Therapeutic Down-Modulators of Staphylococcal Superantigen-Induced Inflammation and Toxic Shock

Abstract
:1. Staphylococcal Exotoxins as Superantigens
1.1. Overview
1.2. Physical properties of staphylococcal superantigens
1.3. Human diseases caused by staphylococcal superantigens
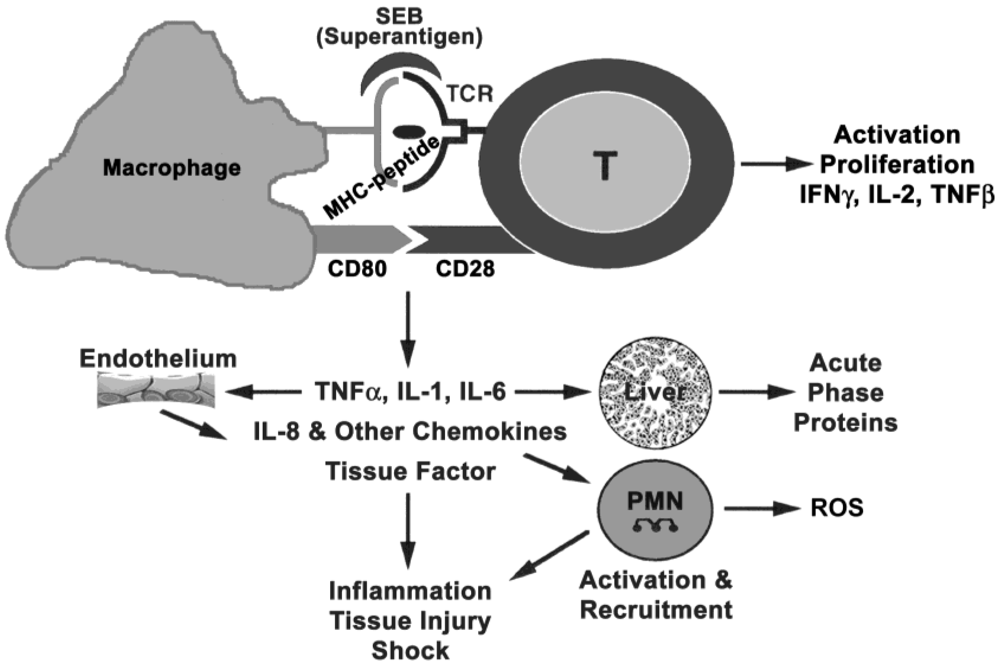
2. Superantigen Binding to Host Cells
2.1. Binding to MHC class II
2.2. Binding to TCR
2.3. Co-stimulatory molecules on host cells
3. Immune Activation
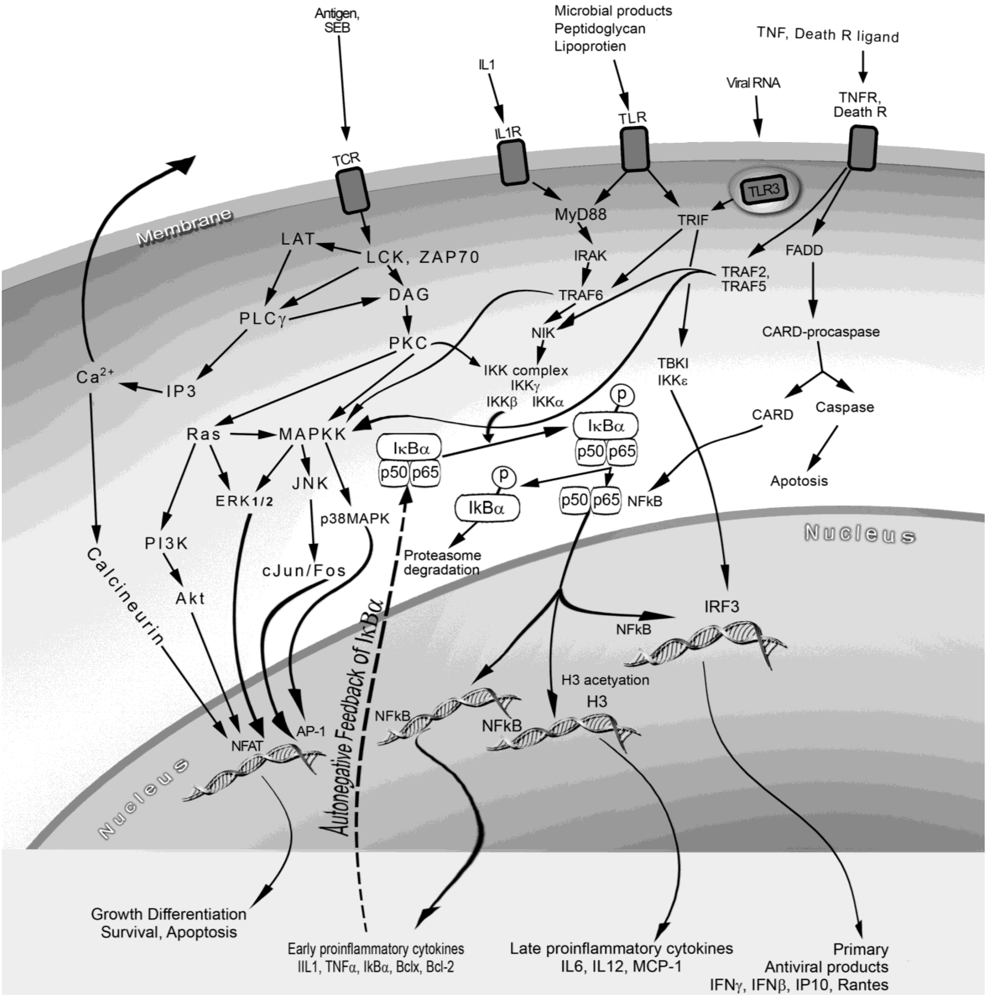
3.1. Signal transduction

3.2. In vitro cellular response
3.3. Signaling and biological effects of proinflammatory mediators

4. Animal Models
4.1. Host response, administration route and dose effects
4.2. Emetic response models
4.3. Murine models of toxic shock using potentiating agents
4.4. Transgenic mouse models
4.5. Murine models using only SEB
5. Therapeutics for Superantigen-Induced Shock
5.1. Influence of animal models on efficacy of therapeutics
5.2. Antibodies against superantigens
5.3. Inhibitors of cell receptor-toxin interaction
5.4. Inhibitors of SEB signal transduction
5.5. Inhibitors of cytokine induction

{kind=link}
{kind=link}
| Pharmacologic agent | Target | Biological effects against SEB |
|---|---|---|
| Rapamycin FDA-approved for prevention of renal graft rejection | Immunophilin FK506BP12 | Blocked SEB-induced MCP-1 and IL-6 in vitro and in vivo [125]. |
| Protected mice from lethality even when administered 24 h after SEB. | ||
| Dexamethasone FDA-approved for treating inflammatory diseases | NF-κB | Inhibited SEB-induced proinflammatory cytokines and chemokines in PBMC [72] and adhesion molecules (ICAM, ELAM, VCAM) on endothelial cells [121]. |
| Reduced serum levels of cytokines, attenuated hypothermia due to SEB, improved survival of mice [72,97]. | ||
| Pentoxifylline FDA-approved for treating peripheral arterial disease | Phosphodiesterase | Attenuated SEB-induced proinflammatory cytokines and chemokines in PBMC [17,97]. |
| Blocked cytokine release in vivo and prevented SEB-induced lethal shock in SEB + LPS murine models [97]. | ||
| Pirfenidone | Inhibition of TGFβ (exact mechanism unknown) | Inhibited SEB-stimulated cytokines in vitro and in vivo [96]. |
| Improved survival of mice [96]. | ||
| Niacinamide | Nitric oxide synthase | Inhibited serum IL-2 and IFNγ [94]. |
| Prevented death of mice from SEB-mediated shock [94]. | ||
| D609 | Phospholipase C | Blocked SEB-stimulated cytokines, chemokines and proliferation in human PBMC [48]. |
| Improved survival of mice [103]. |
5.6. Inhibitors of cytokine signaling
6. Summary
Acknowledgements
Declaim
References
- Chesney, P.J.; Davis, P.J.; Purdy, W.K.; Wand, P.J.; Chesney, R.W. Clinical manifestations of toxic shock syndrome. JAMA 1981, 246, 741–748. [Google Scholar]
- Marrack, P.; Kappler, J. The staphylococcal enterotoxins and their relatives. Science 1990, 248, 705–709. [Google Scholar]
- Kotzin, B.L.; Leung, D.Y.M.; Kappler, J.; Marrack, P. Superantigens and their potential role in human disease. Adv. Immunol. 1993, 54, 99–166. [Google Scholar]
- Kotb, M. Bacterial pyrogenic exotoxins as superantigens. Clin. Microbiol. Rev. 1995, 8, 411–426. [Google Scholar]
- Fraser, J.; Arcus, V.; Kong, P.; Baker, E.; Proft, T. Superantigens—powerful modifiers of the immune system. Mol. Med. Today 2000, 6, 125–132. [Google Scholar]
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: an update. Ann. Rev. Microbiol. 2001, 55, 77–104. [Google Scholar]
- Proft, T.; Fraser, J.D. Bacterial superantigens. Clin. Exp. Immunol. 2003, 133, 299–306. [Google Scholar]
- Fraser, J.D.; Proft, T. The bacterial superantigen and superantigen-like proteins. Immunol. Rev. 2008, 225, 226–243. [Google Scholar]
