Overview of Bacterial Protein Toxins from Pathogenic Bacteria: Mode of Action and Insights into Evolution


Abstract
:1. Introduction
2. Interaction of Toxigenic Bacteria with the Host
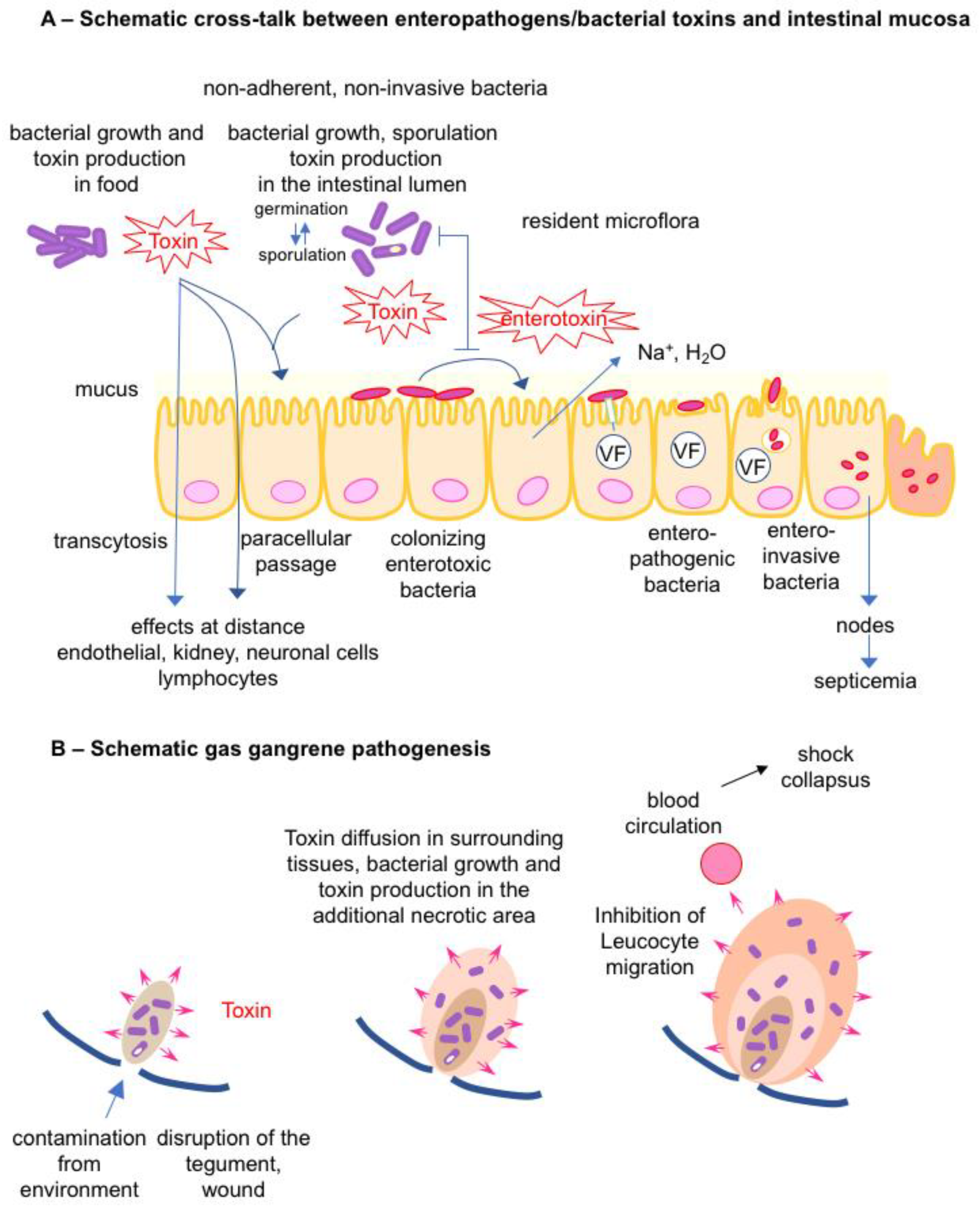
2.1. Cross Talk between Bacterial Toxins/Toxigenic Bacteria and the Intestinal Mucosa
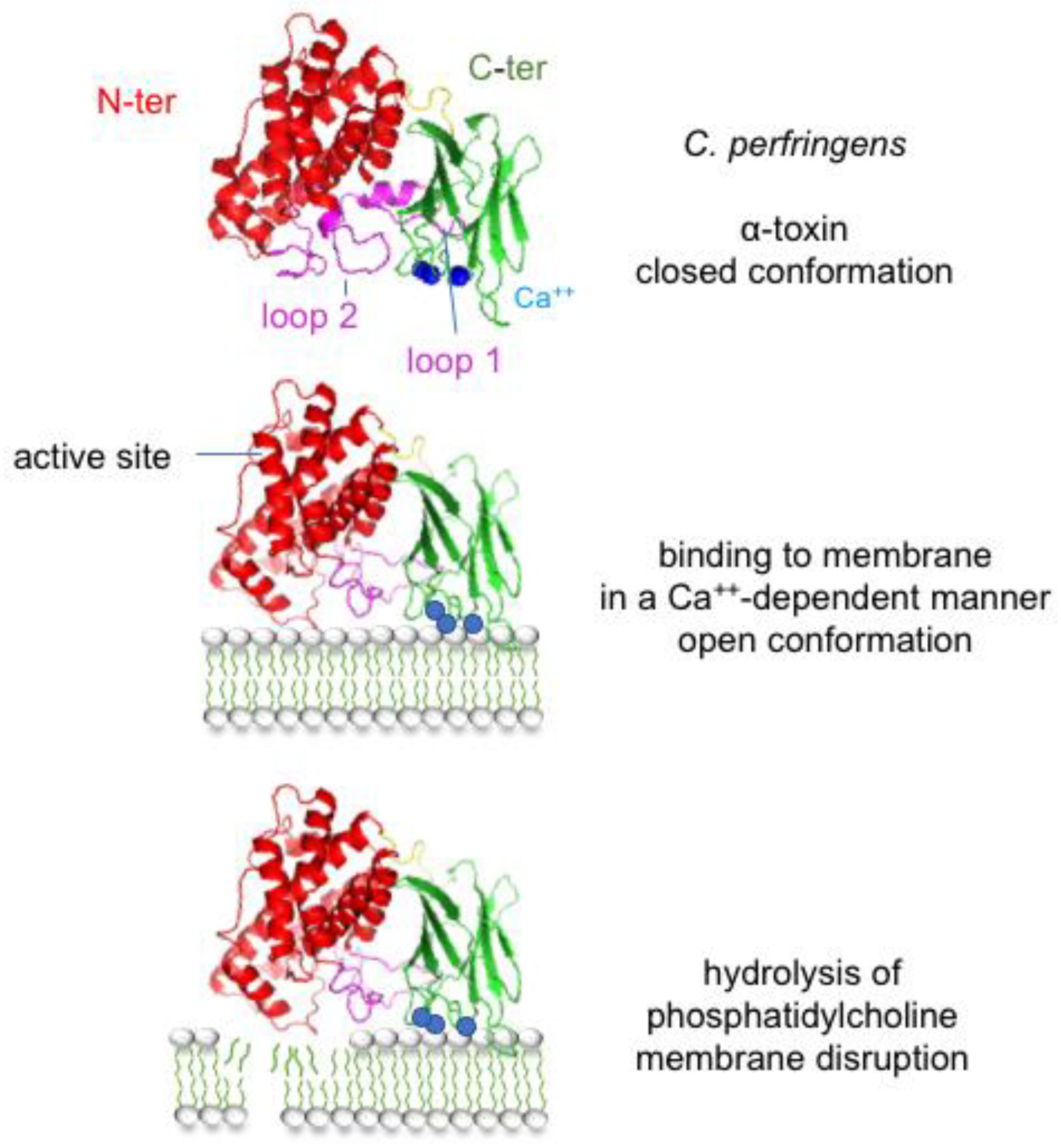
2.2. Bacterial Toxins/Toxigenic Bacteria and Gas Gangrene
3. Diversity in Bacterial Protein Toxins


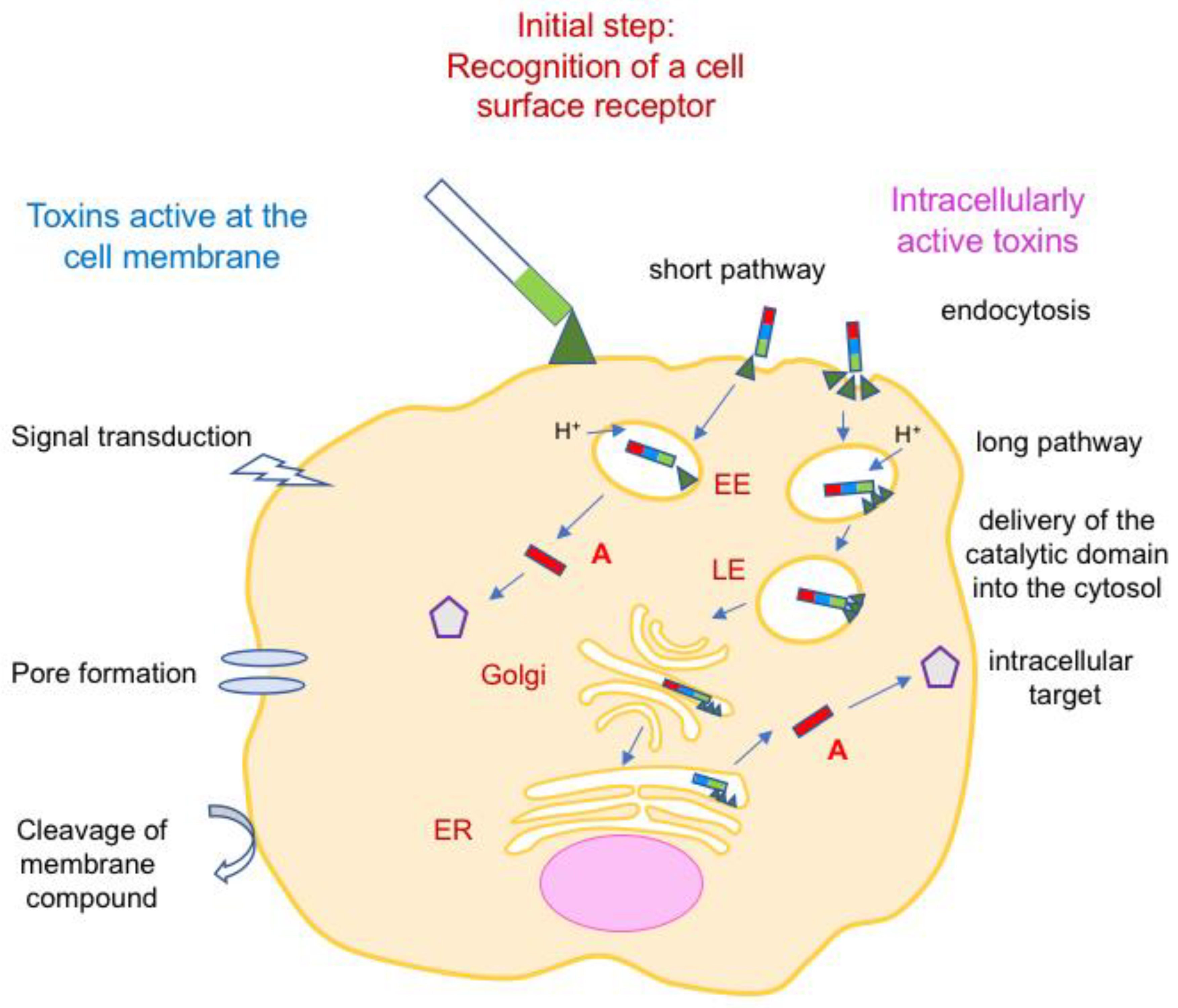
4. Bacterial Protein Toxins Active at the Cell Membrane
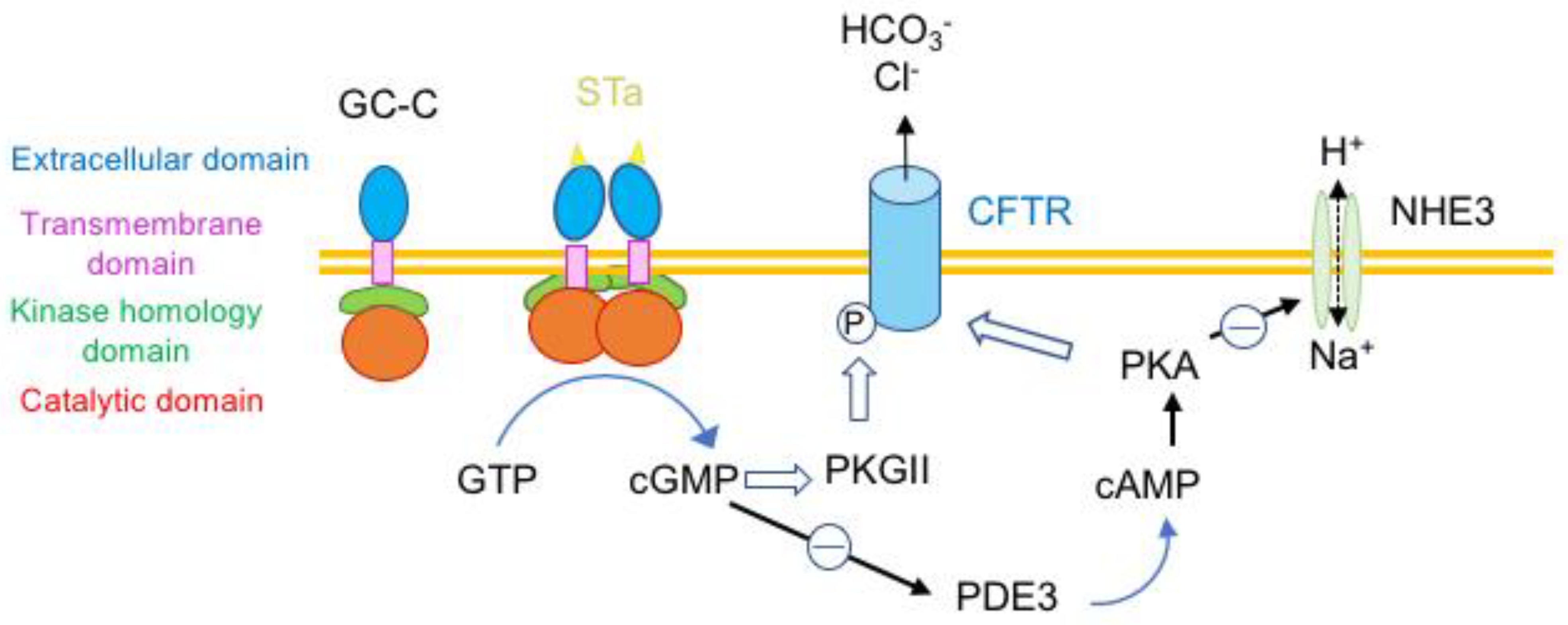
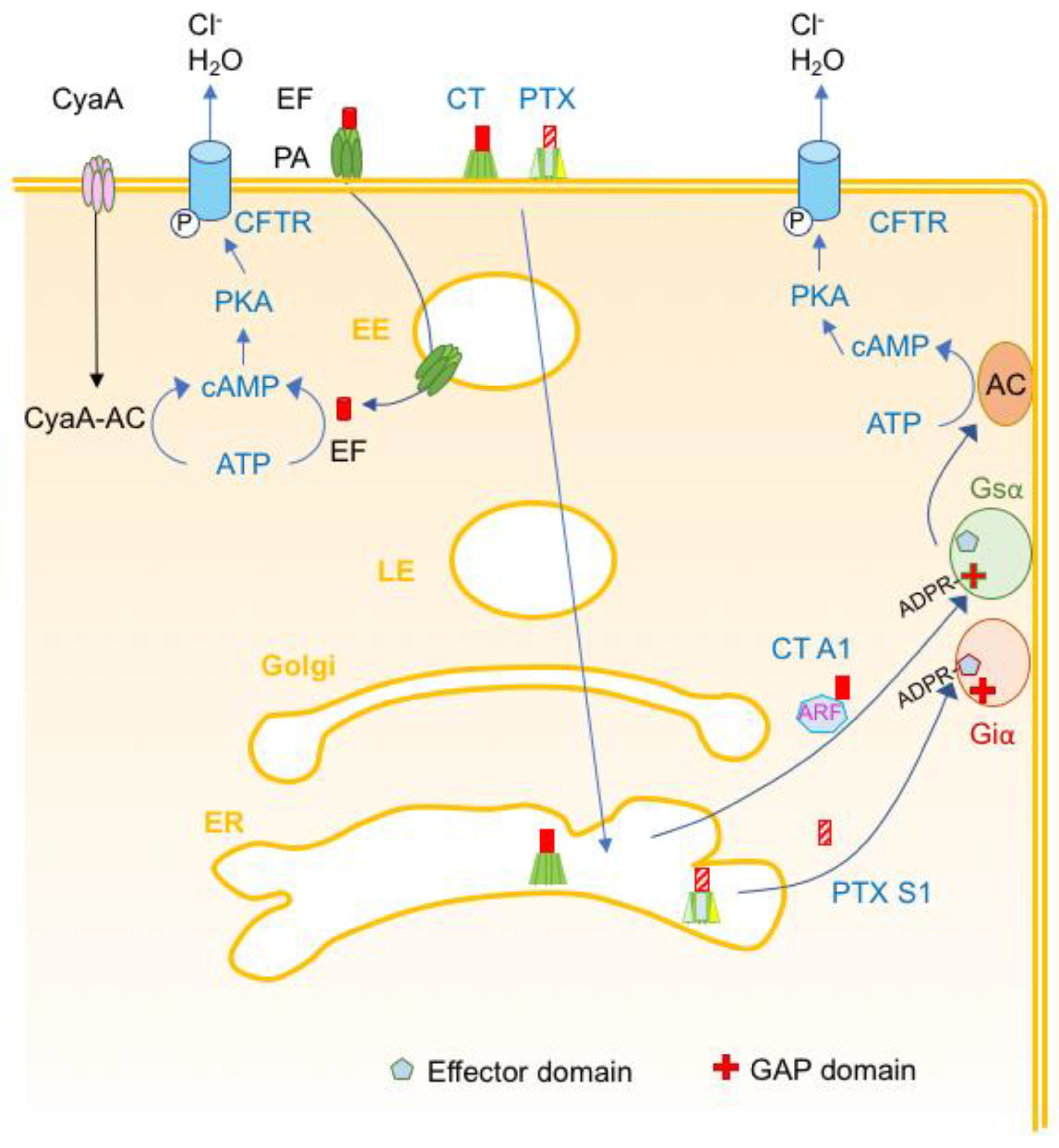
4.1. Toxins Transducing a Transmembrane Signal and Hormone-Like Toxins
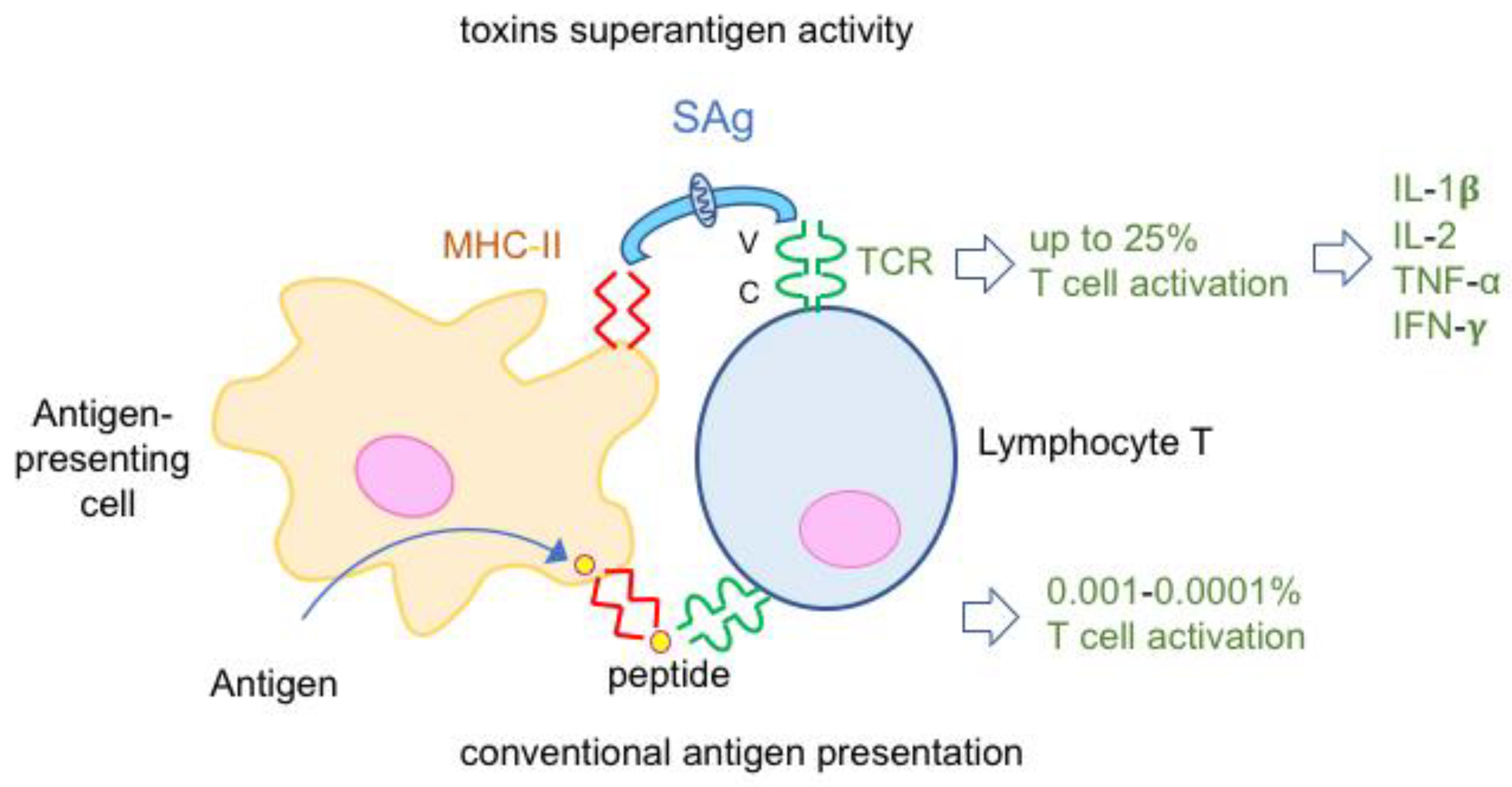
4.2. Toxins Superantigens
4.3. Membrane-Damaging Toxins
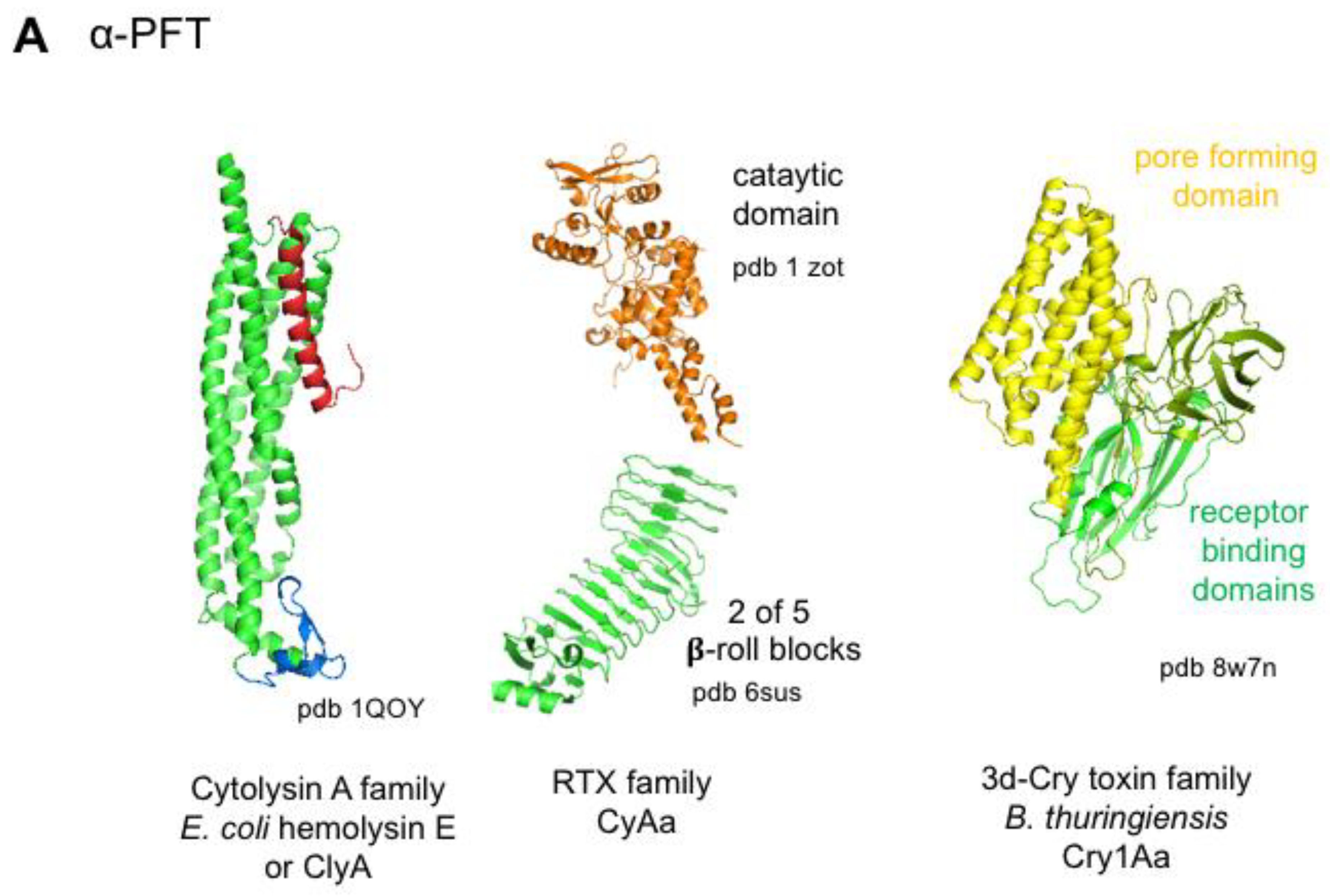
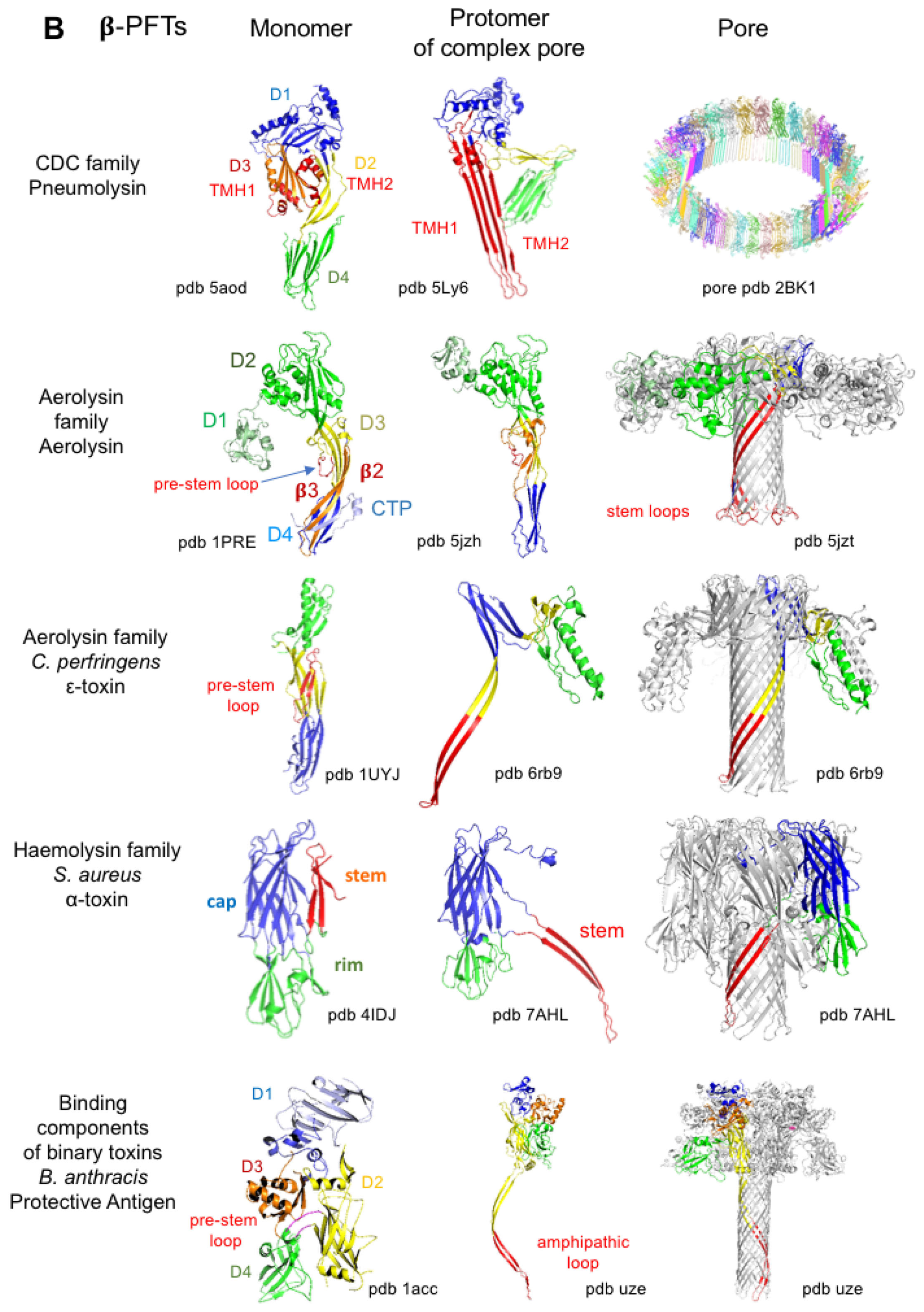
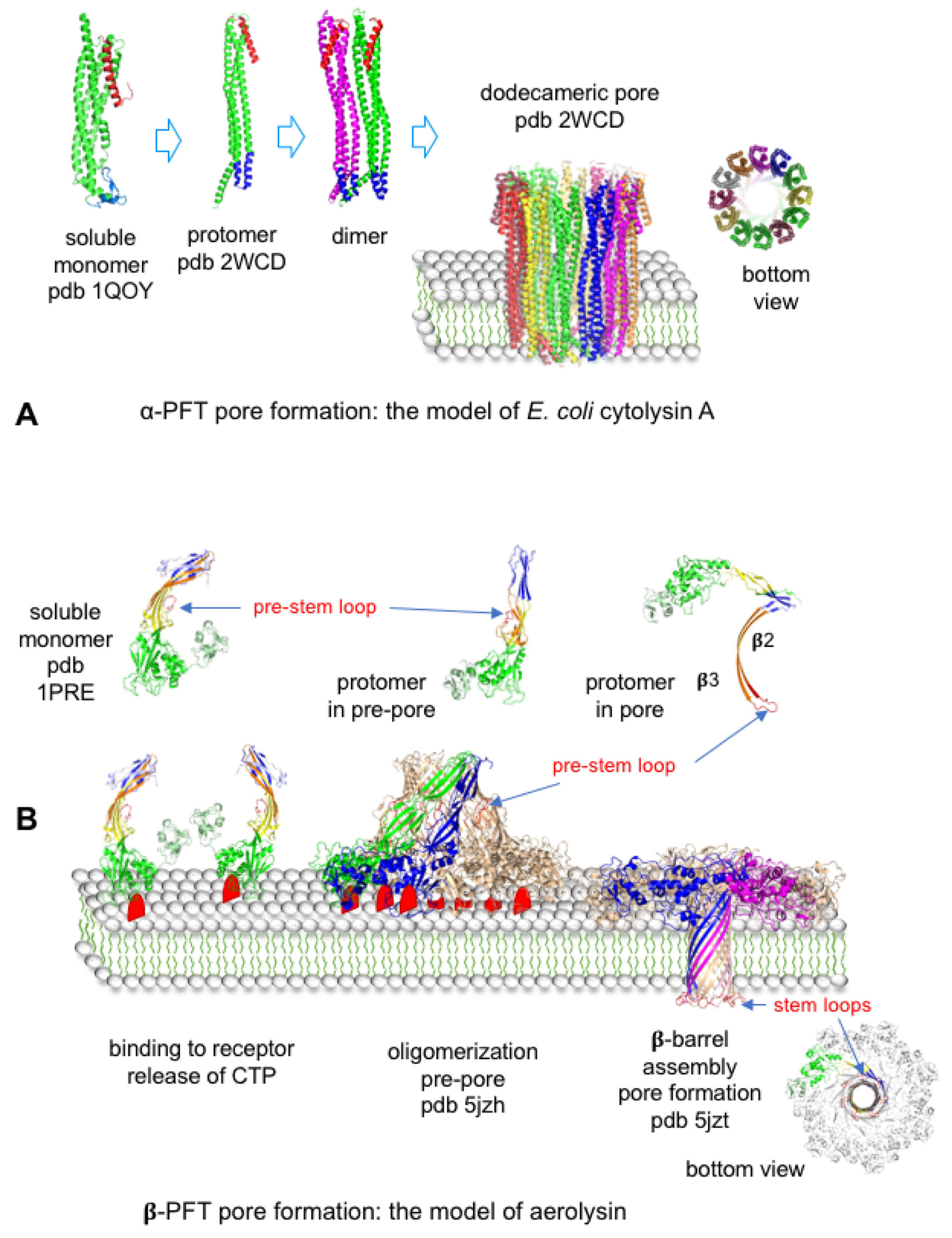
4.3.1. Bacterial Pore-Forming Toxins

| Toxin Family | Toxins | Toxin Producing Organism | Receptor | Oligomers (Number of Monomers) | Pore Size | Disease/Activity | References |
|---|---|---|---|---|---|---|---|
| α-Pore-Forming Toxins | |||||||
| Cytolysin A | Hemolysin E (or ClyA) | Escherichia coli | CD11/CD18 integrin | 12 | 3.5 nm | [44] | |
| Non-hemolytic enterotoxin (NHeA) (tripartite toxin: NheA, NheB, NheC) | Bacillus cereus | cholesterol | ? | ~2 nm | food poisoning | [93,94,95] | |
| hemolysin BL (Hbl-B) (tripartite toxin: Hbl-B, Hbl-L1, Hbl-L2) | Bacillus cereus | LITAF, CDIP1 | 7–8 | 1–2 nm | hemolysis, enterotoxicity | [96,97,98] | |
| RTX | α-hemolysin (HlyA) | Escherichia coli | CD11/CD18 integrin, glycophorin | ? | 1.1–2 nm | virulence factor associated with CNF in uropathogenic E. coli | [46,99,100] |
| Adenylate cyclase (CyaA) | Bordetella pertussis | CD11b/CD18 integrin | variable 12? | 0.6–0.8 nm | Translocation of the catalytic domain, whooping cough | [50,101,102,103] | |
| MARTX | Vibrio vulnificus Aeromonas hydrophila | ? | 1.8 nm | delivery of effector domains, intestinal tissue destruction | [26,53,104] | ||
| 3d-Cry Toxins | Three domain Crystal (Cry) Toxins | Bacillus thuringiensis | cadherin, ABC transporter subfamily C2, aminopeptidase, alkaline phosphatase | 4 | 4–5 nm | insecticidal activity | [55,56,57,58] |
| β-Pore-Forming Toxins | |||||||
| Cholesterol-dependent cytolysins (CDCs) | Perfringolysin O (PFO) | Clostridium perfringens | cholesterol, glycans | 40–50 | 25–45 nm | myonecrosis, gangrene | [66,105,106] |
| Botunolysin | Clostridium botulinum | cholesterol | 30–50 | hemolysis | [65] | ||
| Tetanolysin | Clostridum tetani | cholesterol | 30–50 | hemolysis | [65] | ||
| Streptolysin O (SLO) | Streptococcus pyogenes | cholesterol, glycans | 50–80 | 30 nm | hemolysis | [106,107] | |
| Listeriolysin O (LLO) | Listeria monocytogenes | cholesterol, glycans | 30–50 | 30 nm | Listeria vacuolar escape | [106,108] | |
| Intermedilysin (ILY) | Streptococcus intermedius | cholesterol, CD59, N-linked glycan | 30–50 | 25–30 nm | tissue destruction | [106,109,110] | |
| Pneumolysin (PLY) | Streptococcus pneumoniae | cholesterol, glycans | 44 | 26 nm | pneumonia, meningitis, otitis | [106,111] | |
| Anthrolysin (ALO) | Bacillus anthracis | cholesterol, glycans | 30–50 | [65,106] | |||
| Aerolysin | Aerolysin | Aeromonas sp. | GPI-anchored proteins | 7 | 0.7–1.7 nm | [112,113,114,115] | |
| ε-toxin (ETX) | Clostridium perfringens | HAVCR1, MAL | 7 | 1–2.4 nm | animal enterotoxemia | [116,117,118] | |
| Enterotoxin (CPE) | Clostridium perfringens | Claudins | 7 | 1.4 nm | food poisoning | [119,120,121,122] | |
| α-toxin | Clostridium septicum | GPI-anchored proteins | 6, 7 | 1.2–1.6 nm | myonecrosis, gangrene | [123,124,125] | |
| Cry toxins of ETX-MTX subfamily, Mosquitocidal toxin (MTX) | Bacillus thuringiensis Bacillus sphaericus | insect gut receptor | insecticidal activity | [126] | |||
| Hemolysin or S. aureus α-toxin | α-toxin | Staphylococcus aureus | Phosphatidylcholine, sphingomyelin, ADAM10 disintegrin | 6, 7 | 1.4–3 nm | skin necrosis, soft tissue infections | [127] |
| Panton-Valentine leucocidin LukS-LukF | Staphylococcus aureus | C5aR | 8 | 1.9–2.1 nm | necrotizing pneumonia | [72,128,129,130] | |
| γ-hemolysin (HlgA/HlgB-HlgC) | Staphylococcus aureus | CXCR1, CXCR2, CCR2, C5aR | 8 | 2.5–3 nm | skin, soft tissue infections | [72,131,132,133] | |
| Leucocidin LukA-LukB (LukGH) | Staphylococcus aureus | CD11b, HVCN1 | 8 | 3 nm | soft tissue infections | [134,135,136] | |
| Leucocidin LukE-LukD | Staphylococcus aureus | CCR2, CCR5, CXCR1, CXCR2, DARC | 8 | 1.9–2.1 nm | soft tissue infections | [137,138,139,140] | |
| Beta-toxin | Clostridium perfringens | Platelet endotheial cell adhesion molecule-1 (CD31) | 8 | 1.2–2.0 nm | necrotic enteritis | [141,142] | |
| Net-B | Clostridium perfringens | cholesterol | 7 | 1.6 nm | necrotic enteritis | [143] | |
| Delta-toxin | Clostridium perfringens | monosialo-ganglioside (GM2) | 7 | 4 nm | [144,145] | ||
| CctA | Clostridium chauvoei | myonecrosis, blackleg | [146] | ||||
| Vibrio cholerae cytolysin (VCC) | Vibrio cholerae | Glycoconjugates | 7 | 2.5 nm | hemolysis enterotoxicity | [147,148,149] | |
| Vibrio vulnificus hemolysin | Vibrio vulnificus | gangliosides, N-acetyl-D-galactosamine, N-acetyl-D-lactosamine | 7 | apoptosis | [150,151] | ||
| Binding components of binary toxins | Protective Antigen (PA) | Bacillus anthracis | capillary morphogenesis protein 2 (CMG2), tumor endothelial marker 8 (TEM8) | 7 | 1.2 nm | Translocation of the enzymatic components EF, LF | [89,90,152,153,154] |
| Iota toxin B component (Ib) | Clostridium perfringens | lipolysis-stimulated lipoprotein receptor | 7 | 1 nm | Translocation of Ia | [92,155,156] | |
| C2 toxin B component (C2-II) | Clostridium botulinum C and D | 7 | 1–2 nm | Translocation of C2-I necrotic enteritis | [116] | ||
| Clostridium difficile transferase (CDTb) | Clostridioides difficile | lipolysis-stimulated lipoprotein receptor | 7 | Translocation of CDTa pseudomembranous colitis | [155,157] | ||
| Vegetative insecticidal protein B component (VIP1) | Bacillus thuringiensis | insect midgut membrane receptor | 7? | Translocation of enzymatic component | [158,159] | ||



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4.3.2. Enzymatically Active Toxins against Membrane Compound(s)
Bacterial Phospholipases and Sphingomyelinases
Collagenases and Proteases
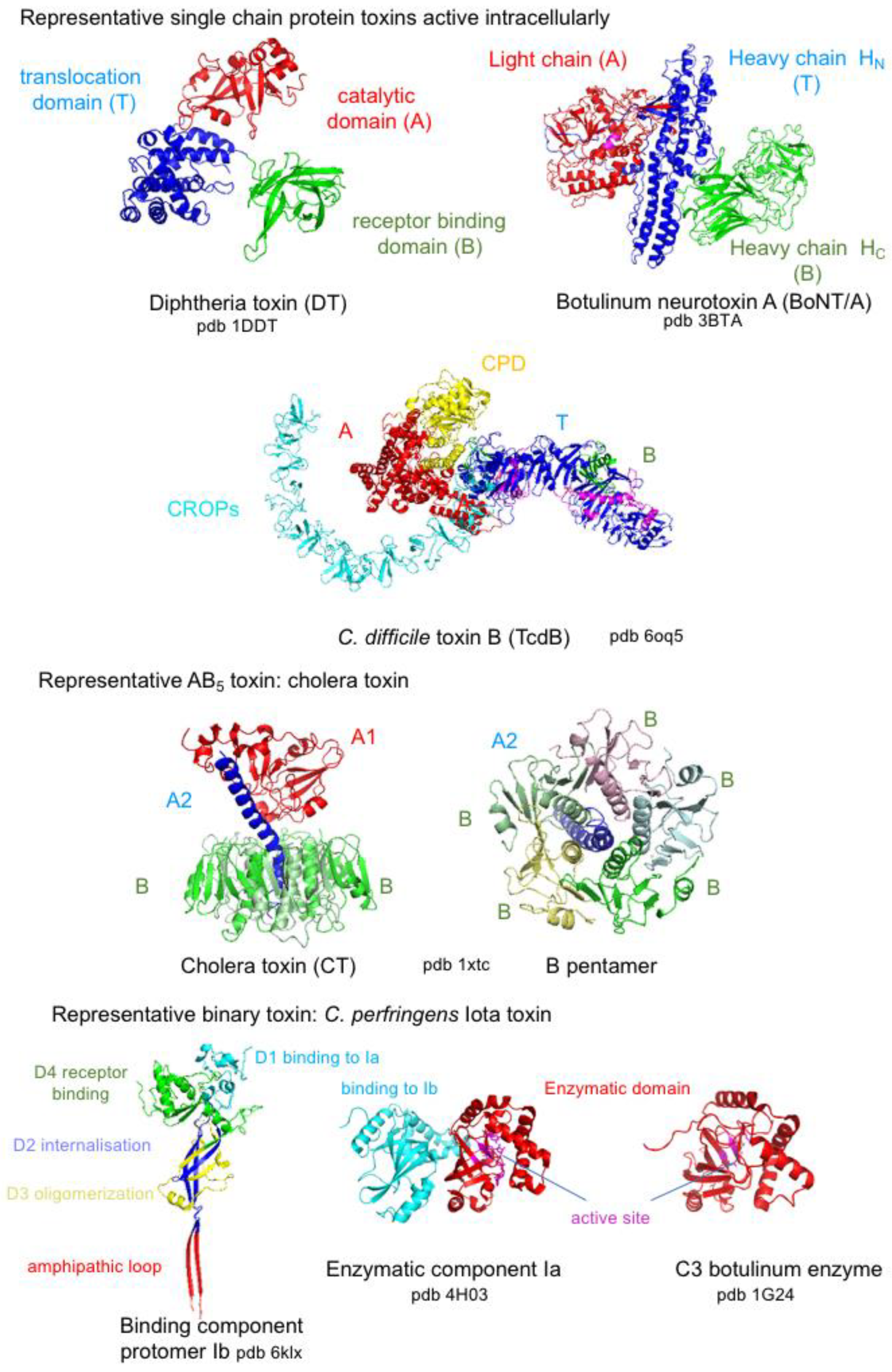
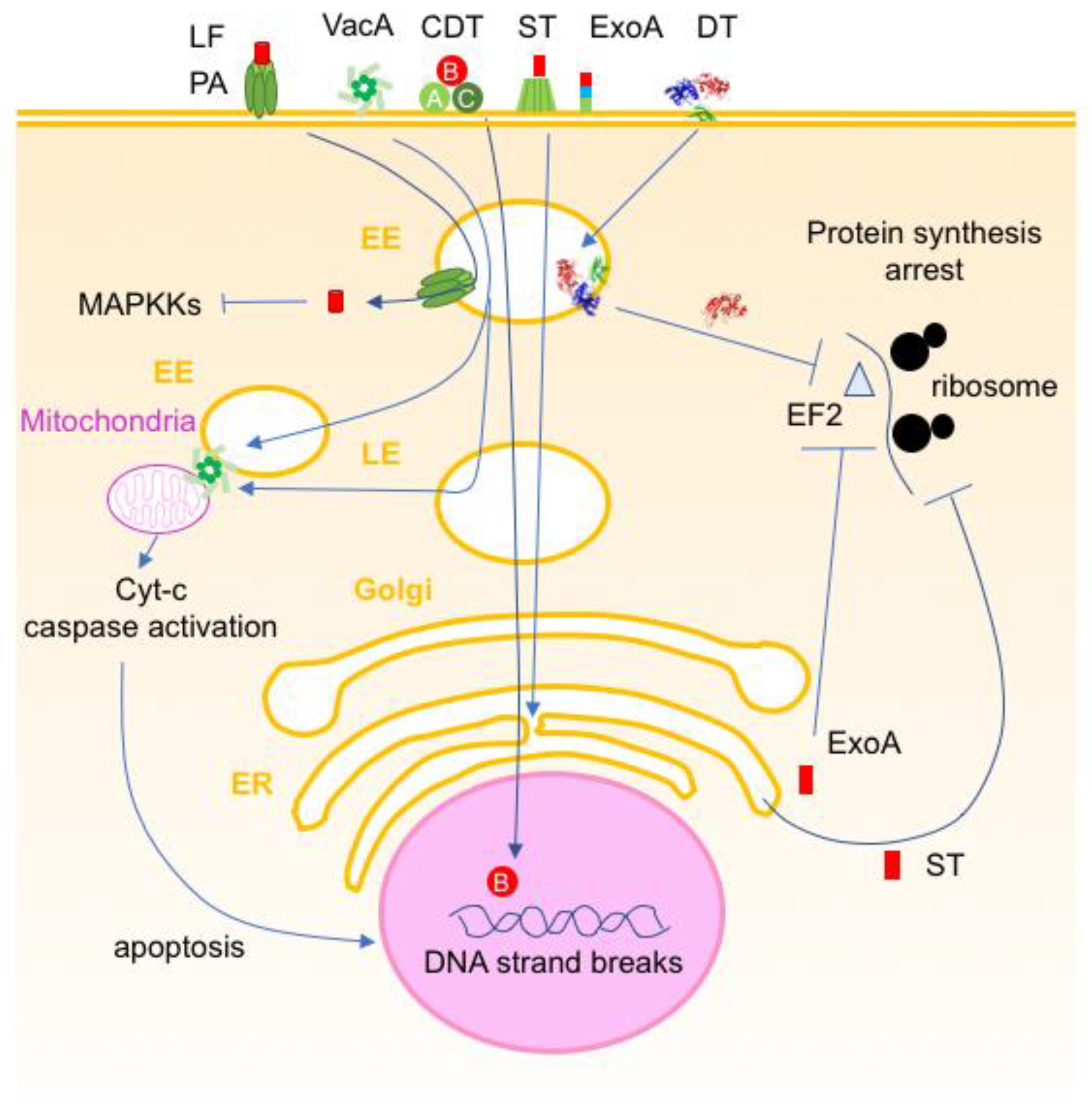
5. Bacterial Protein Toxins Active Intracellularly
5.1. Toxins Inducing Cell Death


5.2. Toxins Perturbing Cell Homeostasis

5.3. Toxins Targeting the Actin Cytoskeleton
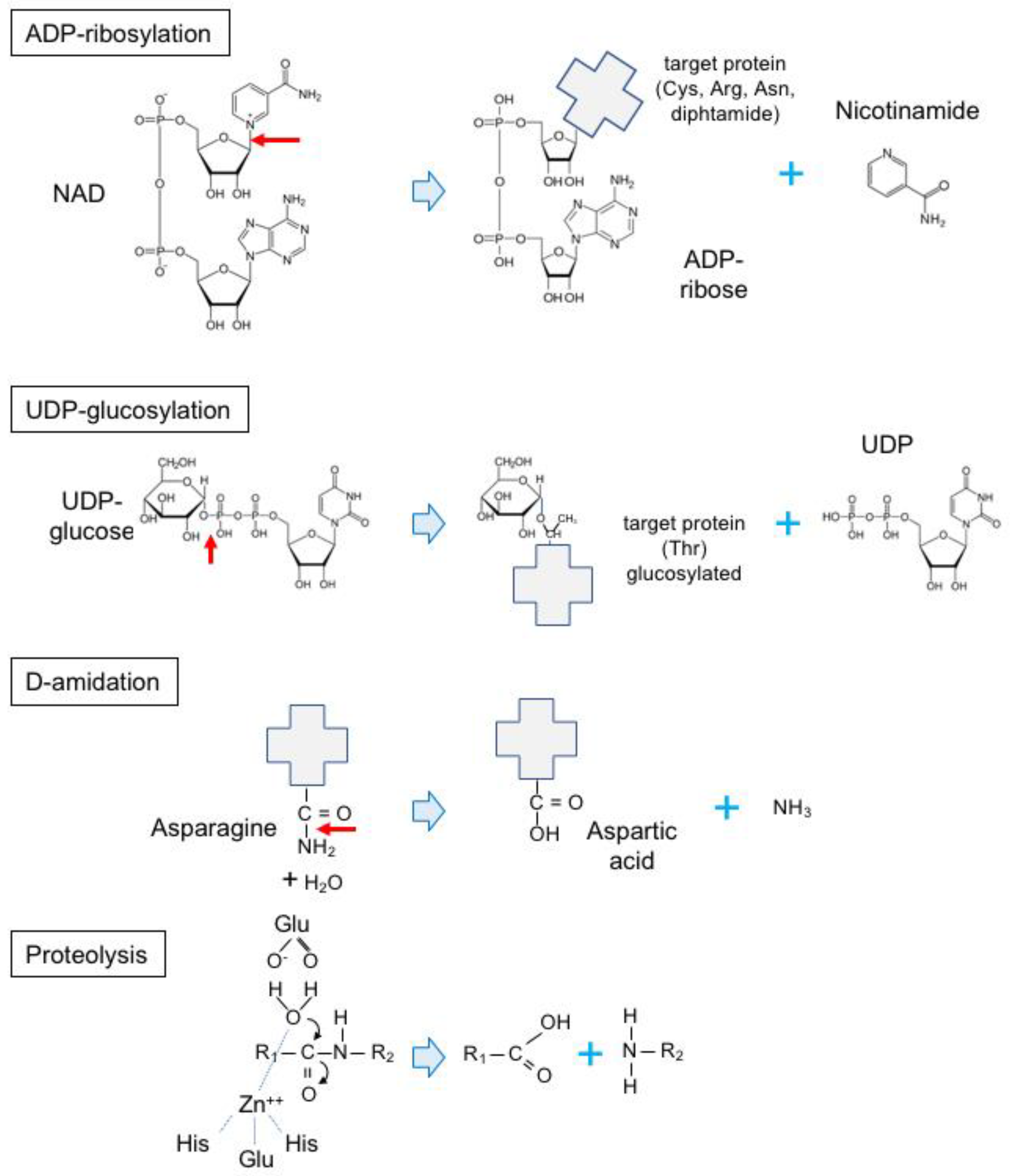
5.3.1. Actin ADP-Ribosylating Toxins
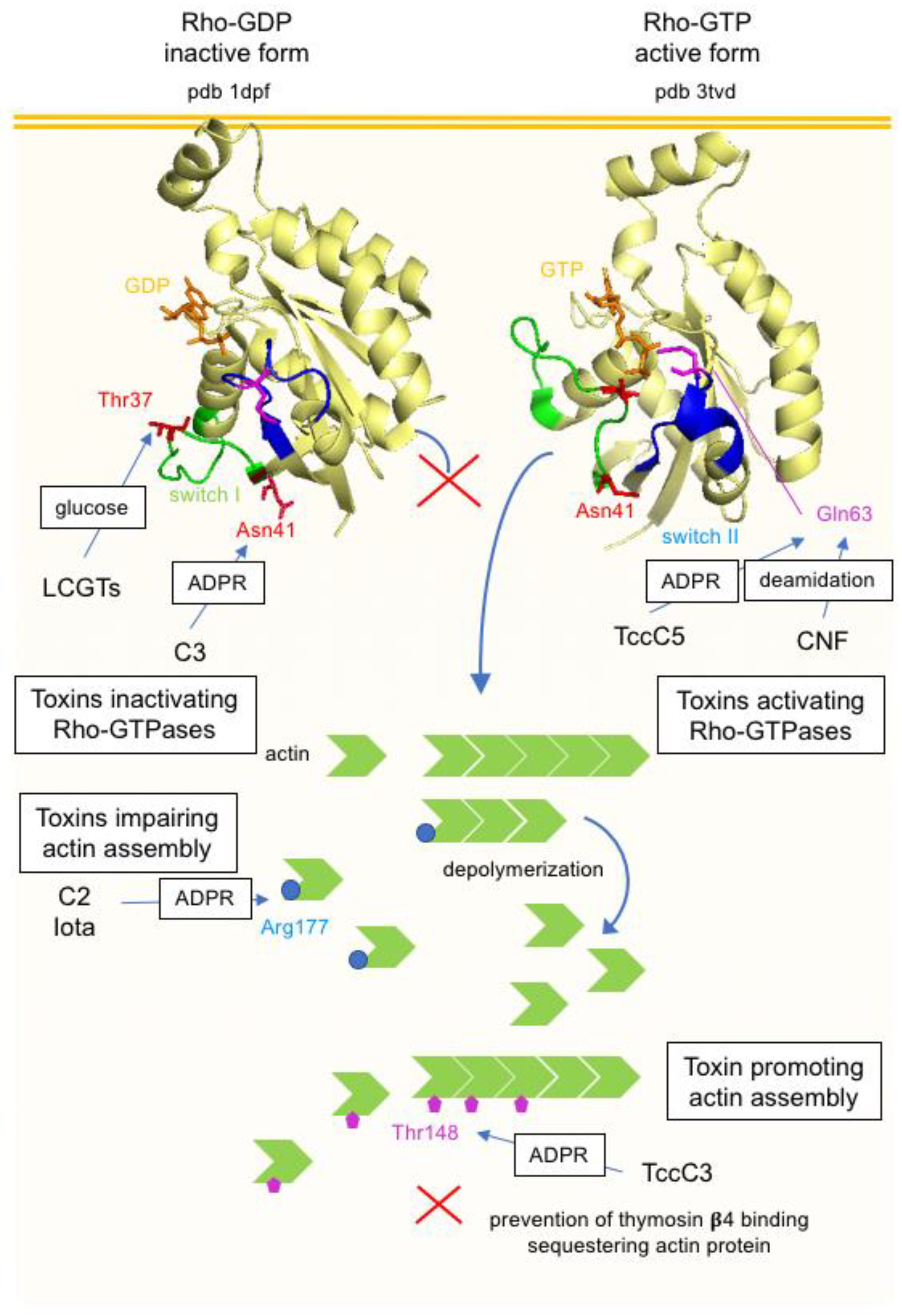
5.3.2. Toxins Modifying Rho-Family GTPases
5.4. Neurotoxins Impairing Neurotransmitter Release
6. Insights into the Evolution of Bacterial Protein Toxins
7. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Green, E.R.; Mecsas, J. Bacterial Secretion Systems: An Overview. Microbiol. Spectr. 2016, 4, 213–239. [Google Scholar] [CrossRef] [PubMed]
- Rapisarda, C.; Fronzes, R. Secretion Systems Used by Bacteria to Subvert Host Functions. Curr. Issues Mol. Biol. 2018, 25, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Galan, J.E. Common themes in the design and function of bacterial effectors. Cell Host Microbe 2009, 5, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Doré, J.; Corthier, G. The human intestinal microbiota. Gastroenterol. Clin. Biol. 2010, 34, S7–S15. [Google Scholar] [CrossRef] [PubMed]
- Thriene, K.; Michels, K.B. Human Gut Microbiota Plasticity throughout the Life Course. Int. J. Environ. Res. Public Health 2023, 20, 1463. [Google Scholar] [CrossRef] [PubMed]
- Yurist-Doutsch, S.; Arrieta, M.C.; Vogt, S.L.; Finlay, B.B. Gastrointestinal microbiota-mediated control of enteric pathogens. Annu. Rev. Genet. 2014, 48, 361–382. [Google Scholar]
- Ducarmon, Q.R.; Zwittink, R.D.; Hornung, B.V.H.; van Schaik, W.; Young, V.B.; Kuijper, E.J. Gut Microbiota and Colonization Resistance against Bacterial Enteric Infection. Microbiol. Mol. Biol. Rev. 2019, 83, e00007-19. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K.; Schwan, C.; Jank, T. Clostridium difficile Toxin Biology. Annu. Rev. Microbiol. 2017, 71, 281–307. [Google Scholar] [CrossRef] [PubMed]
- Perelle, S.; Gibert, M.; Bourlioux, P.; Corthier, G.; Popoff, M.R. Production of a complete binary toxin (actin-specific ADP-ribosylating toxin) by Clostridium difficile CD196. Infect. Immun. 1997, 65, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Young, V.B. Interactions Between the Gastrointestinal Microbiome and Clostridium difficile. Annu. Rev. Microbiol. 2015, 69, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Buddle, J.E.; Fagan, R.P. Pathogenicity and virulence of Clostridioides difficile. Virulence 2023, 14, 2150452. [Google Scholar] [CrossRef]
- Freedman, J.C.; Shrestha, A.; McClane, B.A. Clostridium perfringens Enterotoxin: Action, Genetics, and Translational Applications. Toxins 2016, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, M.; Heikinheimo, A.; Lahti, P.; Korkeala, H. Novel insights into the epidemiology of Clostridium perfringens type A food poisoning. Food Microbiol. 2011, 28, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Heredia, N.L.; Labbé, R.G. Clostridium perfringens. In Guide to Foodborne Pathogens; Labbé, R.G., Garcia, S., Eds.; Wiley Inter-Science: New York, NY, USA, 2001; pp. 133–141. [Google Scholar]
