1. Introduction
Numerous peptides found in animal venom have become invaluable tools for the study of the physiological role and structure of the channels and receptors. Peptide toxins with nanomolar affinities for corresponding targets proved to be useful agents for discriminating between different types of native ion channel currents. Regarding the relatively small size of peptide toxins, these can be obtained by chemical synthesis or recombinant methods, which represent an advantage in comparison to extraction from large quantities of venom which is often hardly accessible. Moreover, their target specificity and relatively high biological stability makes them a very rewarding class of molecular probes for use in the investigation of channels and receptors.
Understanding the importance and impact of ion channels as therapeutic targets may lead to the discovery of a new class of medications. This explains the particular interest in clinical application of toxins; some of them or their synthetic analogues are in clinical phases of trial. Others are already on the market, protected by international patents, where their properties are used to treat numerous channel-related diseases, such as: neurological disorders, cancer and cardiovascular system anomalies [
1,
2].
Even though only a very small percentage of venom compounds are characterized pharmacologically, the range of toxins affecting various types of channels and receptors is immensely broad. The voltage-sensor trapping mechanism may be a common mode of action for toxins acting on all voltage-gated ion channels. Much effort in this field is dedicated to the pharmacology of toxins that modify channel’s properties. The main usefulness of peptide toxins concerns their high specificity towards the corresponding voltage-gated calcium channels; this feature allows a distinction to be drawn from calcium currents, and is, in some cases, the only available pharmacological criterion of difference.
5. A Special Focus on ω-Agatoxins
American funnel web spiders Agelenopsis aperta, which belong to family of Agelenidae, have gained great interest in neuropharmacology, due to the properties of their venom. The biological activity of spider venoms towards insects was a very first motivation for investigation of spider toxins. Studies which were performed in order to find new insecticides [
63] revealed that the substances present in the venom of Agelenopsis aperta alter the properties of voltage-sensitive calcium channels [
64]. The venom of Agelenopsis aperta contains several fractions classified in three groups: α-agatoxins, which provoke a postsynaptic block of a transmitter-activated receptor channel, µ-agatoxins that are presynaptic modulators of insect sodium channels [
65], and ω-agatoxins, which are presynaptic antagonists of voltage-gated calcium channels. ω-Agatoxins have diverse specificity against various subtypes of calcium channels and show selectivity for calcium channels of various zoological groups (mammalian and insects) [
66,
67].
5.1. Type I and II ω-Agatoxins
ω-Agatoxins of type I are heterodimeric proteins, derived from a single polypeptide chain precursor with five disulfide bonds. Post-translational removal of a heptapeptide leads to the mature form, consisting of two peptide chains: a main 66 amino acid chain possessing four internal disulfide bonds connected via an interchain disulfide linkage to a tripeptide (
Table 9). The mature form of ω-agatoxin I has an apparent molecular weight of 7.5 kDa and possesses five tryptophan residues. Up to date, three isoforms of type I ω-agatoxins were identified: ω-agatoxin IA, ω-agatoxin IB and ω-agatoxin IC; all of them have a similar size and share 77% similarity in amino acid sequence with conserved localization of cysteine residues. Type I ω-agatoxins are potent blockers of insect presynaptic calcium channels [
66]. Type II ω-agatoxins target different binding sites than type I ω-agatoxins, but on the same type of calcium channels [
68]. When type I and II ω-agatoxins are applied together they trigger additive effects and abolish all transmitter release [
66]. Structurally, type II ω-agatoxins seems to be large monomeric proteins consisting of 92 amino acid residues, but their full sequence remains unpublished to date (
Table 9). Structural features discovered to date, disclose a 43% sequence similarity with the ω-agatoxins I family and identical location of some cysteine residues [
64].
Table 9.
Sequences of ω-agatoxines I and II [
64].
Table 9.
Sequences of ω-agatoxines I and II [64].
| ω-Agatoxins | Sequences | Swiss-Prot |
|---|
| IA | AKALPPGSVCDGNESDCKCYGKWHKCRCPWKWHFTGEGPCTCEKGMKHTCITKLHCPNKAEWGLDW SPC | P15969 |
| IB | ERGLPEGAECDGNESDCKCAGAWIKCRCPPMWHING | P15970 |
| IIB | GCIEIGGDCDGYQEKSYCQCCRNNGFCS | P15971 |
5.2. Type III ω-Agatoxins
Subsequent studies on
Agelenopsis aperta venom allowed identification of an additional type of ω-agatoxins [
27]. Type III ω-agatoxins are 76 amino acid peptides (
Table 10), containing 12 cysteine residues forming six internal disulfide bonds, and have an amidated C-terminus [
69]. At least seven isoforms of ω-agatoxin IIIA were discovered, having similar amino acid sequences and potencies for channel inhibition. ω-Agatoxin IIIA isoforms are the biggest class of ω-agatoxins with an overall mass around 8.5 kDa [
70].