- Choi, Y.; Kotzin, B.; Hernon, L.; Callahan, J.; Marrack, P.; Kappler, J. Interaction of Staphylococcus aureus toxin "superantigens" with human T cells. Proc. Natl. Acad. Sci. USA 1989, 86, 8941–8945. [Google Scholar]
- Jupin, C.; Anderson, S.; Damais, C.; Alouf, J.E.; Parant, M. Toxic shock syndrome toxin 1 as an inducer of human tumor necrosis factors and gamma interferon. J. Exp. Med. 1988, 167, 752–761. [Google Scholar]
- Parsonnet, J. Mediators in the pathogenesis of toxic shock syndrome: overview. Rev. Infect. Dis. 1989, 11, S263–S269. [Google Scholar]
- Fischer, H.; Dohlsten, M.; Andersson, U.; Hedlund, P.; Ericsson, P.; Hansson, J.; Sjogren, H.O. Production of TNF-α and TNF-β by staphylococcal enterotoxin A activated human T cells. J. Immunol. 1990, 144, 4663–4668. [Google Scholar]
- Trede, N.S.; Geha, R.S.; Chatila, T. Transcriptional activation of IL-1 beta and tumor necrosis factor-alpha genes by MHC class II ligands. J. Immunol. 1991, 146, 2310–2315. [Google Scholar]
- See, R.H.; Kum, W.W.; Chang, A.H.; Goh, S.H.; Chow, A.W. Induction of tumor necrosis factor and interleukin-1 by purified staphylococcal toxic shock syndrome toxin 1 requires the presence of both monocytes and T lymphocytes. Infect. Immun. 1992, 60, 2612–2618. [Google Scholar] [PubMed]
- Krakauer, T. Inhibition of toxic shock syndrome toxin-induced cytokine production and T cell activation by interleukin 10, interleukin 4, and dexamethasone. J. Infect. Dis. 1995, 172, 988–992. [Google Scholar]
- Tessier, P.A.; Naccache, P.H.; Diener, K.R.; Gladue, R.P.; Neotem, K.S.; Clark-Lewis, I.; McColl, S.R. Induction of acute inflammation in vivo by staphylococcal superantigens. II. Critical role for chemokines, ICAM-1, and TNF-alpha. J. Immunol. 1998, 161, 1204–1211. [Google Scholar] [PubMed]
- Krakauer, T. The induction of CC Chemokines in human peripheral blood mononuclear cells by staphylococcal exotoxins and its prevention by pentoxifylline. J. Leukco. Biol. 1999, 66, 158–164. [Google Scholar]
- Krakauer, T. Immune response to staphylococcal superantigens. Immunol. Res. 1999, 20, 163–173. [Google Scholar]
- Mattsson, E.; Herwald, H.; Egsten, A. Superantigen from Staphylococcus aureus induce procoagulant activity and monocyte tissue factor expression in whole blood and mononuclear cells via IL-1β. J. Thromb. Haemost. 2003, 1, 2569–2575. [Google Scholar]
- Krakauer, T.; Vilcek, J.; Oppenheim, J.J. Proinflammatory cytokines: TNF and IL-1 families, chemokines, TGFß and others. In Fundamental Immunology, 4th; Paul, W., Ed.; Lippincott-Raven: Philadelphia, PA, USA, 1998; pp. 775–811. [Google Scholar]
- Vial, T.; Descotes, J. Immune-mediated side-effects of cytokines in human. Toxicology 1995, 105, 31–57. [Google Scholar]
- Papageorgiou, A.C.; Acharya, K.R. Microbial superantigens: from structure to function. Trends Microbiol. 2000, 8, 369–375. [Google Scholar]
- Monday, S.R.; Bohach, G.A. Properties of Staphylococcus aureus enterotoxins and toxic shock syndrome toxin-1. In The Comprehensive Sourcebook of Bacterial Protein Toxins; Alouf, J.E., Freer, J.H., Eds.; Academic: London, UK, 1999; pp. 589–610. [Google Scholar]
- Kappler, J.W.; Herman, A.; Clements, J.; Marrack, P. Mutations defining functional regions of the superantigen staphylococcal enterotoxin B. J. Exp. Med. 1992, 175, 387–396. [Google Scholar]
- Holmberg, S.D.; Blake, P.A. Staphylococcal food poisoning in the United States. New facts and old misconceptions. JAMA 1984, 251, 487–489. [Google Scholar] [PubMed]
- Abdelnour, A.T.; Bremell, T.; Tarkowski, A. TSST-1 contributes to the arthritogenecity of Staphylococcus aureus. J. Infect. Dis. 1994, 170, 94–99. [Google Scholar]
- Schwab, J.H.; Brown, R.R.; Anderle, S.K.; Schlievert, P.M. Superantigen can reactivate bacterial cell wall-induced arthritis. J. Immunol. 1993, 150, 4151–4159. [Google Scholar]