- Rasetti-Escargueil, C.; Lemichez, E.; Popoff, M.R. Public Health Risk Associated with Botulism as Foodborne Zoonoses. Toxins 2020, 12, 17. [Google Scholar] [CrossRef]
- Shirey, T.B.; Dykes, J.K.; Lúquez, C.; Maslanka, S.E.; Raphael, B.H. Characterizing the fecal microbiota of infants with botulism. Microbiome 2015, 3, 54. [Google Scholar] [CrossRef] [PubMed]
- Nesil, P.; Erbas, O. Foodborne and Infant Botulism Linkage with the Gut Microbiome’s Impact on the Immune System and Mental Function. J. Exp. Basic Med. Sci. 2021, 2, 365–374. [Google Scholar]
- Khan, I.; Bai, Y.; Zha, L.; Ullah, N.; Ullah, H.; Shah, S.R.H.; Sun, H.; Zhang, C. Mechanism of the Gut Microbiota Colonization Resistance and Enteric Pathogen Infection. Front. Cell. Infect. Microbiol. 2021, 11, 716299. [Google Scholar] [CrossRef]
- Belotserkovsky, I.; Sansonetti, P.J. Shigella and Enteroinvasive Escherichia Coli. Curr. Top. Microbiol. Immunol. 2018, 416, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Pizarro-Cerdá, J.; Cossart, P. Listeria monocytogenes: Cell biology of invasion and intracellular growth. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Li, Q. Mechanisms for the Invasion and Dissemination of Salmonella. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 2655801. [Google Scholar] [CrossRef]
- Luk, C.H.; Enninga, J.; Valenzuela, C. Fit to dwell in many places—The growing diversity of intracellular Salmonella niches. Front. Cell. Infect. Microbiol. 2022, 12, 989451. [Google Scholar] [CrossRef]
- Bryant, A.E.; Bayer, C.R.; Hayes-Schroer, S.M.; Stevens, D. Activation of platelet gpIIIa by phospholipase C from Clostridium perfringens involves store-operated calcium entry. J. Infect. Dis. 2003, 187, 408–417. [Google Scholar] [CrossRef]
- Stevens, D.L.; Aldape, M.J.; Bryant, A.E. Life-threatening clostridial infections. Anaerobe 2012, 18, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.L.; Bryant, A.E. The role of Clostridial toxins in the pathogenesis of gas gangrene. Clin. Infect. Dis. 2002, 35, S93–S100. [Google Scholar] [CrossRef] [PubMed]
- Gavin, H.E.; Satchell, K.J. MARTX toxins as effector delivery platforms. Pathog. Dis. 2015, 73, ftv092. [Google Scholar] [CrossRef]
- Jank, T.; Aktories, K. Structure and mode of action of clostridial glucosylating toxins: The ABCD model. Trends Microbiol. 2008, 16, 222–229. [Google Scholar] [CrossRef]
- Dubreuil, J.D. Escherichia coli heat-stable enterotoxins. In The Comprehensive Sourcebook of Bacterial Protein Toxins; Alouf, J., Ladant, D., Popoff, M.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 874–910. [Google Scholar]
- Song, J. Bacterial AB toxins and host-microbe interactions. Adv. Microb. Physiol. 2022, 81, 67–109. [Google Scholar] [CrossRef] [PubMed]
- Piot, N.; van der Goot, F.G.; Sergeeva, O.A. Harnessing the Membrane Translocation Properties of AB Toxins for Therapeutic Applications. Toxins 2021, 13, 36. [Google Scholar] [CrossRef]
- Johannes, L.; Parton, R.G.; Bassereau, P.; Mayor, S. Building endocytic pits without clathrin. Nat. Rev. Mol. Cell Biol. 2015, 16, 311–321. [Google Scholar] [CrossRef]
- Pezeshkian, W.; Gao, H.; Arumugam, S.; Becken, U.; Bassereau, P.; Florent, J.C.; Ipsen, J.H.; Johannes, L.; Shillcock, J.C. Mechanism of Shiga Toxin Clustering on Membranes. ACS Nano 2017, 11, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Pezeshkian, W.; Nåbo, L.J.; Ipsen, J.H. Cholera toxin B subunit induces local curvature on lipid bilayers. FEBS Open Bio 2017, 7, 1638–1645. [Google Scholar] [CrossRef]
- Lin, J.E.; Valentino, M.; Marszalowicz, G.; Magee, M.S.; Li, P.; Snook, A.E.; Stoecker, B.A.; Chang, C.; Waldman, S.A. Bacterial heat-stable enterotoxins: Translation of pathogenic peptides into novel targeted diagnostics and therapeutics. Toxins 2010, 2, 2028–2054. [Google Scholar] [CrossRef] [PubMed]
- Weiglmeier, P.R.; Rösch, P.; Berkner, H. Cure and curse: E. coli heat-stable enterotoxin and its receptor guanylyl cyclase C. Toxins 2010, 2, 2213–2229. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.J.; Fraser, J.D.; Proft, T. Bacterial superantigens and superantigen-like toxins. In The Comprehensive Sourcebook of Bacterial Protein Toxins, 4th ed.; Alouf, J., Ladant, D., Popoff, M.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 911–974. [Google Scholar]
- Proft, T.; Fraser, J.D. Streptococcal Superantigens: Biological properties and potential role in disease. In Streptococcus pyogenes: Basic Biology to Clinical Manifestations; Ferretti, J.J., Stevens, D.L., Fischetti, V.A., Eds.; University of Oklahoma Health Sciences Center: Oklahoma City, Oklahoma, 2016; pp. 1–72. [Google Scholar] [PubMed]
- Ahmad-Mansour, N.; Loubet, P.; Pouget, C.; Dunyach-Remy, C.; Sotto, A.; Lavigne, J.P.; Molle, V. Staphylococcus aureus Toxins: An Update on Their Pathogenic Properties and Potential Treatments. Toxins 2021, 13, 677. [Google Scholar] [CrossRef] [PubMed]
- Noli Truant, S.; Redolfi, D.M.; Sarratea, M.B.; Malchiodi, E.L.; Fernández, M.M. Superantigens, a Paradox of the Immune Response. Toxins 2022, 14, 800. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.L.; Otto, M.; Cheung, G.Y.C. Basis of Virulence in Enterotoxin-Mediated Staphylococcal Food Poisoning. Front. Microbiol. 2018, 9, 436. [Google Scholar] [CrossRef] [PubMed]
- Etter, D.; Schelin, J.; Schuppler, M.; Johler, S. Staphylococcal Enterotoxin C-An Update on SEC Variants, Their Structure and Properties, and Their Role in Foodborne Intoxications. Toxins 2020, 12, 584. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.L.; Nakane, A. Mechanisms of staphylococcal enterotoxin-induced emesis. Eur. J. Pharmacol. 2014, 722, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Alouf, J.E. Molecular features of the cytolytic pore forming bacterial protein toxins. Folia Microbiol. 2003, 48, 5–16. [Google Scholar] [CrossRef]
- Roderer, D.; Glockshuber, R. Assembly mechanism of the α-pore-forming toxin cytolysin A from Escherichia coli. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160211. [Google Scholar] [CrossRef]
- Wallace, A.J.; Stillman, T.J.; Atkins, A.; Jamieson, S.J.; Bullough, P.A.; Green, J.; Artymiuk, P.J. E. coli hemolysin E (hlyE, ClyA, SheA): X-ray crystal structure of the toxin and observation of membrane pores by electron microscopy. Cell 2000, 100, 265–276. [Google Scholar] [CrossRef]
- Benz, R. Channel formation by RTX-toxins of pathogenic bacteria: Basis of their biological activity. Biochim. Biophys. Acta 2016, 1858, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Linhartová, I.; Bumba, L.; Mašín, J.; Basler, M.; Osička, R.; Kamanová, J.; Procházková, K.; Adkins, I.; Hejnová-Holubová, J.; Sadílková, L.; et al. RTX proteins: A highly diverse family secreted by a common mechanism. FEMS Microbiol. Rev. 2010, 34, 1076–1112. [Google Scholar] [CrossRef]
- Ostolaza, H.; González-Bullón, D.; Uribe, K.B.; Martín, C.; Amuategi, J.; Fernandez-Martínez, X. Membrane Permeabilization by Pore-Forming RTX Toxins: What Kind of Lesions Do These Toxins Form? Toxins 2019, 11, 354. [Google Scholar] [CrossRef]
- Chenal, A.; Sotomayor Perez, A.C.; Ladant, D. Structure and function of RTX toxins. In The Comprehensive Sourcebook of Bacterial Protein Toxins; Alouf, J., Ladant, D., Popoff, M.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 677–718. [Google Scholar]
- Chenal, A.; Ladant, D. Bioengineering of Bordetella pertussis Adenylate Cyclase Toxin for Antigen-Delivery and Immunotherapy. Toxins 2018, 10, 302. [Google Scholar] [CrossRef]
- Knapp, O.; Benz, R. Membrane Activity and Channel Formation of the Adenylate Cyclase Toxin (CyaA) of Bordetella pertussis in Lipid Bilayer Membranes. Toxins 2020, 12, 169. [Google Scholar] [CrossRef]
- Kim, B.S. The Modes of Action of MARTX Toxin Effector Domains. Toxins 2018, 10, 507. [Google Scholar] [CrossRef]
- Satchell, K.J. Structure and function of MARTX toxins and other large repetitive RTX proteins. Annu. Rev. Microbiol. 2011, 65, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Gavin, H.E.; Satchell, K.J. Distinct roles of the repeat-containing regions and effector domains of the Vibrio vulnificus multifunctional-autoprocessing repeats-in-toxin (MARTX) toxin. mBio. 2015, 6, e00324-15. [Google Scholar] [CrossRef] [PubMed]
- Endo, H. Molecular and Kinetic Models for Pore Formation of Bacillus thuringiensis Cry Toxin. Toxins 2022, 14, 433. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, S.; Gómez, I.; Soberón, M.; Bravo, A. A major conformational change of N-terminal helices of Bacillus thuringiensis Cry1Ab insecticidal protein is necessary for membrane insertion and toxicity. FEBS J. 2023, 290, 2692–2705. [Google Scholar] [CrossRef]
- Pacheco, S.; Quiliche, J.P.J.; Gómez, I.; Sánchez, J.; Soberón, M.; Bravo, A. Rearrangement of N-Terminal α-Helices of Bacillus thuringiensis Cry1Ab Toxin Essential for Oligomer Assembly and Toxicity. Toxins 2020, 12, 647. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Surya, W.; Boonserm, P. Channel Formation in Cry Toxins: An Alphafold-2 Perspective. Int. J. Mol. Sci. 2023, 24, 16809. Available online: https://www.mdpi.com/1422-0067/24/23/16809 (accessed on 15 February 2024). [CrossRef] [PubMed]