Table 10.
Sequences of ω-agatoxines III [
69,
70].
Table 10.
Sequences of ω-agatoxines III [69,70].
| ω-Agatoxins | Sequences | Swiss-Prot |
|---|
| IIIA | SCIDIGGDCDGEKDDCQCCRRNGYCSCYSLFGYLKSGCKCVVGTSAEFQGICRRKARQCYNSDPDKCESHNKPKRR | P33034 |
| IIIB | SCIDFGGDCDGEKDDCQCCRSNGYCSCYNLFGYLKSGCKCEVGTSAEFRRICRRKAKQCYNSDPDKCVSVYKPKRR | P81744 |
| IIIC | NCIDFGGDCDGEKDDCQCCXRNGYCSCYNLFGYLKRGCKXEVG | P81745 |
| IIID | SCIKIGEDCDGDKDDCQCCRTNGYCSXYXLFGYLKSG | P81746 |
It was found that type III ω-agatoxins are not very specific peptides. They inhibit all known neuronal HVA calcium currents L-, N-, P/Q- and R-type, but with different affinities. This is probably due to a common binding site on each channel [
71]. Type III ω-agatoxins inhibit P/Q-type calcium currents by up to 40% and are the only peptide ligands that completely block cardiac L-type calcium channels with high affinity [
72]. Still, type III ω-agatoxins do not bind to T-type voltage gated calcium channels and, thus, are used for the isolation of pure fractions of these channels.
A binding site location and a mechanism of current blocking were proposed for type III ω-agatoxins [
71]. Taking into consideration that ω-agatoxin IIIA inhibit, in a competitive manner, the binding of ω-conotoxin MVIIC to the N-type calcium channels, and knowing that the binding site of ω-conotoxin is located on the extracellular side of the channel, by analogy, the same position for interaction of ω-agatoxin IIIA was suggested. The simplest possibility of the blocking mechanism is that ω-agatoxin IIIA acts as a leaky lid near the outside of the pore, which reduces calcium current. ω-Agatoxin IIIA is a much larger peptide than ω-conotoxin MVIIC, thus probably fits less tightly within the pore vestibule. This way of action explains why ω-agatoxins IIIA produce incomplete blocking of calcium ion flows while completely eliminating access of ω-conotoxin MVIIC. Taken together, results suggest that ω-agatoxins IIIA inhibit calcium current by blocking the channel pore, whether by direct occlusion or through binding sites that are connected allosterically [
61].
5.3. Type IV ω-Agatoxins
With the aim of obtaining a mammalian calcium channel antagonist, fractions isolated from the venom of
Agelenopsis aperta were tested on chicken and rat synaptosomes; this approach resulted in the discovery of type IV ω-agatoxins [
73]. Amino acid sequences of IV ω-agatoxins are different from those of types I, II and III ω-agatoxins. Up to now, only three type IV ω-agatoxins have been identified: ω-agatoxins IVA, IVB (also called ω-agatoxin TK) and IVC; however, these are the most important of all four types of ω-agatoxins regarding pharmacological research on P/Q-type calcium channels and on regulation of glutamate release [
74]. The major advantage of ω-agatoxins IV is their availability for neurophysiology studies; this is the only type of ω-agatoxin successfully synthesized in a biologically active form [
75,
76].
5.3.1. Structure of Type IV ω-Agatoxins
All type IV ω-agatoxins are 48 amino acid peptides containing eight cysteines forming four disulfide bridges. Sequence homology between ω-agatoxins IVA and IVB reaches 71% and is represented in
Table 11. As for ω-agatoxin IVC, its amino acid sequence is nearly identical with that of ω-agatoxin IVB, possessing the only conformational difference on serine 46, which is L-form, comparing to D-serine in ω-agatoxin IVB.
Table 11.
Sequences of ω-agatoxins IVA, IVB (containing the D-serine) and IVC.
Table 11.