- Brocke, S.; Hausmann, S.; Steinmam, L.; Wucherpfennig, K.W. Microbial peptides and superantigens in the pathogenesis of autoimmune diseases of the central nervous system. Semin. Immunol. 1998, 10, 57–67. [Google Scholar]
- Yarwood, J.M.; Leung, D.Y.; Schlievert, P.M. Evidence for the involvement of bacterial superantigens in psoriasis, atopic dermatitis, and Kawasaki syndrome. FEMS Microbiol. Lett. 2000, 192, 1–7. [Google Scholar]
- Origuchi, T.; Eguchi, K.; Kawabe, Y.; Yamashita, I.; Mizokami, A.; Ida, H.; Nagataki, S. Increased levels of serum IgM antibody to staphylococcal enterotoxin B in patients with rheumatoid arthritis. Ann. Rheum. Dis. 1995, 54, 713–732. [Google Scholar]
- Meissner, H.C.; Leung, D.Y.M. Superantigens, conventional antigens and the etiology of Kawasaki syndrome. Pediatr. Infect. Dis. J. 2000, 19, 91–94. [Google Scholar]
- Meissner, H.C.; Leung, D.Y.M. Kawasaki Syndrome: Where Are the Answers? Pediatrics 2003, 112, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Mollick, J.A.; Chintagumpala, M.; Cook, R.G.; Rich, R.R. Staphylococcal exotoxin activation of T cells. Role of exotoxin-MHC class II binding affinity and class II isotype. J. Immunol. 1991, 146, 463–468. [Google Scholar] [PubMed]
- Herrmann, T.; Acolla, R.S.; MacDonald, H.R. Different staphylococcal enterotoxins bind preferentially to distinct MHC class II isotypes. Eur. J. Immunol. 1989, 19, 2171–2174. [Google Scholar]
- Herman, A.; Croteau, G.; Sekaly, R.P.; Kappler, J.; Marrack, P. HLA-DR alleles differ in their ability to present staphylococcal enterotoxins to T cells. J. Exp. Med. 1990, 172, 709–712. [Google Scholar]
- Chintagumpala, M.M.; Mollick, J.A.; Rich, R.R. Staphylococcal toxins bind to different sites on HLA-DR. J. Immunol. 1991, 147, 3876–3882. [Google Scholar]
- Scholl, P.; Sekaly, R.; Diez, A.; Glimcher, L.; Geha, R. Binding of toxic shock syndrome toxin-1 to murine major histocompatibility complex class II molecules. Eur. J. Immunol. 1990, 20, 1911–1916. [Google Scholar]
- Thibodeau, J.; Cloutier, I.; Lavoie, P.M.; Labrecque, N.; Mourad, W.; Jardetzky, T.; Sekaly, R.P. Subsets of HLA-DR1 molecules defined by SEB and TSST-1 binding. Science 1994, 266, 1874–1878. [Google Scholar]
- Ulrich, R.G.; Bavari, B.; Olson, M.A. Staphylococcal enterotoxins A and B share a common structural motif for binding class II major histocompatibility complex molecules. Nat. Struct. Biol. 1995, 2, 554–560. [Google Scholar]
- Hudson, K.R.; Tiedemann, R.E.; Urban, R.G.; Lowe, S.C.; Strominger, J.L.; Fraser, J.D. Staphylococcal enterotoxin A has two cooperative binding sites on major histocompatibility complex class II. J. Exp. Med. 1995, 182, 711–720. [Google Scholar]
- Tiedemann, R.E; Urban, R.J.; Strominger, J.L.; Fraser, J.D. Isolation of HLA-DR1.(staphylococcal enterotoxins A)2 trimers in solution. Proc. Natl. Acad. Sci. USA 1995, 92, 12156–12159. [Google Scholar] [CrossRef]
- Pless, D.D.; Ruthel, G.; Reinke, E.K.; Ulrich, R.G.; Bavari, S. Persistence of zinc-binding bacterial superantigens at the surface of antigen-presenting cells contributes to the extreme potency of these superantigens as T-cell activators. Infect. Immun. 2005, 73, 5358–5366. [Google Scholar]
- Linsley, P.S.; Ledbetter, J.A. The role of the CD28 receptor during T cell responses to antigen. Ann. Rev. Immunol. 1993, 11, 191–212. [Google Scholar]
- Fraser, J.; Newton, M.; Weiss, A. CD28 and T-cell antigen receptor signal transduction coordinately regulates interleukin 2 gene expression in response to superantigen stimulation. J. Exp. Med. 1992, 175, 1131–1134. [Google Scholar]
- Krakauer, T. Costimulatory receptors for the superantigen staphyloccoccal enterotoxin B on human vascular endothelial cells and T cells. J. Leukco. Biol. 1994, 56, 458–463. [Google Scholar]