- Nicolson, G.L. The Fluid-Mosaic Model of Membrane Structure: Still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochim. Biophys. Acta 2014, 1838, 1451–1466. [Google Scholar] [CrossRef] [PubMed]
- Christie, M.P.; Johnstone, B.A.; Tweten, R.K.; Parker, M.W.; Morton, C.J. Cholesterol-dependent cytolysins: From water-soluble state to membrane pore. Biophys. Rev. 2018, 10, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef]
- Dunstone, M.A.; Tweten, R.K. Packing a punch: The mechanism of pore formation by cholesterol dependent cytolysins and membrane attack complex/perforin-like proteins. Curr. Opin. Struct. Biol. 2012, 22, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Iacovache, I.; Bischofberger, M.; van der Goot, F.G. Structure and assembly of pore-forming proteins. Curr. Opin. Struct. Biol. 2010, 20, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R. Clostridial pore-forming toxins: Powerful virulence factors. Anaerobe 2014, 30, 220–238. [Google Scholar] [CrossRef] [PubMed]
- Heuck, A.P.; Moe, P.C.; Johnson, B.B. The cholesterol-dependent cytolysin family of gram-positive bacterial toxins. Subcell. Biochem. 2010, 51, 551–577. [Google Scholar]
- Tweten, R.K.; Hotze, E.M.; Wade, K.R. The Unique Molecular Choreography of Giant Pore Formation by the Cholesterol-Dependent Cytolysins of Gram-Positive Bacteria. Annu. Rev. Microbiol. 2015, 69, 323–340. [Google Scholar] [CrossRef]
- Johnstone, B.A.; Joseph, R.; Christie, M.P.; Morton, C.J.; McGuiness, C.; Walsh, J.C.; Böcking, T.; Tweten, R.K.; Parker, M.W. Cholesterol-dependent cytolysins: The outstanding questions. IUBMB Life 2022, 74, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Rossjohn, J.; Polekhina, G.; Feil, S.C.; Morton, C.J.; Tweten, R.K.; Parker, M.W. Structures of perfringolysin O suggest a pathway for activation of cholesterol-dependent cytolysins. J. Mol. Biol. 2007, 367, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.N.; Peterson, B.N.; Portnoy, D.A. Listeriolysin O: A phagosome-specific cytolysin revisited. Cell. Microbiol. 2019, 21, e12988. [Google Scholar] [CrossRef] [PubMed]
- Quereda, J.J.; Morel, C.; Lopez-Montero, N.; Ziveri, J.; Rolland, S.; Grenier, T.; Aulner, N.; Danckaert, A.; Charbit, A.; Enninga, J.; et al. A Role for Taok2 in Listeria monocytogenes Vacuolar Escape. J. Infect Dis. 2022, 225, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Madden, J.C.; Ruiz, H.; Caparon, M. Cytolysin-mediated translocation (CMT): A functional equivalent of type III secretion in Gram-positive bacteria. Cell 2001, 104, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Prévost, G.; Tawk, M.Y.; Zimmermann-Meise, G.; Jover, E. The staphylococcal alpha-toxin and leucotoxins. In The Comprehensive Sourcebook of Bacterial Protein Toxins; Alouf, J., Ladant, D., Popoff, M.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 739–772. [Google Scholar]
- Oliveira, D.; Borges, A.; Simões, M. Staphylococcus aureus Toxins and Their Molecular Activity in Infectious Diseases. Toxins 2018, 10, 252. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.; Uzal, F.A.; McClane, B.A. The interaction of Clostridium perfringens enterotoxin with receptor claudins. Anaerobe 2016, 41, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.; McClane, B. Clostridium perfringens enterotoxin. In The Comprehensive Sourcebook of Bacterial Protein Toxins, 4th ed.; Alouf, J., Ladant, D.R.P.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 815–838. [Google Scholar]
- Popoff, M.R.; Stiles, B.; Poulain, B. Clostridium perfringens Epsilon Toxin: Structural and Mechanistic Insights. In Microbial Toxins; Gopalakrishnakone, P., Stiles, B., Alape-Giron, A., Dubreuil, J.D., Mandal, M., Eds.; Springer: Dordrecht, The Netherlands, 2018; pp. 53–72. [Google Scholar] [CrossRef]
- Bischofberger, M.; Iacovache, I.; van der Goot, F.G. Pathogenic pore-forming proteins: Function and host response. Cell Host Microbe 2012, 12, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Bouillot, S.; Reboud, E.; Huber, P. Functional Consequences of Calcium Influx Promoted by Bacterial Pore-Forming Toxins. Toxins 2018, 10, 387. [Google Scholar] [CrossRef]
- Fennessey, C.M.; Ivie, S.E.; McClain, M.S. Coenzyme depletion by members of the aerolysin family of pore-forming toxins leads to diminished ATP levels and cell death. Mol. BioSystems 2012, 8, 2097–2105. [Google Scholar] [CrossRef]
- Gurcel, L.; Abrami, L.; Girardin, S.; Tschopp, J.; van der Goot, F.G. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 2006, 126, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Chassin, C.; Bens, M.; de Barry, J.; Courjaret, R.; Bossu, J.L.; Cluzeaud, F.; Ben Mkaddem, S.; Gibert, M.; Poulain, B.; Popoff, M.R.; et al. Pore-forming epsilon toxin causes membrane permeabilization and rapid ATP depletion-mediated cell death in renal collecting duct cells. Am. J. Physiol. Renal. Physiol. 2007, 293, F927–F937. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Maier, E.; Gibert, M.; Popoff, M.R.; Benz, R. Clostridium perfringens epsilon-toxin induces a rapid change in cell membrane permeability to ions and forms channels in artificial lipid bilayers. J. Biol. Chem. 2001, 276, 15736–15740. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.; Mehdizadeh Gohari, I.; McClane, B.A. RIP1, RIP3, and MLKL Contribute to Cell Death Caused by Clostridium perfringens Enterotoxin. mBio 2019, 10, e02985-19. [Google Scholar] [CrossRef] [PubMed]
- Krantz, B.A.; Finkelstein, A.; Collier, R.J. Protein translocation through the anthrax toxin transmembrane pore is driven by a proton gradient. J. Mol. Biol. 2006, 355, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Machen, A.J.; Fisher, M.T.; Freudenthal, B.D. Anthrax toxin translocation complex reveals insight into the lethal factor unfolding and refolding mechanism. Sci. Rep. 2021, 11, 13038. [Google Scholar] [CrossRef] [PubMed]
- Neumeyer, T.; Tonello, F.; Dal Molin, F.; Schiffler, B.; Benz, R. Anthrax edema factor, voltage-dependent binding to the protective antigen ion channel and comparison to LF binding. J. Biol. Chem. 2006, 281, 32335–32343. [Google Scholar] [CrossRef] [PubMed]
- Kintzer, A.F.; Thoren, K.L.; Sterling, H.J.; Dong, K.C.; Feld, G.K.; Tang, I.I.; Zhang, T.T.; Williams, E.R.; Berger, J.M.; Krantz, B.A. The protective antigen component of anthrax toxin forms functional octameric complexes. J. Mol. Biol. 2009, 392, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Neumeyer, T.; Tonello, F.; Dal Molin, F.; Schiffler, B.; Orlik, F.; Benz, R. Anthrax lethal factor (LF) mediated block of the anthrax protective antigen (PA) ion channel: Effect of ionic strength and voltage. Biochemistry 2006, 45, 3060–3068. [Google Scholar] [CrossRef] [PubMed]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A.T. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar] [CrossRef]
- Scobie, H.M.; Rainey, G.J.; Bradley, K.A.; Young, J.A. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar] [CrossRef] [PubMed]
- Blocker, D.; Pohlmann, K.; Haug, G.; Bachmeyer, C.; Benz, R.; Aktories, K.; Barth, H. Clostridium botulinum C2 toxin: Low pH-induced pore formation is required for translocation of the enzyme component C2I into the cytosol of host cells. J. Biol. Chem. 2003, 278, 37360–37367. [Google Scholar] [CrossRef] [PubMed]
- Knapp, O.; Benz, R.; Gibert, M.; Marvaud, J.C.; Popoff, M.R. Interaction of Clostridium perfringens iota-toxin with lipid bilayer membranes. J. Biol. Chem. 2002, 277, 6143–6152. [Google Scholar] [CrossRef] [PubMed]
- Fagerlund, A.; Lindbäck, T.; Storset, A.K.; Granum, P.E.; Hardy, S.P. Bacillus cereus Nhe is a pore-forming toxin with structural and functional properties similar to the ClyA (HlyE, SheA) family of haemolysins, able to induce osmotic lysis in epithelia. Microbiology 2008, 154, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Didier, A.; Dietrich, R.; Heilkenbrinker, U.; Waltenberger, E.; Jessberger, N.; Märtlbauer, E.; Benz, R. Formation of small transmembrane pores: An intermediate stage on the way to Bacillus cereus non-hemolytic enterotoxin (Nhe) full pores in the absence of NheA. Biochem. Biophys. Res. Commun. 2016, 469, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Ganash, M.; Phung, D.; Sedelnikova, S.E.; Lindbäck, T.; Granum, P.E.; Artymiuk, P.J. Structure of the NheA component of the Nhe toxin from Bacillus cereus: Implications for function. PLoS ONE 2013, 8, e74748. [Google Scholar] [CrossRef] [PubMed]
- Jessberger, N.; Dietrich, R.; Schauer, K.; Schwemmer, S.; Märtlbauer, E.; Benz, R. Characteristics of the Protein Complexes and Pores Formed by Bacillus cereus Hemolysin BL. Toxins 2020, 12, 672. [Google Scholar] [CrossRef] [PubMed]
- Madegowda, M.; Eswaramoorthy, S.; Burley, S.K.; Swaminathan, S. X-ray crystal structure of the B component of Hemolysin BL from Bacillus cereus. Proteins 2008, 71, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zuo, Z.; Sastalla, I.; Liu, C.; Jang, J.Y.; Sekine, Y.; Li, Y.; Pirooznia, M.; Leppla, S.H.; Finkel, T.; et al. Sequential CRISPR-Based Screens Identify LITAF and CDIP1 as the Bacillus cereus Hemolysin BL Toxin Host Receptors. Cell Host Microbe 2020, 28, 402–410.e405. [Google Scholar] [CrossRef]
- Menestrina, G.; Moser, C.; Pellet, S.; Welch, R. Pore-formation by Escherichia coli hemolysin (HlyA) and other members of the RTX toxins family. Toxicology 1994, 87, 249–267. [Google Scholar] [CrossRef]
