Tentacles Are Recruited into Its Venom Arsenal" Toxins 16, no. 2: 85. https://doi.org/10.3390/toxins16020085
APA StyleHoepner, C. M., Stewart, Z. K., Qiao, R., Fobert, E. K., Prentis, P. J., Colella, A., Chataway, T., Burke da Silva, K., & Abbott, C. A. (2024). Proteotransciptomics of the Most Popular Host Sea Anemone Entacmaea quadricolor Reveals Not All Toxin Genes Expressed by Tentacles Are Recruited into Its Venom Arsenal. Toxins, 16(2), 85. https://doi.org/10.3390/toxins16020085