- Mehindate, K.; al-Daccak, R.; Damdoumi, F.; Mourad, W. Synergistic effect between CD40 and class II signals overcomes the requirement for class II dimerization in superantigen-induced cytokine gene expression. Eur.J. Immunol. 1996, 26, 2075–2080. [Google Scholar]
- Saha, B.; Jaklic, B.; Harlan, D.M.; Gray, G.S.; June, C.H.; Abe, R. Toxic shock syndrome toxin-1 induced death is prevented by CTLA4Ig. J. Immunol. 1996, 157, 3869–3875. [Google Scholar]
- Carlsson, R.; Fischer, H.; Sjogren, H.O. Binding of staphylococcal enterotoxin A to accessory cells is a requirement for its ability to activate human T cells. J. Immunol. 1998, 140, 2484–2488. [Google Scholar]
- Tiedemann, R.E.; Fraser, J.D. Cross-linking of MHC class II molecules by staphylococcal enterotoxin A is essential for antigen-presenting cell and T cell activation. J. Immunol. 1996, 157, 3958–3966. [Google Scholar]
- Anderson, M.R.; Tary-Lehmann, M. Staphylococcal enterotoxin-B-induced lethal shock in mice is T-cell-dependent, but disease susceptibility is defined by the non-T-cell compartment. Clin. Immunol. 2001, 98, 85–94. [Google Scholar]
- Chatila, T.; Geha, R.S. Signal transduction by microbial superantigens via MHC class II molecules. Immunol. Rev. 1993, 131, 43–59. [Google Scholar]
- Chatila, T.; Wood, N.; Parsonnet, J.; Geha, R.S. Toxic shock syndrome toxin-1 induces inositol phospholipid turnover, protein kinase C translocation, and calcium mobilization in human T cells. J. Immunol. 1988, 140, 1250–1255. [Google Scholar]
- Scholl, P.R.; Trede, N.; Chatila, T.A.; Geha, R.S. Role of protein tyrosine phosphorylation in monokine induction by the staphylococcal superantigen toxic shock syndrome toxin-1. J. Immunol. 1992, 148, 2237–2241. [Google Scholar]
- Krakauer, T. Small nonpeptide inhibitors of staphylococcal superantigen-induced cytokine production and toxic shock. In Superantigens: Molecular Basis for Their Role in Human Diseases; Kotb, M., Fraser, J.D., Eds.; ASM: Washington, DC, USA, 2007; pp. 229–224. [Google Scholar]
- Hewitt, C.; Lamb, J.; Hayball, J.; Hill, M.; Owen, M.; O’Hehir, R. MHC Independent clonal T cell anergy by direct interaction of staphylococcal enterotoxin B with the T-cell antigen receptor. J. Exp. Med. 1992, 175, 1493–1499. [Google Scholar]
- Scheuber, P.H.; Denzlinger, C.; Wilker, D.; Beck, G.; Kepper, D.; Hammer, D.K. Staphylococcal enterotoxin B as a nonimmunological mast cell stimulus in primates: the role of endogenous cysteinyl leukotrienes. Int. Arch. Allergy Appl. Immunol. 1987, 82, 289–291. [Google Scholar]
- Mourad, W.; Mehindate, K.; Schall, T.; McColl, S. Engagement of MHC class II molecules by superantigen induces inflammatory cytokine gene expression in human rheumatoid fibroblast-like synoviocytes. J. Exp. Med. 1992, 175, 613–616. [Google Scholar]
- Hamad, A.R.; Marrack, P.; Kappler, J.W. Transcytosis of staphylococcal superantigen toxins. J. Exp. Med. 1997, 185, 1447–1454. [Google Scholar]
- Peterson, M.L.; Ault, K.; Kremer, M.J.; Klingelhutz, A.J.; Davis, C.C.; Squier, C.A.; Schlievert, P.M. The innate immune system is activated by stimulation of vaginal epithelial cells with Staphylococcus aureus and toxic shock syndrome toxin 1. Infect. Immun. 2005, 73, 2164–2174. [Google Scholar]
- McKay, D.M. Bacterial superantigens: provocateurs of gut dysfunction and inflammation? Trends Immunol. 2001, 22, 497–501. [Google Scholar]
- Krakauer, T. Stimulant-dependent modulation of cytokines and chemokines by airway epithelial cells: cross- talk between pulmonary epithelial and peripheral blood mononuclear cells. Clin. Diagn. Lab. Immunol. 2002, 9, 126–131. [Google Scholar]
- Shupp, J.W.; Jett, M.; Pontzer, C.H. Identification of a transcytosis epitope on staphylococcal enterotoxins. Infect. Immun. 2002, 1029, 313–318. [Google Scholar]