- Wiles, T.J.; Mulvey, M.A. The RTX pore-forming toxin α-hemolysin of uropathogenic Escherichia coli: Progress and perspectives. Future Microbiol. 2013, 8, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Vojtova-Vodolanova, J.; Basler, M.; Osicka, R.; Knapp, O.; Maier, E.; Cerny, J.; Benada, O.; Benz, R.; Sebo, P. Oligomerization is involved in pore formation by Bordetella adenylate cyclase toxin. FASEB J. 2009, 23, 2831–2843. [Google Scholar] [CrossRef]
- Benz, R.; Maier, E.; Ladant, D.; Ullmann, A.; Sebo, P. Adenylate cyclase toxin (CyaA) of Bordetella pertussis. Evidence for the formation of small ion-permeable channels and comparison with HlyA of Escherichia coli. J. Biol. Chem. 1994, 269, 27231–27239. [Google Scholar] [CrossRef] [PubMed]
- González-Bullón, D.; Uribe, K.B.; Largo, E.; Guembelzu, G.; García-Arribas, A.B.; Martín, C.; Ostolaza, H. Membrane Permeabilization by Bordetella Adenylate Cyclase Toxin Involves Pores of Tunable Size. Biomolecules 2019, 9, 183. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.R.; Lee, S.E.; Kook, H.; Yeom, J.A.; Na, H.S.; Kim, S.Y.; Chung, S.S.; Choy, H.E.; Rhee, J.H. Vibrio vulnificus RTX toxin kills host cells only after contact of the bacteria with host cells. Cell. Microbiol. 2008, 10, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Shepard, L.; Shatursky, O.; Johnson, A.; Tweten, R. The mechanism of pore assembly for a cholesterol-dependent cytolysin: Formation of a large prepore complex precedes the insertion of the transmembrane β-hairpins. Biochemistry 2000, 39, 10284–10293. [Google Scholar] [CrossRef] [PubMed]
- Shewell, L.K.; Day, C.J.; Jen, F.E.; Haselhorst, T.; Atack, J.M.; Reijneveld, J.F.; Everest-Dass, A.; James, D.B.A.; Boguslawski, K.M.; Brouwer, S.; et al. All major cholesterol-dependent cytolysins use glycans as cellular receptors. Sci. Adv. 2020, 6, eaaz4926. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.; Harris, R.; Freytag, C.; Kehoe, M.; Tranum-Jensen, J.; Bhakdi, S. Assembly mechanism of the oligomeric streptolysin O pore: The early membrane lesion is lined by a free edge of the lipid membrane and is extended gradually during oligomerization. EMBO J. 1998, 17, 1598–1605. [Google Scholar] [CrossRef] [PubMed]
- Köster, S.; van Pee, K.; Hudel, M.; Leustik, M.; Rhinow, D.; Kühlbrandt, W.; Chakraborty, T.; Yildiz, Ö. Crystal structure of listeriolysin O reveals molecular details of oligomerization and pore formation. Nat. Commun. 2014, 5, 3690. [Google Scholar] [CrossRef]
- Giddings, K.S.; Zhao, J.; Sims, P.J.; Tweten, R.K. Human CD59 is a receptor for the cholesterol-dependent cytolysin intermedilysin. Nat. Struct. Mol. Biol. 2004, 11, 1173–1178. [Google Scholar] [CrossRef]
- Polekhina, G.; Giddings, K.S.; Tweten, R.K.; Parker, M.W. Insights into the action of the superfamily of cholesterol-dependent cytolysins from studies of intermedilysin. Proc. Natl. Acad. Sci. USA 2005, 102, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Tilley, S.J.; Orlova, E.V.; Gilbert, R.J.; Andrew, P.W.; Saibil, H.R. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell 2005, 121, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Fivaz, M.; Glauser, P.E.; Parton, R.G.; van der Goot, F.G. A pore-forming toxin interacts with a GPI-anchored protein and causes vacuolation of the endoplasmic reticulum. J. Cell Biol. 1998, 140, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Buckley, J.T.; Postma, J.P.; Tucker, A.D.; Leonard, K.; Pattus, F.; Tsernoglou, D. Structure of the Aeromonas toxin proaerolysin in its water-soluble and membrane-channel states. Nature 1994, 367, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Tsitrin, Y.; Morton, C.J.; el-Bez, C.; Paumard, P.; Velluz, M.C.; Adrian, M.; Dubochet, J.; Parker, M.W.; Lanzavecchia, S.; van der Goot, F.G. Conversion of a transmembrane to a water-soluble protein complex by a single point mutation. Nat. Struct. Biol. 2002, 9, 729–733. [Google Scholar] [CrossRef]
- Chakraborty, T.; Schmid, A.; Notermans, S.; Benz, R. Aerolysin of Aeromonas sobria: Evidence for formation of ion-permeable channels and comparison with alpha-toxin of Staphylococcus aureus. Infect. Immun. 1990, 58, 2127–2132. [Google Scholar] [CrossRef]
- Knapp, O.; Maier, E.; Benz, R.; Geny, B.; Popoff, M.R. Identification of the channel-forming domain of Clostridium perfringens Epsilon-toxin (ETX). Biochim. Biophys. Acta 2009, 1788, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Rumah, K.R.; Ma, Y.; Linden, J.R.; Oo, M.L.; Anrather, J.; Schaeren-Wiemers, N.; Alonso, M.A.; Fischetti, V.A.; McClain, M.S.; Vartanian, T. The Myelin and Lymphocyte Protein MAL Is Required for Binding and Activity of Clostridium perfringens ε-Toxin. PLoS Pathog. 2015, 11, e1004896. [Google Scholar] [CrossRef]
- Savva, C.G.; Clark, A.R.; Naylor, C.E.; Popoff, M.R.; Moss, D.S.; Basak, A.K.; Titball, R.W.; Bokori-Brown, M. The pore structure of Clostridium perfringens epsilon toxin. Nat. Commun. 2019, 10, 2641. [Google Scholar] [CrossRef]
- Katahira, J.; Inoue, N.; Horiguchi, Y.; Matsuda, M.; Sugimoto, N. Molecular cloning and functional characterization of the receptor for Clostridium perfringens enterotoxin. J. Cell Biol. 1997, 136, 1239–1247. [Google Scholar] [CrossRef]
- Benz, R.; Popoff, M.R. Clostridium perfringens Enterotoxin: The Toxin Forms Highly Cation-Selective Channels in Lipid Bilayers. Toxins 2018, 10, 341. [Google Scholar] [CrossRef] [PubMed]
- Briggs, D.C.; Naylor, C.E.; Smedley, J.G., 3rd; Lukoyanova, N.; Robertson, S.; Moss, D.S.; McClane, B.A.; Basak, A.K. Structure of the food-poisoning Clostridium perfringens enterotoxin reveals similarity to the aerolysin-like pore-forming toxins. J. Mol. Biol. 2011, 413, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Kitadokoro, K.; Nishimura, K.; Kamitani, S.; Fukui-Miyazaki, A.; Toshima, H.; Abe, H.; Kamata, Y.; Sugita-Konishi, Y.; Yamamoto, S.; Karatani, H.; et al. Crystal structure of Clostridium perfringens enterotoxin displays features of beta-pore-forming toxins. J. Biol. Chem. 2011, 286, 19549–19555. [Google Scholar] [CrossRef] [PubMed]
- Knapp, O.; Maier, E.; Mkaddem, S.B.; Benz, R.; Bens, M.; Chenal, A.; Geny, B.; Vandewalle, A.; Popoff, M.R. Clostridium septicum alpha-toxin forms pores and induces rapid cell necrosis. Toxicon 2010, 55, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Gordon, V.M.; Nelson, K.L.; Buckley, J.T.; Stevens, V.L.; Tweten, R.K.; Elwood, P.C.; Leppla, S.H. Clostridium septicum alpha-toxin uses glycosylphosphatidylinositol-anchored protein receptors. J. Biol. Chem. 1999, 274, 27274–27280. [Google Scholar] [CrossRef] [PubMed]
- Melton, J.A.; Parker, M.W.; Rossjohn, J.; Buckley, J.T.; Tweten, R.K. The identification and structure of the membrane-spanning domain of the Clostridium septicum alpha toxin. J. Biol. Chem. 2004, 279, 14315–14322. [Google Scholar] [CrossRef] [PubMed]
- Palma, L.; Munoz, D.; Berry, C.; Murillo, J.; Caballero, P. Bacillus thuringiensis toxins: An overview of their biocidal activity. Toxins 2014, 6, 3296–3325. [Google Scholar] [CrossRef]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 1996, 274, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Spaan, A.N.; Henry, T.; van Rooijen, W.J.M.; Perret, M.; Badiou, C.; Aerts, P.C.; Kemmink, J.; de Haas, C.J.C.; van Kessel, K.P.M.; Vandenesch, F.; et al. The staphylococcal toxin Panton-Valentine Leukocidin targets human C5a receptors. Cell Host Microbe 2013, 13, 584–594. [Google Scholar] [CrossRef]
- Guillet, V.; Roblin, P.; Werner, S.; Coraiola, M.; Menestrina, G.; Monteil, H.; Prévost, G.; Mourey, L. Crystal structure of leucotoxin S component: New insight into the Staphylococcal beta-barrel pore-forming toxins. J. Biol. Chem. 2004, 279, 41028–41037. [Google Scholar] [CrossRef]
- Pédelacq, J.D.; Maveyraud, L.; Prévost, G.; Baba-Moussa, L.; González, A.; Courcelle, E.; Shepard, W.; Monteil, H.; Samama, J.P.; Mourey, L. The structure of a Staphylococcus aureus leucocidin component (LukF-PV) reveals the fold of the water-soluble species of a family of transmembrane pore-forming toxins. Structure 1999, 7, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Kawai, Y.; Tanaka, Y.; Hirano, N.; Kaneko, J.; Tomita, N.; Ohta, M.; Kamio, Y.; Yao, M.; Tanaka, I. Crystal structure of the octameric pore of staphylococcal γ-hemolysin reveals the β-barrel pore formation mechanism by two components. Proc. Natl. Acad. Sci. USA 2011, 108, 17314–17319. [Google Scholar] [CrossRef] [PubMed]
- Sugawara-Tomita, N.; Tomita, T.; Kamio, Y. Stochastic assembly of two-component staphylococcal gamma-hemolysin into heteroheptameric transmembrane pores with alternate subunit arrangements in ratios of 3:4 and 4:3. J. Bacteriol. 2002, 184, 4747–4756. [Google Scholar] [CrossRef] [PubMed]
- Hodille, E.; Plesa, A.; Bourrelly, E.; Belmont, L.; Badiou, C.; Lina, G.; Dumitrescu, O. Staphylococcal Panton-Valentine Leucocidin and Gamma Haemolysin Target and Lyse Mature Bone Marrow Leucocytes. Toxins 2020, 12, 725. [Google Scholar] [CrossRef] [PubMed]