Sequences of ω-agatoxins IVA, IVB (containing the D-serine) and IVC.
| ω-Agatoxins | Sequence | Swiss-Prot |
|---|
| IVA | KKKCIAKDYGRCKWGGTPCCRGRGCICSIMGTNCECKPRLIMEGLGLA | P30288 |
| IVB | EDNCIAEDYGKCTWGGTKCCRGRPCRCSMIGTNCECTPRLIMEGLsFA | P37045 |
| IVC | EDNCIAEDYGKCTWGGTKCCRGRPCRCSMIGTNCECTPRLIMEGLSFA | Not available |
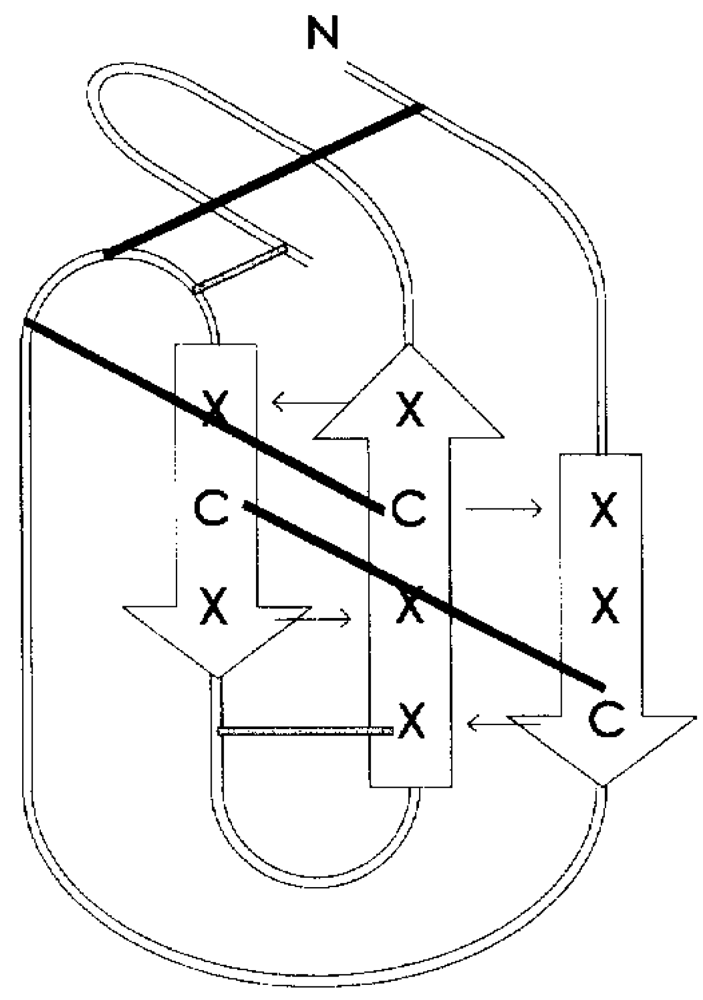
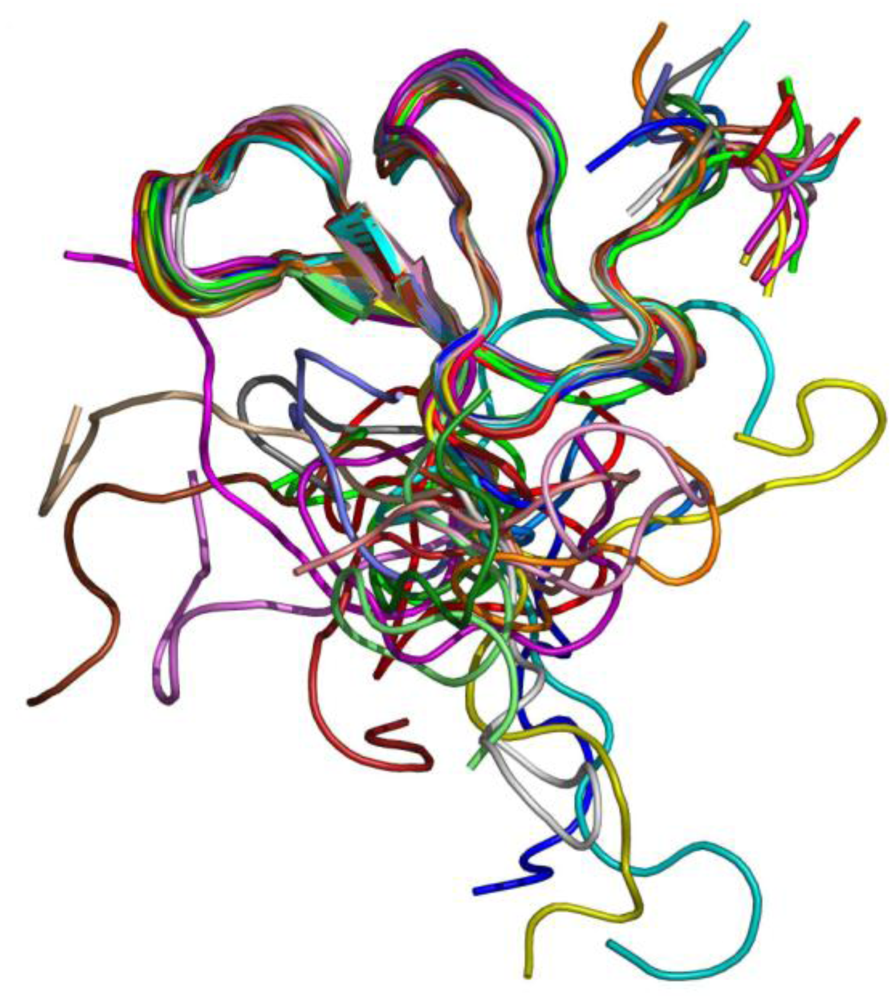
Experiments of two-dimensional ¹H NMR spectroscopy uncovered the spatial arrangement of ω-agatoxins (
Figure 5). Due to its high abundance in spider venom, thus sample accessibility, ω-agatoxin IVB was the first peptide for which a low resolution NMR structure was obtained [
77]. Amino acid sequence similarity between ω-agatoxin IVA and IVB suggests possible common structural features and, indeed, disulfide bonding patterns of both molecules are identical [
78].
Figure 5.
NMR structure of ω-agatoxin IVB [
77,
78,
81].
Figure 5.
NMR structure of ω-agatoxin IVB [
77,
78,
81].
Combination of NMR results with molecular modeling calculations confirm that the molecular scaffold of ω-agatoxins is highly stabilized by the disulfide bridges of identical configuration for both toxins [
79]. Additionally, the components of secondary structure of ω-agatoxin IVA were characterized; residues form a short triple-stranded antiparallel β-sheet and three β-turns. The first β-turn is present in the loop between β-strand and N-terminal fragment. The second and third β-turns are located in the long external loop between the first and second β-strand [
80].
The disulfide bond between the cysteines 12 and 25 interconnects the first and the second β-strands, while the disulfide bonds between cysteines 4-20 and 19-36 connect the external long loop with the N-terminal region and the third β-strand, respectively. The disulfide bond between cysteines 27 and 34 connects the second and third β-strand [
78].