- Pinchuk, I.V.; Beswick, E.J.; Saada, J.I.; Suarez, G.; Winston, J.; Mifflin, R.C.; Di Mari, J.F.; Powell, D.W.; Reyes, V.E. Monocyte chemoattractant protein-1 production by intestinal myofibroblasts in response to staphylococcal enterotoxin A: relevance to staphylococcal enterotoxigenic disease. J. Immunol. 2007, 178, 8097–8106. [Google Scholar]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar]
- Krakauer, T. Molecular therapeutic targets in inflammation: cyclooxygenase and NF-κB. Curr. Drug Targets Inflamm. Allergy 2004, 3, 317–324. [Google Scholar]
- Keystone, E.C.; Ware, C.F. Tumor necrosis factor and anti-tumor necrosis factor therapies. J. Rheumatol. 2010, 85, 27–39. [Google Scholar]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar]
- Wang, X.; Lupardus, P.; LaPorte, S.L.; Garcia, K.C. Structural biology of shared cytokine receptors. Annu. Rev. Immunol. 2009, 27, 27–60. [Google Scholar]
- Luster, A.D.; Alon, R.; von Andrian, U.H. Immune cell migration in inflammation: present and future therapeutic targets. Nat. Immunol. 2005, 6, 1182–1190. [Google Scholar]
- Neumann, B.; Engelhardt, B.; Wagner, H.; Holzmann, B. Induction of acute inflammatory lung injury by staphylococcal enterotoxin B. J. Immunol. 1997, 158, 1862–1871. [Google Scholar]
- Krakauer, T.; Buckley, M.; Huzella, L.M.; Alves, D. Critical timing, location and duration of glucocorticoid administration rescues mice from superantigen-induced shock and attenuates lung injury. Int. Immunopharmacol. 2009, 9, 1168–1174. [Google Scholar]
- Ettinger, R.; Panka, D.J.; Wang, J.K.; Stanger, B.Z.; Ju, S.T.; Marshak-Rothstein, A. Fas ligand-mediated cytotoxicity is directly responsible for apoptosis of normal CD4+ T cells responding to a bacterial superantigen. J. Immunol. 1995, 154, 4302–4308. [Google Scholar]
- Proft, T.; Sriskandan, S.; Yang, L.; Fraser, J.D. Superantigens and streptococcal toxic shock syndrome. Emerg. Infect. Dis. 2003, 9, 1211–1218. [Google Scholar]
- Sriskandan, S.; Unnikrishnan, M.; Krausz, T.; Dewchand, H.; Van Noorden, S.; Cohen, J.; Altmann, D.M. Enhanced susceptibility to superantigen-associated streptococcal sepsis in human leukocyte antigen-DQ transgenic mice. J. Infect. Dis. 2001, 184, 166–173. [Google Scholar]
- Krakauer, T.; Pitt, L.; Hunt, R.E. Detection of IL-6 and IL-2 in serum of rhesus monkeys exposed to a nonlethal dose of staphylococcal enterotoxin B. Mil. Med. 1997, 162, 612–615. [Google Scholar]
- Mattix, M.E.; Hunt, R.E.; Wilhelmsen, C.L.; Johnson, A.J.; Baze, W.B. Aerosolized staphylococcal enterotoxin B-induced pulmonary lesions in rhesus monkeys (Macaca mulatta). Toxicol. Pathol. 1995, 23, 262–268. [Google Scholar]
- Goettelfinger, P.; Lecerf, F.; Berrih-Aknin, S.; German-Fattal, M. Cytokine deviation induced by intrathymic injection of staphylococcal enterotoxin B (SEB). Eur. Cytokine Netw. 2001, 12, 487–500. [Google Scholar]
- Huzella, L.M.; Buckley, M.J.; Alves, D.A.; Stiles, B.G.; Krakauer, T. Central roles for IL-2 and MCP-1 following intranasal exposure to SEB: A new mouse model. Vet. Res. Sci. 2009, 86, 241–247. [Google Scholar]
- Hodoval, L.F.; Morris, E.L.; Crawley, G.J.; Beisel, W.R. Pathogenesis of lethal shock after intravenous staphylococcal enterotoxin B in monkeys. Appl. Microbiol. 1968, 16, 187–192. [Google Scholar]
- Spiekermann, G.M.; Nagler-Anderson, C. Oral adminstration of the bacterial superantigen staphylococcal enterotoxin B induces activation and cytokine production by T cells in murine gut-associated lymphoid tissue. J. Immunol. 1998, 161, 5825–5831. [Google Scholar]
- Alber, G.; Scheuber, P.H.; Reck, B.; Sailer-Kramer, B.; Hartmann, A.; Krammer, D.K. Role of substance P in immediate-type skin reactions induced by staphylococcal enterotoxin B in unsensitized monkeys. J. Allergy Clin. Immunol. 1998, 84, 880–885. [Google Scholar]