- Perelman, S.S.; James, D.B.A.; Boguslawski, K.M.; Nelson, C.W.; Ilmain, J.K.; Zwack, E.E.; Prescott, R.A.; Mohamed, A.; Tam, K.; Chan, R.; et al. Genetic variation of staphylococcal LukAB toxin determines receptor tropism. Nat. Microbiol. 2021, 6, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Badarau, A.; Rouha, H.; Malafa, S.; Logan, D.T.; Håkansson, M.; Stulik, L.; Dolezilkova, I.; Teubenbacher, A.; Gross, K.; Maierhofer, B.; et al. Structure-function analysis of heterodimer formation, oligomerization, and receptor binding of the Staphylococcus aureus bi-component toxin LukGH. J. Biol. Chem. 2015, 290, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Trstenjak, N.; Milić, D.; Graewert, M.A.; Rouha, H.; Svergun, D.; Djinović-Carugo, K.; Nagy, E.; Badarau, A. Molecular mechanism of leukocidin GH-integrin CD11b/CD18 recognition and species specificity. Proc. Natl. Acad. Sci. USA 2020, 117, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Nocadello, S.; Minasov, G.; Shuvalova, L.; Dubrovska, I.; Sabini, E.; Bagnoli, F.; Grandi, G.; Anderson, W.F. Crystal structures of the components of the Staphylococcus aureus leukotoxin ED. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Lubkin, A.; Lee, W.L.; Alonzo, F., 3rd; Wang, C.; Aligo, J.; Keller, M.; Girgis, N.M.; Reyes-Robles, T.; Chan, R.; O’Malley, A.; et al. Staphylococcus aureus Leukocidins Target Endothelial DARC to Cause Lethality in Mice. Cell Host Microbe 2019, 25, 463–470.e469. [Google Scholar] [CrossRef]
- Gravet, A.; Colin, D.A.; Keller, D.; Girardot, R.; Monteil, H.; Prévost, G. Characterization of a novel structural member, LukE-LukD, of the bi-component staphylococcal leucotoxins family. FEBS Lett. 1998, 436, 202–208. [Google Scholar] [CrossRef]
- Reyes-Robles, T.; Alonzo, F., 3rd; Kozhaya, L.; Lacy, D.B.; Unutmaz, D.; Torres, V.J. Staphylococcus aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe 2013, 14, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Bruggisser, J.; Iacovache, I.; Musson, S.C.; Degiacomi, M.T.; Posthaus, H.; Zuber, B. Cryo-EM structure of the octameric pore of Clostridium perfringens β-toxin. EMBO Rep. 2022, 23, e54856. [Google Scholar] [CrossRef]
- Tarek, B.; Bruggisser, J.; Cattalani, F.; Posthaus, H. Platelet Endothelial Cell Adhesion Molecule 1 (CD31) Is Essential for Clostridium perfringens Beta-Toxin Mediated Cytotoxicity in Human Endothelial and Monocytic Cells. Toxins 2021, 13, 893. [Google Scholar] [CrossRef]
- Savva, C.G.; Fernandes da Costa, S.P.; Bokori-Brown, M.; Naylor, C.E.; Cole, A.R.; Moss, D.S.; Titball, R.W.; Basak, A.K. Molecular architecture and functional analysis of NetB, a pore-forming toxin from Clostridium perfringens. J. Biol. Chem. 2013, 288, 3512–3522. [Google Scholar] [CrossRef]
- Huyet, J.; Naylor, C.E.; Savva, C.G.; Gibert, M.; Popoff, M.R.; Basak, A.K. Structural Insights into Delta Toxin Pore Formation. PLoS ONE 2013, 8, e66673. [Google Scholar] [CrossRef]
- Manich, M.; Knapp, O.; Gibert, M.; Maier, E.; Jolivet-Reynaud, C.; Geny, B.; Benz, R.; Popoff, M.R. Clostridium perfringens delta toxin is sequence related to beta toxin, NetB, and Staphylococcus pore-forming toxins, but shows functional differences. PLoS ONE 2008, 3, e3764. [Google Scholar] [CrossRef] [PubMed]
- Frey, J.; Johansson, A.; Burki, S.; Vilei, E.M.; Redhead, K. Cytotoxin CctA, a major virulence factor of Clostridium chauvoei conferring protective immunity against myonecrosis. Vaccine 2012, 30, 5500–5505. [Google Scholar] [CrossRef]
- De, S.; Olson, R. Crystal structure of the Vibrio cholerae cytolysin heptamer reveals common features among disparate pore-forming toxins. Proc. Natl. Acad. Sci. USA 2011, 108, 7385–7390. [Google Scholar] [CrossRef] [PubMed]
- Levan, S.; De, S.; Olson, R. Vibrio cholerae cytolysin recognizes the heptasaccharide core of complex N-glycans with nanomolar affinity. J. Mol. Biol. 2013, 425, 944–957. [Google Scholar] [CrossRef]
- Olson, R.; Gouaux, E. Crystal structure of the Vibrio cholerae cytolysin (VCC) pro-toxin and its assembly into a heptameric transmembrane pore. J. Mol. Biol. 2005, 350, 997–1016. [Google Scholar] [CrossRef]
- Kaus, K.; Lary, J.W.; Cole, J.L.; Olson, R. Glycan specificity of the Vibrio vulnificus hemolysin lectin outlines evolutionary history of membrane targeting by a toxin family. J. Mol. Biol. 2014, 426, 2800–2812. [Google Scholar] [CrossRef]
- Kashimoto, T.; Sugiyama, H.; Kawamidori, K.; Yamazaki, K.; Kado, T.; Matsuda, K.; Kodama, T.; Mukai, T.; Ueno, S. Vibiro vulnificus hemolysin associates with gangliosides. BMC Microbiol. 2020, 20, 69. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, R.O.; Finkelstein, A. Voltage-dependent block of anthrax toxin channels in planar phospholipid bilayer membranes by symetric tetraalkylammonium ions. J. Gen. Physiol. 1990, 96, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Pentelute, B.L.; Collier, R.J.; Zhou, Z.H. Atomic structure of anthrax protective antigen pore elucidates toxin translocation. Nature 2015, 521, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Nassi, S.; Collier, R.J.; Finkelstein, A. PA63 channel of anthrax toxin: An extended beta-barrel. Biochemistry 2002, 41, 1445–1450. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, P.; Carette, J.E.; Bell, G.W.; Schwan, C.; Guttenberg, G.; Brummelkamp, T.R.; Aktories, K. Lipolysis-stimulated lipoprotein receptor (LSR) is the host receptor for the binary toxin Clostridium difficile transferase (CDT). Proc. Natl. Acad. Sci. USA 2011, 108, 16422–16427. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Yoshida, T.; Kawamoto, A.; Mitsuoka, K.; Iwasaki, K.; Tsuge, H. Cryo-EM structures reveal translocational unfolding in the clostridial binary iota toxin complex. Nat. Struct. Mol. Biol. 2020, 27, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Sheedlo, M.J.; Jensen, J.L.; Lacy, D.B. Structural insights into the transition of Clostridioides difficile binary toxin from prepore to pore. Nat. Microbiol. 2020, 5, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Aktories, K.; Popoff, M.R.; Stiles, B.G. Binary bacterial toxins: Biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol. Mol. Biol. Rev. 2004, 68, 373–402. [Google Scholar] [CrossRef]
- Gupta, M.; Kumar, H.; Kaur, S. Vegetative Insecticidal Protein (Vip): A Potential Contender from Bacillus thuringiensis for Efficient Management of Various Detrimental Agricultural Pests. Front. Microbiol. 2021, 12, 659736. [Google Scholar] [CrossRef]
- Degiacomi, M.T.; Iacovache, I.; Pernot, L.; Chami, M.; Kudryashev, M.; Stahlberg, H.; van der Goot, F.G.; Dal Peraro, M. Molecular assembly of the aerolysin pore reveals a swirling membrane-insertion mechanism. Nat. Chem. Biol. 2013, 9, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R.; Bouvet, P. Genetic characteristics of toxigenic Clostridia and toxin gene evolution. Toxicon 2013, 75, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Naylor, C.E.; Eaton, J.T.; Howells, A.; Justin, N.; Moss, D.S.; Titball, R.W.; Basak, A.K. Structure of the key toxin in gas gangrene. Nat. Struct. Biol. 1998, 5, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Ellemor, D.M.; Boyd, R.L.; Emmins, J.J.; Rood, J.I. Synergistic effects of alpha-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect. Immun. 2001, 69, 7904–7910. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Bryant, A.E.; Stevens, D.L.; Rood, J.I. Virulence studies on chromosomal alpha-toxin and theta-toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of alpha-toxin in Clostridium perfringens-mediated gas gangrene. Mol. Microbiol. 1995, 15, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.T.; Naylor, C.E.; Howells, A.M.; Moss, D.S.; Titball, R.W.; Basak, A.K. Crystal structure of the C. perfringens alpha-toxin with the active site closed by a flexible loop region. J. Mol. Biol. 2002, 319, 275–281. [Google Scholar] [CrossRef]
- MacFarlane, M.G.; Knight, B.C.J.G. The biochemistry of bacterial toxins. I. Lecithinase activity of C. welchii toxins. Biochem. J. 1941, 35, 884–902. [Google Scholar] [CrossRef]
- Sakurai, J.; Nagahama, M.; Oda, M. Clostridium perfringens alpha-toxin: Characterization and mode of action. J. Biochem. 2004, 136, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Titball, R.W.; Naylor, C.E.; Basak, A.K. The Clostridium perfringens α-toxin. Anaerobe 1999, 5, 51–64. [Google Scholar] [CrossRef]
- Takehara, M.; Takagishi, T.; Seike, S.; Ohtani, K.; Kobayashi, K.; Miyamoto, K.; Shimizu, T.; Nagahama, M. Clostridium perfringens α-Toxin Impairs Innate Immunity via Inhibition of Neutrophil Differentiation. Sci. Rep. 2016, 6, 28192. [Google Scholar] [CrossRef]
- Flores-Diaz, M.; Alape-Giron, A. Role of Clostridium perfringens phospholipase C in the pathogenesis of gas gangrene. Toxicon 2003, 42, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Bryant, A.E.; Stevens, D.L. Clostridial toxins in the pathogenesis of gas gangrene. In The Comprehensive Sourcebook of Bacterial Protein Toxins, 4th ed.; Alouf, J., Ladant, D., Popoff, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 977–994. [Google Scholar]
- Suzaki, A.; Hayakawa, S. Clinical and Microbiological Features of Fulminant Haemolysis Caused by Clostridium perfringens Bacteraemia: Unknown Pathogenesis. Microorganisms 2023, 11, 824. [Google Scholar] [CrossRef]
- Suzaki, A.; Ohtani, K.; Komine-Aizawa, S.; Matsumoto, A.; Kamiya, S.; Hayakawa, S. Pathogenic Characterization of Clostridium perfringens Strains Isolated from Patients with Massive Intravascular Hemolysis. Front. Microbiol. 2021, 12, 713509. [Google Scholar] [CrossRef] [PubMed]
- Titball, R.W. Bacterial phospholipases C. Microbiol. Rev. 1993, 57, 347–366. [Google Scholar] [CrossRef]
- Flores-Diaz, M.; Monturiol-Gross, L.; Alape-Giron, A. Membrane-damaging and cytotoxic sphingomyelinases and phospholipases. In The Comprehensive Sourcebook of Bacterial Protein Toxins; Alouf, J., Ladant, D., Popoff, M.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 627–676. [Google Scholar]