ω-Agatoxin IVA contains only two aromatic residues, tyrosine and tryptophan in positions 9 and 14, respectively, while ω-agatoxin IVB possess three aromatic residues, two of which are homologous to ω-agatoxin IVA and an additional phenylalanine at position 47, located in the C-terminal region. The side chain of tryptophan 14 is located on the upper side of the second β-strand. It should be noted that tyrosine and tryptophan are amphiphilic amino acids that can interact with both hydrophilic and hydrophobic environments [
81]. Residues isoleucine in position 5, glycine in 10 and cysteines in position 12, 19, 25, 34 and 36 are located in the peptide core, which is inaccessible for solvents and proteases.
In summary, ω-agatoxins IVA and IVB possess identical disulfide patterns, forming four loop regions, a small three-stranded antiparallel β-sheet, a solvent inaccessible hydrophobic core and disordered N- and C-terminal segments.
5.3.2. Important ω-Agatoxins Residues for Channel Binding
Experiments performed with ω-agatoxins led to several conclusions concerning the characteristic amino acids or whole segments of toxin that are involved in interactions with P-type calcium channels. As a result, different models of toxin binding were proposed. Determination of amino acid sequence and NMR/molecular modeling studies revealed that the basic amino acids residues (arginines in positions 21, 23, 39) form a positively charged surface located close to the hydrophobic C-terminus tail and are conserved in both ω-agatoxins IVA and IVB. Consequently, it was suggested that the positively charged toxin core interacts with calcium channel’s negatively charged residues located near to the external pore [
77]. Complementary electrostatic attractions between the toxin and the extracellular loops near the entrance of the pore may contribute to binding affinity, while the toxin’s hydrophobic tail enhances the blockage of the channel by establishing additional molecular contacts with the channel protein [
80].