- Hu, D.L.; Zhu, G.; Mori, F.; Omoe, K.; Okada, M.; Wakabayashi, K.; Kaneko, S.; Shinagawa, K.; Nakane, A. Staphylococcal enterotoxin induces emesis through increasing serotonin release in intestine and it is downregulated by cannabinoid receptor 1. Cell Microbiol. 2007, 9, 2267–2277. [Google Scholar]
- Stiles, B.G.; Bavari, S.; Krakauer, T.; Ulrich, R.G. Toxicity of staphylococcal enterotoxins potentiated by lipopolysaccharide: major histocompatibility complex class II molecule dependency and cytokine release. Infect. Immun. 1993, 61, 5333–5338. [Google Scholar]
- Sugiyama, H.; McKissic, E.M.; Bergdoll, M.S.; Heller, B. Enhancement of bacterial endotoxin lethality by staphylococcal enterotoxin. J. Infect. Dis. 1964, 4, 111–118. [Google Scholar]
- Miethke, T.; Wahl, C.; Heeg, K.; Echtenacher, B.; Krammer, P.H.; Wagner, H. T cell-mediated lethal shock triggered in mice by the superantigen staphylococcal enterotoxin B: critical role of tumor necrosis factor. J. Exp. Med. 1992, 175, 91–98. [Google Scholar]
- Miethke, T.; Wahl, C.; Heeg, K.; Echtenacher, B.; Krammer, P.H.; Wagner, H. Superantigen mediated shock: a cytokine release syndrome. Immunobiology 1993, 189, 270–284. [Google Scholar]
- Sarawar, S.R.; Blackman, B.A.; Doherty, P.C. Superantigen shock in mice with an inapparent viral infection. J. Infect. Dis. 1994, 170, 1189–1194. [Google Scholar]
- Chen, J.Y.; Qiao, Y.; Komisar, J.L.; Baze, W.B.; Hsu, I.C.; Tseng, J. Increased susceptibility to staphylococcal enterotoxin B intoxication in mice primed with actinomycin D. Infect. Immun. 1994, 62, 4626–4631. [Google Scholar]
- Zhang, W.J.; Sarawar, S.; Nguyen, P.; Daly, K.; Rehig, J.E.; Doherty, P.C.; Woodland, D.L.; Blackman, M.A. Lethal synergism between influenza infection and staphylococcal enterotoxin B in mice. J. Immunol. 1996, 157, 5049–5060. [Google Scholar] [PubMed]
- Blank, C.; Luz, A.; Bendigs, S.; Erdmann, A.; Wagner, H.; Heeg, K. Superantigen and endotoxin synergize in the induction of lethal shock. Eur. J. Immunol. 1997, 27, 825–833. [Google Scholar]
- Khan, A.A.; Priya, S.; Saha, B. IL-2 regulates SEB induced toxic shock syndrome in BALB/c mice. PLoS One 2009, 4, e8473. [Google Scholar]
- Matthys, P.; Mitera, T.; Heremans, H.; Van Damme, J.; Billiau, A. Anti-gamma interferon and anti-interleukin-6 antibodies affect staphylococcal enterotoxin B-induced weight loss, hypoglycemia, and cytokine release in D-galactosamine-sensitized and unsensitized mice. Infect. Immun. 1995, 63, 1158–1164. [Google Scholar]
- LeClaire, R.D.; Kell, W.; Bavari, S.; Smith, T.; Hunt, R.E. Protective effects of niacinamide in staphylococcal enterotoxin B induced toxicity. Toxicology 1996, 107, 69–81. [Google Scholar]
- Krakauer, T.; Stiles, B.G. Pentoxifylline inhibits staphylococcal superantigen induced toxic shock and cytokine release. Clin. Diagn. Lab. Immunol. 1999, 6, 594–598. [Google Scholar]
- Hale, M.L.; Margolin, S.B.; Krakauer, T.; Roy, C.J.; Stiles, B.G. Pirfenidone blocks in vitro and in vivo effects of staphylococcal enterotoxin B. Infect. Immun. 2002, 70, 2989–2994. [Google Scholar] [CrossRef] [PubMed]
- Krakauer, T.; Buckley, M. Dexamethasone attenuates staphylococcal enterotoxin B-induced hypothermic response and protects mice from superantigen-induced toxic shock. Antimicrob. Agents Chemother. 2006, 50, 391–395. [Google Scholar]
- Krakauer, T.; Buckley, M.; Fisher, D. Proinflammatory mediators of toxic shock and their Correlation to lethality. Mediators Inflamm. 2010. [Google Scholar]
- Bean, A.G.; Freiberg, R.A.; Andrade, S.; Menon, S.; Zlotnik, A. Interleukin 10 protects mice against staphylococcal enterotoxin B-induced lethal shock. Infect. Immun. 1993, 61, 4937–4939. [Google Scholar]