- Monturiol-Gross, L.; Villalta-Romero, F.; Flores-Díaz, M.; Alape-Girón, A. Bacterial phospholipases C with dual activity: Phosphatidylcholinesterase and sphingomyelinase. FEBS Open Bio 2021, 11, 3262–3275. [Google Scholar] [CrossRef] [PubMed]
- Songer, J.G. Bacterial phospholipases and their role in virulence. Trends Microbiol. 1997, 5, 156–161. [Google Scholar] [CrossRef]
- Matsushita, O.; Koide, T.; Kobayashi, R.; Nagata, K.; Okabe, A. Substrate recognition by the collagen-binding domain of Clostridium histolyticum class I collagenase. J. Biol. Chem. 2001, 276, 8761–8770. [Google Scholar] [CrossRef]
- Bauer, R.; Wilson, J.J.; Philominathan, S.T.; Davis, D.; Matsushita, O.; Sakon, J. Structural comparison of ColH and ColG collagen-binding domains from Clostridium histolyticum. J. Bacteriol. 2013, 195, 318–327. [Google Scholar] [CrossRef]
- Eckhard, U.; Schönauer, E.; Brandstetter, H. Structural basis for activity regulation and substrate preference of clostridial collagenases G., H, and T. J. Biol. Chem. 2013, 288, 20184–20194. [Google Scholar] [CrossRef]
- Hu, Y.; Webb, E.; Singh, J.; Morgan, B.A.; Gainor, J.A.; Gordon, T.D.; Siahaan, T.J. Rapid determination of substrate specificity of Clostridium histolyticum beta-collagenase using an immobilized peptide library. J. Biol. Chem. 2002, 277, 8366–8371. [Google Scholar] [CrossRef]
- Philominathan, S.T.; Koide, T.; Matsushita, O.; Sakon, J. Bacterial collagen-binding domain targets undertwisted regions of collagen. Protein Sci. 2012, 21, 1554–1565. [Google Scholar] [CrossRef]
- Matsushita, O.; Jung, C.M.; Katayama, S.; Minami, J.; Takahashi, Y.; Okabe, A. Gene duplication and multiplicity of collagenases in Clostridium histolyticum. J. Bacteriol. 1999, 181, 923–933. [Google Scholar] [CrossRef]
- Matsushita, O.; Jung, C.M.; Minami, J.; Katayama, S.; Nishi, N.; Okabe, A. A study of the collagen-binding domain of a 116 kDa Clostridium histolyticum collagenase. J. Biol. Chem. 1998, 273, 3643–3648. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, O.; Yoshihara, K.; Katayama, S.; Minami, J.; Okabe, A. Purification and characterization of a Clostridium perfringens 120-kilodalton collagenase and nucleotide sequence of the corresponding gene. J. Bacteriol. 1994, 176, 149–156. [Google Scholar] [CrossRef]
- Haghi, F.; Goli, E.; Mirzaei, B.; Zeighami, H. The association between fecal enterotoxigenic B. fragilis with colorectal cancer. BMC Cancer 2019, 19, 879. [Google Scholar] [CrossRef] [PubMed]
- Valguarnera, E.; Wardenburg, J.B. Good Gone Bad: One Toxin Away from Disease for Bacteroides fragilis. J. Mol. Biol. 2020, 432, 765–785. [Google Scholar] [CrossRef] [PubMed]
- Goulas, T.; Arolas, J.L.; Gomis-Rüth, F.X. Structure, function and latency regulation of a bacterial enterotoxin potentially derived from a mammalian adamalysin/ADAM xenolog. Proc. Natl. Acad. Sci. USA 2011, 108, 1856–1861. [Google Scholar] [CrossRef]
- Sears, C.L. Enterotoxigenic Bacteroides fragilis: A rogue among symbiotes. Clin. Microbiol. Rev. 2009, 2009, 349–369. [Google Scholar] [CrossRef]
- Wu, S.; Lim, K.C.; Huang, J.; Saidi, R.F.; Sears, C.L. Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proc. Natl. Acad. Sci. USA 1998, 95, 14979–14984. [Google Scholar] [CrossRef]
- Ko, S.H.; Choi, J.H.; Kim, J.M. Bacteroides fragilis Enterotoxin Induces Autophagy through an AMPK and FoxO3-Pathway, Leading to the Inhibition of Apoptosis in Intestinal Epithelial Cells. Toxins 2023, 15, 544. [Google Scholar] [CrossRef]
- Jeon, J.I.; Ko, S.H.; Kim, J.M. Intestinal Epithelial Cells Exposed to Bacteroides fragilis Enterotoxin Regulate NF-κB Activation and Inflammatory Responses through β-Catenin Expression. Infect. Immun. 2019, 87, e00312–e00319. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.J.; Zhang, M.; Franco, A.; Sears, C.L. Bacteroides fragilis toxin stimulates intestinal epithelial cell shedding and gamma-secretase-dependent E-cadherin cleavage. J. Cell Sci. 2007, 120, 1944–1952. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.; Thiele Orberg, E.; Geis, A.L.; Chan, J.L.; Fu, K.; DeStefano Shields, C.E.; Dejea, C.M.; Fathi, P.; Chen, J.; Finard, B.B.; et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe 2018, 23, 203–214.e205. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.R. Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins 2011, 3, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Pirazzini, M.; Azarnia Tehran, D.; Zanetti, G.; Rossetto, O.; Montecucco, C. Hsp90 and Thioredoxin-Thioredoxin Reductase enable the catalytic activity of Clostridial neurotoxins inside nerve terminals. Toxicon 2018, 147, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Basak, S.; Chen, P.; Zhang, C.; Perry, K.; Tian, S.; Yu, C.; Dong, M.; Huang, L.; Bowen, M.E.; et al. Structure and conformational dynamics of Clostridioides difficile toxin A. Life Sci. Alliance 2022, 5, e202201383. [Google Scholar] [CrossRef] [PubMed]
- Chumbler, N.M.; Rutherford, S.A.; Zhang, Z.; Farrow, M.A.; Lisher, J.P.; Farquhar, E.; Giedroc, D.P.; Spiller, B.W.; Melnyk, R.A.; Lacy, D.B. Crystal structure of Clostridium difficile toxin A. Nat. Microbiol. 2016, 1, 15002. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhan, X.; Luo, J.; Li, D.; Zhou, R.; Zhang, J.; Pan, Z.; Zhang, Y.; Jia, T.; Zhang, X.; et al. Structural dynamics of the CROPs domain control stability and toxicity of Paeniclostridium sordellii lethal toxin. Nat. Commun. 2023, 14, 8426. [Google Scholar] [CrossRef] [PubMed]
- Kenworthy, A.K.; Schmieder, S.S.; Raghunathan, K.; Tiwari, A.; Wang, T.; Kelly, C.V.; Lencer, W.I. Cholera Toxin as a Probe for Membrane Biology. Toxins 2021, 13, 543. [Google Scholar] [CrossRef]
- Wernick, N.L.; Chinnapen, D.J.; Cho, J.A.; Lencer, W.I. Cholera toxin: An intracellular journey into the cytosol by way of the endoplasmic reticulum. Toxins 2010, 2, 310–325. [Google Scholar] [CrossRef]
- Ernst, K. Requirement of Peptidyl-Prolyl Cis/Trans isomerases and chaperones for cellular uptake of bacterial AB-type toxins. Front. Cell. Infect. Microbiol. 2022, 12, 938015. [Google Scholar] [CrossRef] [PubMed]
- Ernst, K.; Schnell, L.; Barth, H. Host Cell Chaperones Hsp70/Hsp90 and Peptidyl-Prolyl Cis/Trans Isomerases Are Required for the Membrane Translocation of Bacterial ADP-Ribosylating Toxins. Curr. Top. Microbiol. Immunol. 2017, 406, 163–198. [Google Scholar] [CrossRef] [PubMed]
- Lemichez, E.; Bomsel, M.; Devilliers, G.; vanderSpek, J.; Murphy, J.R.; Lukianov, E.V.; Olsnes, S.; Boquet, P. Membrane translocation of diphtheria toxin fragment A exploits early to late endosome trafficking machinery. Mol. Microbiol. 1997, 23, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.J. Diphtheria toxin: Mode of action and structure. Bacteriol. Rev. 1975, 39, 54–85. [Google Scholar] [CrossRef] [PubMed]
- Chenal, A.; Prongidi-Fix, L.; Perier, A.; Aisenbrey, C.; Vernier, G.; Lambotte, S.; Haertlein, M.; Dauvergne, M.T.; Fragneto, G.; Bechinger, B.; et al. Deciphering membrane insertion of the diphtheria toxin T domain by specular neutron reflectometry and solid-state NMR spectroscopy. J. Mol. Biol. 2009, 391, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Gillet, D.; Barbier, J. Diphtheria toxin. In The Comprehensive Sourcebook of Bacterial Protein Toxins; Alouf, J., Ladant, D.R.P.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 111–132. [Google Scholar]
- Jørgensen, R.; Wang, Y.; Visschedyk, D.; Merrill, A.R. The nature and character of the transition state for the ADP-ribosyltransferase reaction. EMBO Rep. 2008, 9, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Michalska, M.; Wolf, P. Pseudomonas Exotoxin A: Optimized by evolution for effective killing. Front. Microbiol. 2015, 6, 963. [Google Scholar] [CrossRef] [PubMed]
- Rolsma, S.; Frank, D.W.; Barbieri, J.T. Pseudomonas aeruginosa toxins. In The Comprehensive Sourcebook of Bacterial Protein Toxins; Alouf, J., Ladant, D., Popoff, M.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 133–160. [Google Scholar]
- Johannes, L. Shiga Toxin-A Model for Glycolipid-Dependent and Lectin-Driven Endocytosis. Toxins 2017, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tian, S.; Thaker, H.; Dong, M. Shiga Toxins: An Update on Host Factors and Biomedical Applications. Toxins 2021, 13, 222. [Google Scholar] [CrossRef]
- Collier, R.J. Membrane translocation by anthrax toxin. Mol. Aspects Med. 2009, 30, 413–422. [Google Scholar] [CrossRef]
- Liu, S.; Moayeri, M.; Leppla, S.H. Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol. 2014, 22, 317–325. [Google Scholar] [CrossRef] [PubMed]
- van der Goot, G.; Young, J.A. Receptors of anthrax toxin and cell entry. Mol. Aspects Med. 2009, 30, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, J.; Shanahan, F. The normal intestinal microbiota. Curr. Opin. Infect. Dis. 2007, 20, 508–513. [Google Scholar] [CrossRef]
- Young, J.A.; Collier, R.J. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 2007, 76, 243–265. [Google Scholar] [CrossRef] [PubMed]
- Frisan, T. Bacterial genotoxins: The long journey to the nucleus of mammalian cells. Biochim. Biophys. Acta 2016, 1858, 567–575. [Google Scholar] [CrossRef]
- Martin, O.C.B.; Frisan, T. Bacterial Genotoxin-Induced DNA Damage and Modulation of the Host Immune Microenvironment. Toxins 2020, 12, 63. [Google Scholar] [CrossRef]