The remarkable difference between ω-agatoxins IVA and IVB is the sequence of amino acids at the N-terminus, which alters charge distribution in the whole molecule; in fact, the two peptides have opposite charges. Whereas the N-terminus of ω-agatoxin IVA is very basic, in ω-agatoxin IVB it is strongly acidic. Electrophysiological analysis of P-type channel inhibition by these two toxins shows that their affinities are similar (2-4 mM). However, kinetics of channel inhibition and dissociation are significantly slower for ω-agatoxin IVB. Experiments indicate that the N-terminus peptide is important in determining the rates at which the toxin binds to and unbinds from the channel. The model explaining the obtained result suggests that the N-terminus sequence of ω-agatoxins may serve to enhance the toxin approach to the outer vestibule of the calcium channel prior to receptor binding. Nevertheless, inhibition potency of the toxins remains unaffected [
78].
As biological examinations of a tryptophan 14 modified analogue revealed incomplete channel blocking, it was proposed that this residue might be important for toxin binding to the channel [
75]. Tryptophan’s side chain is positioned on the upper side of the second β-strand, thus well exposed for toxin-channel interactions [
81]. Currently, there is no defined mechanism explaining how ω-agatoxin IV docks to the channel protein. One cannot help noticing that the model of channel inhibition includes tryptophan 14 as a residue involved in direct binding interactions. In a recent paper presented by our research team, on various synthesized analogues, it was proved that the tryptophan in position 14 is indeed the key-residue for the toxin-channel binding [
82].
5.3.3. Importance of Charge for Toxin Interaction
Amino acid sequence examinations revealed that ω-agatoxin IVA is a highly basic molecule, with 10 positively and three negatively charged residues, whereas ω-agatoxin IVB, with six positively charged and six negatively charged residues, is neutral. Taking into consideration that the C- and N-termini are not modified and their participation in the molecule charge is null, the ω-agatoxin IVA net charge is +7 and ω-agatoxin IVB possesses a net charge equal to zero. With such a large difference in the overall charge of the peptides, it could be expected that ω-agatoxin IVB might interact with calcium channels somewhat differently than ω-agatoxin IVA; moreover, the unbinding of ω-agatoxin IVA is strongly voltage-dependent, which might confirm the charge-binding relation. In fact, the properties of calcium channel inhibition by the two toxins are remarkably similar; both are highly selective for the P-type calcium channels; however, their kinetic rate of inhibition is different [
77].
5.3.4. Effect of Serine 46 Isomerization in ω-Agatoxin IVB
The post-translational conversion of L- to D-serine described for ω-agatoxin IVB is an evolutionary strategy of stability and activity improvement. Existence of D-amino acids alters the structure of a toxin and is a result of modification of ω-agatoxin IVC via an isomerase [
83]. The isomerase activity protects the ω-agatoxin IVB’s C-terminus from degradation; enhanced stability may explain why ω-agatoxin IVB is about 10-times more abundant in venom than the other toxins of its class. A study on both D and L isomers of ω-agatoxins IVB and IVC demonstrated the crucial impact of isomerization on toxin selectivity and stabilization against proteolysis [
84,
85]. The inhibitory effect comparison of two synthetic ω-agatoxins, one with D-serine: ω-agatoxin IVB, and the second with L-serine: ω-agatoxin IVC, revealed that the L-isomer is 90-fold less potent [
86].
5.3.5. Action Mode and Location of the Binding Site
ω-Agatoxins IV are the most potent blockers of P-type calcium channels in both insect and mammalian central neurons. However, the ω-agatoxins IVA and IVB constitute a unique component of Agelenopsis aperta venom that has remarkable specificity for P-type calcium channels in the mammalian central nervous system. These channels occur throughout the brain, but are particularly developed in Purkinje cells of the cerebellum and are directly involved in glutamate release.