- Florquin, S.; Amraoui, Z.; Abramowicz, D.; Goldman, M. Systemic release and protective role of IL-10 in staphylococcal enterotoxin B-induced shock in mice. J. Immunol. 1994, 153, 2618–2623. [Google Scholar]
- Sundstedt, A.; Hoiden, L.; Rosendahl, A.; Kalland, T.; van Rooijen, N.; Dohlsten, M. Immunoregulatory role of IL-10 during superantigen-induced-hyporesponsiveness in vivo. J. Immunol. 1997, 158, 180–186. [Google Scholar]
- Miller, C.; Ragheb, J.; Schwartz, R. Anergy and cytokine-mediated suppression as distinct superantigen-induced tolerance mechanisms in vivo. J. Exp. Med. 1999, 190, 53–64. [Google Scholar]
- McCormack, J.E.; Callahan, J.E.; Kappler, J.; Marrack, P. Profound deletion of mature T cells in vivo by chronic exposure to endogenous superantigen. J. Immunol. 1993, 150, 3785–3792. [Google Scholar]
- DaSilva, L.; Welcher, B.; Ulrich, R.; Aman, J.; David, C.S.; Bavari, S. Humanlike immune response of human leukocyte antigen-DR3 transgenic mice to staphylocococal enterotoxins: a novel model for superantigen vaccines. J. Infect. Dis. 2002, 185, 1754–1760. [Google Scholar]
- Rajagopalan, G.; Sen, M.M.; David, C.S. In vitro and in vivo evaluation of staphylococcal superantigen peptide antagonists. Infect. Immun. 2004, 72, 6733–6737. [Google Scholar]
- Roy, C.J.; Warfield, K.L.; Welcher, B.C.; Gonzales, R.F.; Larsen, T.; Hanson, J.; David, C.S.; Krakauer, T.; Bavari, S. Human leukocyte antigen-DQ8 transgenic mice: a model to examine the toxicity of aerosolized staphylococcal enterotoxin B. Infect. Immun. 2005, 73, 2452–2460. [Google Scholar]
- Yeung, R.S.; Penninger, J.M.; Kundig, J.; Khoo, W.; Ohashi, P.S.; Kroemer, G.; Mak, T.W. Human CD4 and human major histocompatibility complex class II (DQ6) transgenic mice: supersensitivity to superantigen-induced septic shock. Eur.J. Immun. 1996, 26, 1074–1082. [Google Scholar]
- Savransky, V.; Rostaphshov, V.; Pinelis, D.; Polotsky, Y.; Korolev, S.; Komisar, J.; Fegeding, K. Murine lethal toxic shock caused by intranasal administration of staphylococcal enterotoxin B. Toxicol. Pathol. 2003, 31, 373–378. [Google Scholar]
- Stebbings, R.; Findlay, L.; Edwards, C.; Eastwood, D.; Bird, C.; North, D.; Mistry, Y.; Dilger, P.; Liefooghe, E.; Cludts, I.; et al. "Cytokine Storm" in the Phase I Trial of Monoclonal Antibody TGN1412: Better Understanding the Causes to Improve PreClinical Testing of Immunotherapeutics. J. Immunol. 2007, 179, 3325–3331. [Google Scholar] [PubMed]
- Darenberg, J.; Soderquist, B.; Normark, B.H.; Norrby-Teglund, A. Differences in potency of intravenous polyspecific immunoglobulin G against streptococcal and staphylococcal superantigens: implications for therapy of toxic shock syndrome. Clin. Infect. Dis. 2004, 38, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Bavari, S.; Ulrich, R.G.; LeClaire, R.D. Cross-reactive antibodies prevent the lethal effects of Staphylococcus aureus superantigens. J. Infect. Dis. 1999, 180, 1365–1369. [Google Scholar]
- Bavari, S.; Dyas, B.; Ulrich, R.G. Superantigen vaccines: a comparative study of genetically attenuated receptor-binding mutants of staphylococcal enterotoxin A. J. Infect. Dis. 1996, 174, 338–345. [Google Scholar]
- Arad, G.; Levy, R.; Hillman, D.; Kaempfer, R. Superantigen antagonist protects against lethal shock and defines a new domain for T-cell activation. Nat. Med. 2000, 6, 414–421. [Google Scholar]
- Visvanathan, K.; Charles, A.; Bannan, J.; Pugach, P.; Kashfi, K.; Zabriskie, J.B. Inhibition of bacterial superantigens by peptides and antibodies. Infect. Immun. 2001, 69, 875–884. [Google Scholar]
- Wang, S.; Li, Y.; Xiong, H.; Cao, J. A broad-spectrum inhibitory peptide against staphylococcal enterotoxin superantigen SEA, SEB and SEC. Immunol. Lett. 2008, 121, 167–172. [Google Scholar]