- Taieb, F.; Petit, C.; Nougayrède, J.P.; Oswald, E. The Enterobacterial Genotoxins: Cytolethal Distending Toxin and Colibactin. EcoSal Plus 2016, 7. [Google Scholar] [CrossRef]
- Du, L.; Song, J. Delivery, structure, and function of bacterial genotoxins. Virulence 2022, 13, 1199–1215. [Google Scholar] [CrossRef]
- Caso, G.C.; McClain, M.S.; Erwin, A.L.; Truelock, M.D.; Campbell, A.M.; Leasure, C.S.; Nagel, M.; Schey, K.L.; Lacy, D.B.; Ohi, M.D.; et al. Functional Properties of Oligomeric and Monomeric Forms of Helicobacter pylori VacA Toxin. Infect. Immun. 2021, 89, e0034821. [Google Scholar] [CrossRef]
- Foegeding, N.J.; Caston, R.R.; McClain, M.S.; Ohi, M.D.; Cover, T.L. An Overview of Helicobacter pylori VacA Toxin Biology. Toxins 2016, 8, 173. [Google Scholar] [CrossRef]
- Ricci, V.; Sommi, P.; Boquet, P. Helicobacter pylori vacuolating toxin. In The Comprehensive Sourcebook of Bacterial Protein Toxins; Alouf, J., Ladant, D.R.P.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 515–557. [Google Scholar]
- Sanchez, J.; Holmgren, J. Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cell. Mol. Life Sci. 2008, 65, 1347–1360. [Google Scholar] [CrossRef]
- Kabbani, A.M.; Raghunathan, K.; Lencer, W.I.; Kenworthy, A.K.; Kelly, C.V. Structured clustering of the glycosphingolipid GM1 is required for membrane curvature induced by cholera toxin. Proc. Natl. Acad. Sci. USA 2020, 117, 14978–14986. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Bader, C.; Teter, K. The manipulation of cell signaling and host cell biology by cholera toxin. Cell. Signal. 2022, 100, 110489. [Google Scholar] [CrossRef] [PubMed]
- Heggelund, J.E.; Bjornestad, V.A.; Krengel, U. Vibrio cholerae and Escherichia coli heat-labile enterotoxins and beyond. In The Comprehensive Sourcebook of Bacterial Protein Toxins; Alouf, J.E., Ladant, D., Popoff, M.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 195–229. [Google Scholar]
- Locht, C.; Antoine, R. The History of Pertussis Toxin. Toxins 2021, 13, 623. [Google Scholar] [CrossRef] [PubMed]
- Locht, C.; Coutte, L.; Mielcarek, N. The ins and outs of pertussis toxin. FEBS J. 2011, 278, 4668–4682. [Google Scholar] [CrossRef] [PubMed]
- Takehara, M.; Takagishi, T.; Seike, S.; Oda, M.; Sakaguchi, Y.; Hisatsune, J.; Ochi, S.; Kobayashi, K.; Nagahama, M. Cellular Entry of Clostridium perfringens Iota-Toxin and Clostridium botulinum C2 Toxin. Toxins 2017, 9, 247. [Google Scholar] [CrossRef] [PubMed]
- Ernst, K.; Sailer, J.; Braune, M.; Barth, H. Intoxication of mammalian cells with binary clostridial enterotoxins is inhibited by the combination of pharmacological chaperone inhibitors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 941–954. [Google Scholar] [CrossRef]
- Stiles, B.G.; Pradhan, K.; Fleming, J.M.; Samy, R.P.; Barth, H.; Popoff, M.R. Clostridium and Bacillus binary enterotoxins: Bad for the bowels, and eukaryotic being. Toxins 2014, 6, 2626–2656. [Google Scholar] [CrossRef]
- Papatheodorou, P.; Aktories, K. Receptor-Binding and Uptake of Binary Actin-ADP-Ribosylating Toxins. Curr. Top. Microbiol. Immunol. 2017, 406, 119–133. [Google Scholar] [CrossRef]
- Aktories, K.; Schmidt, G.; Lang, A.E. Photorhabdus luminescens Toxins TccC3 and TccC5: Insecticidal ADP-Ribosyltransferases that Modify Threonine and Glutamine. Curr. Top. Microbiol. Immunol. 2015, 384, 53–67. [Google Scholar]
- Aktories, K.; Weller, U.; Chhatwal, G.S. Clostridium botulinum type C produces a novel ADP-ribosyltransferase distinct from botulinum C2 toxin. FEBS Lett. 1987, 212, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Chardin, P.; Boquet, P.; Madaule, P.; Popoff, M.R.; Rubin, E.J.; Gill, D.M. The mammalian G protein rhoC is ADP-ribosylated by Clostridium botulinum exoenzyme C3 and affects actin microfilaments in Vero Cells. EMBO J. 1989, 8, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Rubin, E.J.; Gill, D.M.; Boquet, P.; Popoff, M.R. Functional modification of a 21-kilodalton G protein when ADP-ribosylated by exoenzyme C3 of Clostridium botulinum. Mol. Cell. Biol. 1988, 8, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Fischer, S.; Möglich, A.; Förtsch, C. Clostridial C3 Toxins Target Monocytes/Macrophages and Modulate Their Functions. Front. Immunol. 2015, 6, 339. [Google Scholar] [CrossRef] [PubMed]
- Fellermann, M.; Huchler, C.; Fechter, L.; Kolb, T.; Wondany, F.; Mayer, D.; Michaelis, J.; Stenger, S.; Mellert, K.; Möller, P.; et al. Clostridial C3 Toxins Enter and Intoxicate Human Dendritic Cells. Toxins 2020, 12, 563. [Google Scholar] [CrossRef] [PubMed]
- Rohrbeck, A.; Höltje, M.; Adolf, A.; Oms, E.; Hagemann, S.; Ahnert-Hilger, G.; Just, I. The Rho ADP-ribosylating C3 exoenzyme binds cells via an Arg-Gly-Asp motif. J. Biol. Chem. 2017, 292, 17668–17680. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Gerhard, R.; Maeda, A.; Amano, M.; Kaibuchi, K.; Aktories, K.; Just, I. Entrapment of Rho ADP-ribosylated by Clostridium botulinum C3 exoenzyme in the Rho-guanine nucleotide dissociation inhibitor-1 complex. J. Biol. Chem. 2003, 278, 28523–28527. [Google Scholar] [CrossRef]
- Wilde, C.; Aktories, K. The Rho-ADP-ribosylating C3 exoenzyme from Clostridium botulinum and related C3-like transferases. Toxicon 2001, 39, 1647–1660. [Google Scholar] [CrossRef] [PubMed]
- Orrell, K.E.; Melnyk, R.A. Large Clostridial Toxins: Mechanisms and Roles in Disease. Microbiol. Mol. Biol. Rev. 2021, 85, e0006421. [Google Scholar] [CrossRef]
- Popoff, M.R.; Bouvet, P. Clostridial toxins. Future Microbiol. 2009, 4, 1021–1064. [Google Scholar] [CrossRef]
- Uzal, F.A.; Navarro, M.A.; Li, J.; Freedman, J.C.; Shrestha, A.; McClane, B.A. Comparative pathogenesis of enteric clostridial infections in humans and animals. Anaerobe 2018, 53, 11–20. [Google Scholar] [CrossRef] [PubMed]
- LaFrance, M.E.; Farrow, M.A.; Chandrasekaran, R.; Sheng, J.; Rubin, D.H.; Lacy, D.B. Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 7073–7078. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Zhang, J.; Meraner, P.; Tovaglieri, A.; Wu, X.; Gerhard, R.; Zhang, X.; Stallcup, W.B.; Miao, J.; He, X.; et al. Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature 2016, 538, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Zhang, H.; Cai, C.; Zhu, S.; Zhou, Y.; Yang, X.; He, R.; Li, C.; Guo, S.; Li, S.; et al. Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res. 2015, 25, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Beilhartz, G.L.; Kucharska, I.; Raman, S.; Cui, H.; Lam, M.H.Y.; Liang, H.; Rubinstein, J.L.; Schramek, D.; Julien, J.P.; et al. Recognition of Semaphorin Proteins by P. sordellii Lethal Toxin Reveals Principles of Receptor Specificity in Clostridial Toxins. Cell 2020, 182, 345–356.e316. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Tian, S.; Zhang, J.; Liu, Z.; Robinson-McCarthy, L.; Miyashita, S.I.; Breault, D.T.; Gerhard, R.; Oottamasathien, S.; Whelan, S.P.J.; et al. Sulfated glycosaminoglycans and low-density lipoprotein receptor contribute to Clostridium difficile toxin A entry into cells. Nat. Microbiol. 2019, 4, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Liu, Y.; Wu, H.; Liu, H.; Zeng, J.; Choi, M.Y.; Chen, H.; Gerhard, R.; Dong, M. Genome-Wide CRISPR Screen Identifies Semaphorin 6A and 6B as Receptors for Paeniclostridium sordellii Toxin TcsL. Cell Host Microbe 2020, 27, 782–792.e787. [Google Scholar] [CrossRef]
- Schorch, B.; Song, S.; van Diemen, F.R.; Bock, H.H.; May, P.; Herz, J.; Brummelkamp, T.R.; Papatheodorou, P.; Aktories, K. LRP1 is a receptor for Clostridium perfringens TpeL toxin indicating a two-receptor model of clostridial glycosylating toxins. Proc. Natl. Acad. Sci. USA 2014, 111, 6431–6436. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, P.; Minton, N.P.; Aktories, K.; Barth, H. An Updated View on the Cellular Uptake and Mode-of-Action of Clostridioides difficile Toxins. Adv. Exp. Med. Biol. 2024, 1435, 219–247. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K. Bacterial protein toxins that modify host regulatory GTPases. Nat. Rev. Microbiol. 2011, 9, 487–498. [Google Scholar] [CrossRef]
- Aktories, K. From signal transduction to protein toxins-a narrative review about milestones on the research route of C. difficile toxins. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2023, 396, 173–190. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Pauillac, S.; Schelle, I.; Bouvet, P.; Bouchier, C.; Varela-Chavez, C.; Just, I.; Popoff, M.R. Haemorrhagic toxin and lethal toxin from Clostridium sordellii strain VPI9048: Molecular characterization and comparative analysis of substrate specificity of the large clostridial glucosylating toxins. Cell. Microbiol. 2014, 16, 1706–1721. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, R.N.; Lacy, D.B. Toward a structural understanding of Clostridium difficile toxins A and B. Front. Cell. Infect. Microbiol. 2012, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K.; Just, I. Clostridial Rho-inhibiting protein toxins. Curr. Top. Microbiol. Immunol. 2005, 291, 113–145. [Google Scholar] [PubMed]