The mechanism of toxin-channel interaction of ω-agatoxin IVA on P/Q-type calcium channels is voltage-dependent, meaning that the toxin perturbs the gating properties of the channel and does not physically occlude the pore. Binding of the toxin shifts the channel’s activation voltage to positive potentials, which are not reached during normal physiological activity of the cell. The toxin raises the energy barrier for voltage-dependent gating to non-physiological levels. Type IV ω-agatoxins bind with high affinity to closed state channels, leaving the channels in the open state unaffected [
87]. The experiments showed that repetitive application of strongly positive voltage steps led to the opening of the channel and the dissociation of the toxin [
88]. Toxin dissociation occurs because, in contrast to its high affinity binding to the channel in the closed state, the affinity of the channel in its open state is very low. Additionally, strong depolarization may cause channel conformational changes, which are unfavorable from the point of view of spatial arrangement and act allosterically on the toxin binding site [
87]. Thus, ω-agatoxin IVA inhibits the inward calcium current by increasing the voltage required for channel activation, without affecting the permeation of the channel.
In order to establish the binding site’s location on the calcium channel, comparative experiments using ω-agatoxin IVA and ω-grammotoxin SIA were performed. A similar shift of gating on the N- and P-type calcium channels was observed, suggesting similar binding sites for both toxins. The results of electrophysiological studies show that the binding sites for both toxins involve a linker between transmembrane segments 3 and 4 in P-type calcium channels. However, the voltage shifts produced by the two toxins is additive, suggesting that they bind simultaneously to different sites of the channel [
61].
Sequence comparison between the α
1 subunit of voltage-gated potassium, sodium and calcium channels performed in order to establish the similarities between the regions of toxin binding, allowed the amino acids that are probably involved in channel-toxin interactions to be chosen. Subsequent mutation of the channel protein revealed, at least partially, the location of the binding site for ω-agatoxin IVA, which is situated on the external part of the channel near the channel pore. The extracellular loop between transmembrane segments 3 and 4 in domain IV of the channel is directly affected by ω-agatoxin IVA. This places the toxin in direct proximity to the transmembrane segment 4 voltage sensor. Replacement of glutamic acid with lysine, at position 1658 in the channel protein, makes the channel completely resistant to modifications provoked by ω-agatoxin IVA [
89]. These results provide compelling insight into the manner by which gating modification triggered by toxins blocks calcium channels.
5.3.6. Selectivity and Affinity
Selectivity of ω-agatoxin IVA for the P-type calcium channels, which are responsible for the release of glutamate, is well established, with inhibition occurring at concentrations less than 10 nM and a Kd of 2 nM. ω-Agatoxin IVB evokes the same selectivity and similar potency with a Kd of 3 nM, nevertheless, is about eight-times slower regarding the kinetics of current blocking. Given that the kinetics of inhibition is dependent on toxin concentration, the ten-fold higher abundance of ω-agatoxin IVB in venom equilibrates the rate of block, which is comparable to that of ω-agatoxin IVA. As previously mentioned, biological activity of ω-agatoxin IVC, precursor of ω-agatoxin IVB, is much less important than that of ω-agatoxin IVB.
Regarding Q-type calcium channels, the selectivity of ω-agatoxins is less remarkable (K
d ~100 nM), with an effective toxin concentration arising to a micromolar range. Studies of the effect of ω-agatoxin IVA on the N-type calcium currents demonstrated that the selectivity towards P/Q-type currents is not complete. Subsequent investigation showed that ω-agatoxin IVA affects a variety of dihydropyridine-insensitive calcium channels (non L-type) when used at micromolar range. These data suggest that ω-agatoxin IVA is selective at the nanomolar range; nevertheless, its selectivity is diminished at micromolar range, thus its usefulness for functional studies of Q-type calcium channels is limited [
90]. The dissimilarity of selectivity towards P- and Q-type channels is explained by the different structures of the channels, resulting from differential splicing of the genes encoding for the α
1A subunit [
67].
Notwithstanding this limitation, the ω-agatoxins IVA and IVB retain their usefulness as selective and potent blockers of neuronal P-type voltage-gated calcium channels and will remain important tools to relate the properties of recombinant calcium channels to the complexity of their native counterparts.
Since the tryptophan residue located at position 14 of the ω-agatoxin IVB has been suggested as essential for binding, analogues were synthesized. Our recent studies of constraint analogues of the loop containing the tryptophan 14 revealed a potent agonist activity on P/Q-type calcium channels [
82]. The synthesis of such cyclopeptides is currently under development. Furthermore, specific modifications increasing the peptide stability
in vitro and
in vivo are being explored.
6. Conclusion
In this review, a large variety of natural potential tools were presented; the described peptide neurotoxins and their analogues helped in the elucidation of structure/function relationships toward voltage-gated calcium channels. Furthermore, the use of synthetic analogues of these toxins will lead to the development of new therapeutic agents and strategies for treatment of ion channel-related diseases.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}