- Geller-Hong, E.; Möllhoff, M.; Shiflett, P.R.; Gupta, G. Design of chimeric receptor mimics with different TcRVβ isoforms: type-specific inhibition of superantigen pathogenesis. J. Biol. Chem. 2004, 279, 5676–5684. [Google Scholar]
- Rasooly, R.; Do, P.M.; Friedman, M. Inhibition of biological activity of staphylococcal enterotoxin A (SEA) by apple juice and apple polyphenols. J. Agric. Food Chem. 2010, 58, 5421–5426. [Google Scholar]
- Liu, D.; Liu, X.Y.; Robinson, D.; Burnett, C.; Jackson, C.; Seele, L.; Veach, R.A.; Downs, S.; Collins, R.D.; Ballard, R.W.; Hawiger, J. Suppression of staphylococcal enterotoxin B-induced toxicity by a nuclear import inhibitor. J. Biol. Chem. 2004, 279, 19239–19246. [Google Scholar]
- Liu, D.; Zienkiewicz, J.; DiGiandomenico, A.; Hawiger, J. Suppression of acute lung inflammation by intracellular peptide delivery of a nuclear import inhibitor. Mol. Ther. 2009, 17, 796–802. [Google Scholar]
- Tilahun, A.Y.; Theuer, J.E.; Patel, R.; David, C.S.; Rajagopalan, G. Detrimental Effect of the Proteasome Inhibitor, Bortezomib in Bacterial Superantigen- and Lipopolysaccharide-induced Systemic Inflammation. Mol. Ther. 2010, 18, 1143–1154. [Google Scholar]
- Krakauer, T. A sensitive ELISA for measuring the adhesion of leukocytic cells to human endothelial cells. J. Immunol. Meth. 1994, 177, 207–213. [Google Scholar]
- See, R.H.; Chow, A.W. Staphylococcal toxic shock syndrome toxin 1-induced tumor necrosis factor alpha and interleukin-1ß secretion by human peripheral blood monocytes and T lymphocytes is differentially suppressed by protein kinase inhibitors. Infect. Immun. 1992, 60, 3456–3459. [Google Scholar]
- Krakauer, T. Suppression of endotoxin- and staphylococcal exotoxin-induced cytokines and chemokines by a phospholipase C inhibitor in human peripheral blood mononuclear cells. Clin. Diagn. Lab. Immunol. 2001, 8, 449–453. [Google Scholar]
- Tschaikowsky, K.J.; Schmidt, J.; Meisner, M. Modulation of mouse endotoxin shock by inhibition of phosphatidylcholine-specific phospholipase C. J. Pharmacol. Exp. Therap. 1999, 285, 800–804. [Google Scholar]
- Krakauer, T.; Buckley, M.; Issaq, H.J.; Fox, S.D. Rapamycin protects mice from staphylococcal enterotoxin B- induced toxic shock and blocks cytokine release In vitro and in vivo. Antimicrob. Agents Chemother. 2010, 54, 1125–1131. [Google Scholar]
- Flechner, S.M. Sirolimus in kidney transplantation indications and practical guidelines: de novo sirolimus-based therapy without calcineurin inhibitors. Transplantation 2009, 97, S1–S6. [Google Scholar]
- Krakauer, T. Caspase inhibitors attenuate superantigen-induced inflammatory cytokines, chemokines and T-cell proliferation. Clin. Diagn. Lab. Immunol. 2004, 11, 621–624. [Google Scholar]
- Krakauer, T.; Buckley, M. Doxycycline is anti-inflammatory and inhibits staphylococcal exotoxin-induced cytokines and chemokines. Antimicrob. Agents Chemother. 2003, 47, 3630–3633. [Google Scholar]
- Jo, D.; Liu, D.; Yao, S.; Collins, R.D.; Hawiger, J. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat. Med. 2005, 11, 892–898. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krakauer, T. Therapeutic Down-Modulators of Staphylococcal Superantigen-Induced Inflammation and Toxic Shock. Toxins 2010, 2, 1963-1983. https://doi.org/10.3390/toxins2081963
Krakauer T. Therapeutic Down-Modulators of Staphylococcal Superantigen-Induced Inflammation and Toxic Shock. Toxins. 2010; 2(8):1963-1983. https://doi.org/10.3390/toxins2081963
Chicago/Turabian StyleKrakauer, Teresa. 2010. "Therapeutic Down-Modulators of Staphylococcal Superantigen-Induced Inflammation and Toxic Shock" Toxins 2, no. 8: 1963-1983. https://doi.org/10.3390/toxins2081963
APA StyleKrakauer, T. (2010). Therapeutic Down-Modulators of Staphylococcal Superantigen-Induced Inflammation and Toxic Shock. Toxins, 2(8), 1963-1983. https://doi.org/10.3390/toxins2081963