- Kordus, S.L.; Thomas, A.K.; Lacy, D.B. Clostridioides difficile toxins: Mechanisms of action and antitoxin therapeutics. Nat. Rev. Microbiol. 2022, 20, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shao, F. Diverse mechanisms for inflammasome sensing of cytosolic bacteria and bacterial virulence. Curr. Opin. Microbiol. 2016, 29, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Just, I. Functional implications of lethal toxin-catalysed glucosylation of (H/K/N)Ras and Rac1 in Clostridium sordellii-associated disease. Eur. J. Cell Biol. 2011, 90, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Geny, B.; Grassart, A.; Manich, M.; Chicanne, G.; Payrastre, B.; Sauvonnet, N.; Popoff, M.R. Rac1 inactivation by lethal toxin from Clostridium sordellii modifies Focal Adhesions upstream of actin depolymerization. Cell. Microbiol. 2010, 12, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Geny, B.; Khum, H.; Fitting, C.; Zarantonelli, L.; Mazuet, C.; Cayet, N.; Szatanik, M.; Prevost, M.C.; Cavaillon, J.M.; Huerre, M.; et al. Clostridium sordellii lethal toxin kills mice by inducing a major increase in lung vascular permeability. Am. J. Pathol. 2007, 170, 1003–1017. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R.; Geny, B. Rho/Ras-GTPase-dependent and -independent activity of clostridial glucosylating toxins. J. Med. Microbiol. 2011, 60, 1057–1069. [Google Scholar] [CrossRef]
- Piteau, M.; Papatheodorou, P.; Schwan, C.; Schlosser, A.; Aktories, K.; Schmidt, G. Lu/BCAM adhesion glycoprotein is a receptor for Escherichia coli Cytotoxic Necrotizing Factor 1 (CNF1). PLoS Pathog. 2014, 10, e1003884. [Google Scholar] [CrossRef]
- Kim, K.J.; Chung, J.W.; Kim, K.S. 67-kDa laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J. Biol. Chem. 2005, 280, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Lemichez, E.; Munro, P.; Boyer, L. Deamidase toxins. In The Comprehensice Sourcebook of Bacterial Protein Toxins, 4th ed.; Alouf, J., Ladant, D., Popoff, M.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 499–514. [Google Scholar]
- Chaoprasid, P.; Dersch, P. The Cytotoxic Necrotizing Factors (CNFs)-A Family of Rho GTPase-Activating Bacterial Exotoxins. Toxins 2021, 13, 901. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; Mettouchi, A.; Wilson, B.A.; Lemichez, E. CNF1-like deamidase domains: Common Lego bricks among cancer-promoting immunomodulatory bacterial virulence factors. Pathog. Dis. 2018, 76, fty045. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Schmidt, G.; Schlosser, A.; Hey, T.D.; Larrinua, I.M.; Sheets, J.J.; Mannherz, H.G.; Aktories, K. Photorhabdus luminescens toxins ADP-ribosylate actin and RhoA to force actin clustering. Science 2010, 327, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Chapeton-Montes, D.; Plourde, L.; Bouchier, C.; Ma, L.; Diancourt, L.; Criscuolo, A.; Popoff, M.R.; Bruggemann, H. The population structure of Clostridium tetani deduced from its pan-genome. Sci. Rep. 2019, 9, 11220. [Google Scholar] [CrossRef] [PubMed]
- Peck, M.W.; Smith, T.J.; Anniballi, F.; Austin, J.W.; Bano, L.; Bradshaw, M.; Cuervo, P.; Cheng, L.W.; Derman, Y.; Dorner, B.G.; et al. Historical Perspectives and Guidelines for Botulinum Neurotoxin Subtype Nomenclature. Toxins 2017, 9, 38. [Google Scholar] [CrossRef]
- Dong, M.; Masuyer, G.; Stenmark, P. Botulinum and Tetanus Neurotoxins. Annu. Rev. Biochem. 2019, 88, 811–837. [Google Scholar] [CrossRef] [PubMed]
- Pirazzini, M.; Montecucco, C.; Rossetto, O. Toxicology and pharmacology of botulinum and tetanus neurotoxins: An update. Arch. Toxicol. 2022, 03271–03279. [Google Scholar] [CrossRef]
- Poulain, B.; Lemichez, E.; Popoff, M.R. Neuronal selectivity of botulinum neurotoxins. Toxicon 2020, 178, 20–32. [Google Scholar] [CrossRef]
- Rummel, A. Two Feet on the Membrane: Uptake of Clostridial Neurotoxins. Curr. Top. Microbiol. Immunol. 2017, 406, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.A.; Ho, M. Evolutionary aspects of toxin-producing bacteria. In The Comprehensive Sourcebook of Bacterial Protein Toxins, 4th ed.; Alouf, J., Ladant, D., Popoff, M.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 3–39. [Google Scholar]
- Gargi, A.; Reno, M.; Blanke, S.R. Bacterial toxin modulation of the eukaryotic cell cycle: Are all cytolethal distending toxins created equally? Front. Cell. Infect. Microbiol. 2012, 2, 124. [Google Scholar] [CrossRef] [PubMed]
- Verster, K.I.; Tarnopol, R.L.; Akalu, S.M.; Whiteman, N.K. Horizontal Transfer of Microbial Toxin Genes to Gall Midge Genomes. Genome Biol. Evol. 2021, 13, evab202. [Google Scholar] [CrossRef] [PubMed]
- Janezic, S.; Dingle, K.; Alvin, J.; Accetto, T.; Didelot, X.; Crook, D.W.; Lacy, D.B.; Rupnik, M. Comparative genomics of Clostridioides difficile toxinotypes identifies module-based toxin gene evolution. Microb. Genom. 2020, 6, mgen000449. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, M.J.; Tremblay, B.J.; Zeng, J.; Wei, X.; Hodgins, H.; Worley, J.; Bry, L.; Dong, M.; Doxey, A.C. Phylogenomics of 8,839 Clostridioides difficile genomes reveals recombination-driven evolution and diversification of toxin A and B. PLoS Pathog. 2020, 16, e1009181. [Google Scholar] [CrossRef]
- Kumar, R.; Feltrup, T.M.; Kukreja, R.V.; Patel, K.B.; Cai, S.; Singh, B.R. Evolutionary Features in the Structure and Function of Bacterial Toxins. Toxins 2019, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.F.; Mainguy, G.; Gibert, M.; Marvaud, J.C.; Stiles, B.G.; Popoff, M.R. Transcytosis of iota toxin across polarized CaCo-2 cell monolayers. Mol. Microbiol. 2002, 43, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Lencer, W.I.; Constable, C.; Moe, S.; Jobling, M.G.; Webb, H.M.; Ruston, S.; Madara, J.L.; Hirst, T.R.; Holmes, R.K. Targeting of Cholera Toxin and Escherichia coli Heat Labile Toxin in polarized epithelia: Role of COOH-terminal KDEL. J. Cell Biol. 1995, 131, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Chapman, C.M.L.; Kapinos, A.; Rivera-Chávez, F. Modulation of Host-Microbe Metabolism by Cholera Toxin. Infect. Immun. 2023, 91, e0043522. [Google Scholar] [CrossRef]
- Mansfield, M.J.; Doxey, A.C. Genomic insights into the evolution and ecology of botulinum neurotoxins. Pathog. Dis. 2018, 76, 4978416. [Google Scholar] [CrossRef]
- Montecucco, C.; Rasotto, M.B. On botulinum neurotoxin variability. mBio 2015, 6, e02131-14. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Botulinum toxin: State of the art. Mov. Disord. 2017, 32, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Valtierra-de-Luis, D.; Villanueva, M.; Berry, C.; Caballero, P. Potential for Bacillus thuringiensis and Other Bacterial Toxins as Biological Control Agents to Combat Dipteran Pests of Medical and Agronomic Importance. Toxins 2020, 12, 773. [Google Scholar] [CrossRef] [PubMed]







| Bacteria | Toxin/Enzyme | Enzyme Class | Substrate Specificity | Role in Pathogenicity |
|---|---|---|---|---|
| Bacillus anthracis | SMase PI-PLC | SMC, PLC | SM, PI, PC | macrophage associated growth of B. anthracis, bacterial escape from phagosome |
| Bacillus cereus | SMase | SMC | SM | evasion of macrophage |
| PLC | PLC | PC, PE, PS | hemolysis | |
| Clostridium novyi | γ-toxin | PLC | PC, PE, PI, LPC, PG, SM | myonecrosis |
| Clostridium perfringens | α-toxin | PLC, SMC | PC, PE, PI, PG, PS, SM | gas gangrene |
| Corynebacterium pseudotuberculosis | SMD | SM, LPC | platelet aggregation, intravascular coagulation, endothelial hyperpermeability, bacterial spreading | |
| Helicobacter pylori | SMC | SM | apoptosis in gastric cells | |
| PldA1 | PLA2 | PC | hemolysis, bacterial survival and growth at low pH | |
| Legionella pneumophila | PlaB | PLA2 | PC, PG | hemolysis, destruction of lung surfactant |
| VipD/PatA | PLA2 | PC, LPC | establishment of bacterial niche | |
| Listeria monocytogenes | PLC-B | PLC, SMC | PC, PE, PS, SM | bacterial escape from phagosome bacterial cell to cell spreading |
| PLC-A | PLC | PI | ||
| Mycobacterium tuberculosis | PLC-A, PLC-B, PLC-C, PLC-D | acid phosphatases | PC, SM | pulmonary infection, necrosis of alveolar macrophages |
| Pseudomonas aeruginosa | PLC-H | acid phosphatases | PC, LPC, PE, PG, SM | cytotoxic to human monocytes and endothelial cells, hemolysis, platelet aggregation and thrombosis |
| Pseudomonas aeruginosa | ExoU | PLA2 | PC, PE, PA, PI | focal adhesion disruption, cytoskeleton alteration, inflammatory response |
| Salmonella enterica serovar Typhimurium | SseJ | PLA1 and GCATase | endosomal tabulation and establishment of bacterial intracellular niche | |
| Staphylococcus aureus | β-toxin | SMC | SM, LPC | bacterial survival in neutrophils, cytotoxic to neutrophils and monocytes |
| PI-LPI | PLC | PI, LPI | bacterial survival in neutrophils | |
| Streptococcus pyogenes | SlaA | PLA2 | PC, PE, PS, LPC | bacterial adherence to host cells, cytotoxicity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popoff, M.R. Overview of Bacterial Protein Toxins from Pathogenic Bacteria: Mode of Action and Insights into Evolution. Toxins 2024, 16, 182. https://doi.org/10.3390/toxins16040182
Popoff MR. Overview of Bacterial Protein Toxins from Pathogenic Bacteria: Mode of Action and Insights into Evolution. Toxins. 2024; 16(4):182. https://doi.org/10.3390/toxins16040182
Chicago/Turabian StylePopoff, Michel R. 2024. "Overview of Bacterial Protein Toxins from Pathogenic Bacteria: Mode of Action and Insights into Evolution" Toxins 16, no. 4: 182. https://doi.org/10.3390/toxins16040182
APA StylePopoff, M. R. (2024). Overview of Bacterial Protein Toxins from Pathogenic Bacteria: Mode of Action and Insights into Evolution. Toxins, 16(4), 182. https://doi.org/10.3390/toxins16040182