The Potential Contributions of Lethal and Edema Toxins to the Pathogenesis of Anthrax Associated Shock

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Lethal and Edema Toxin Structure and Intracellular Effects
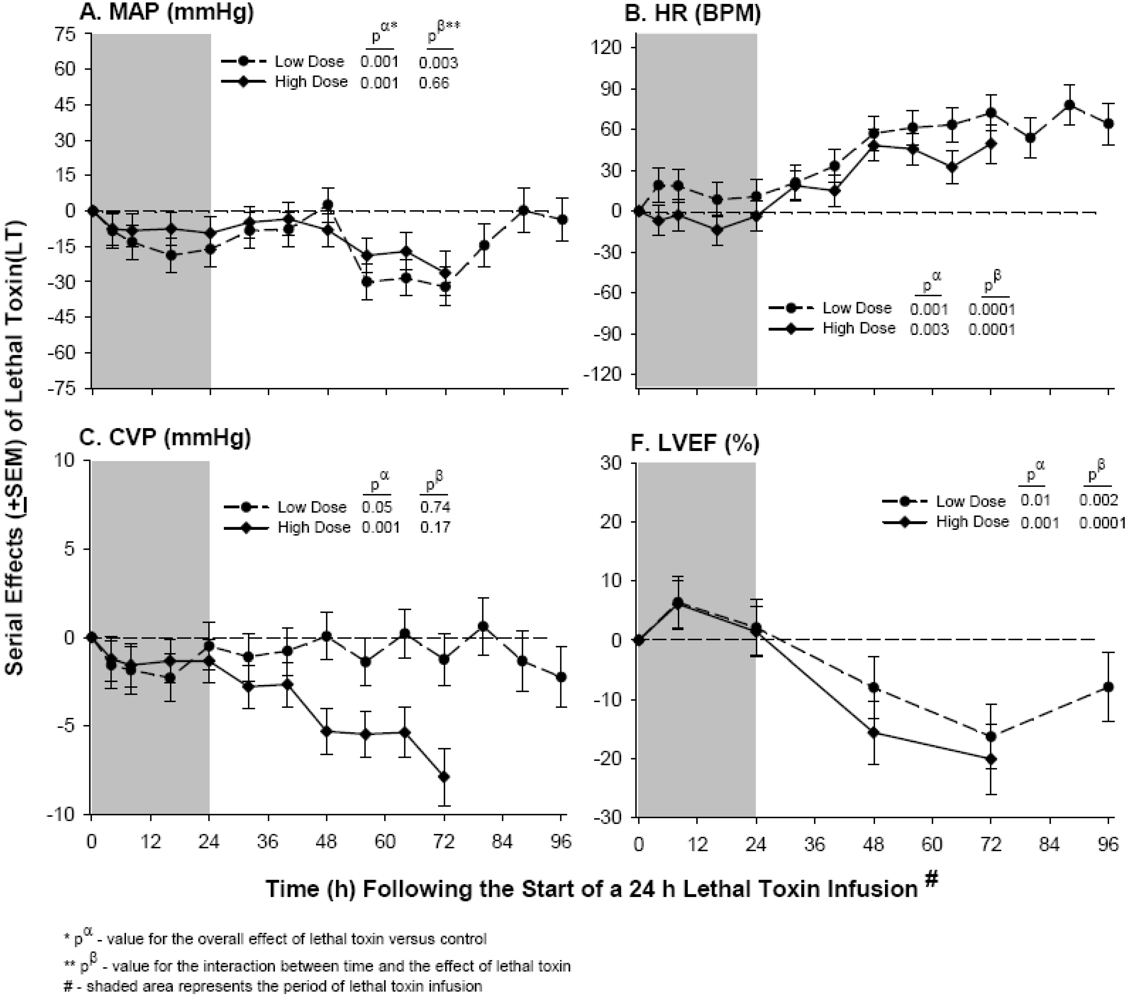
3. Lethal Toxin


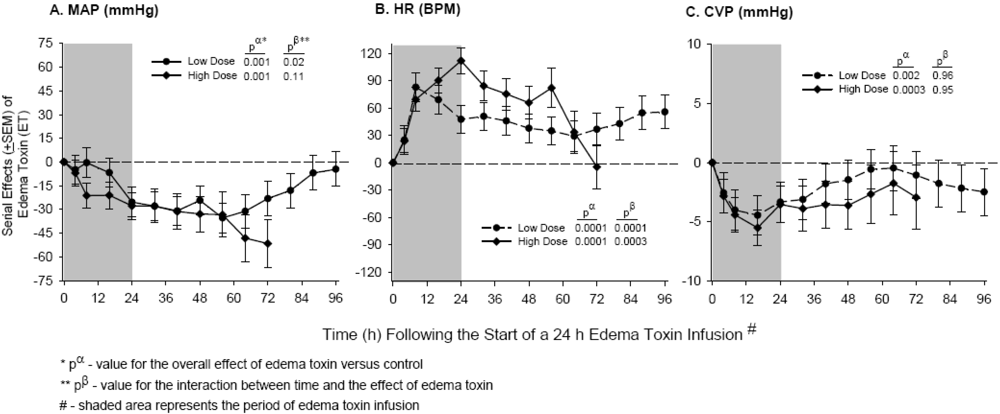
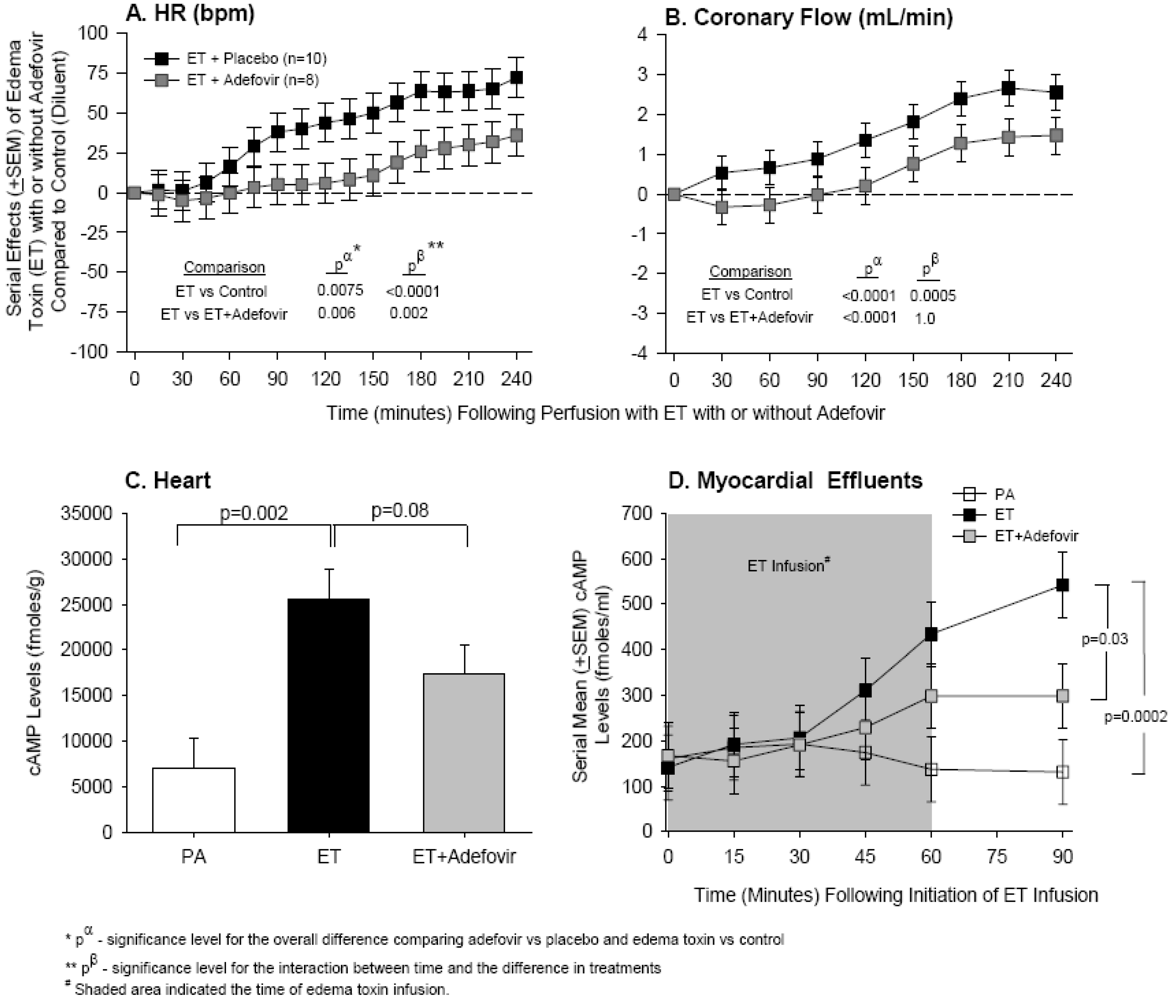
4. Edema Toxin




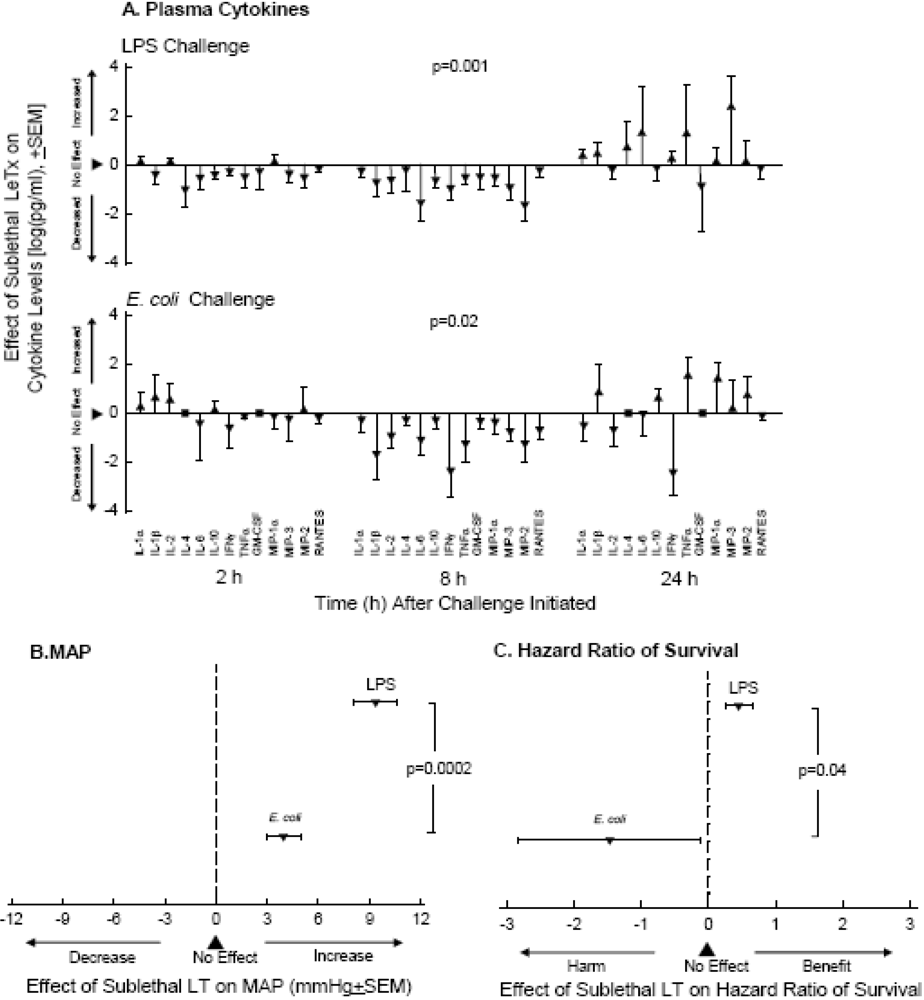
5. Other Components that May Aggravate Toxin Associated Shock
6. Conclusions
Conflict of Interest
References
- Jernigan, D.B.; Raghunathan, P.L.; Bell, B.P.; Brechner, R.; Bresnitz, E.A.; Butler, J.C.; Cetron, M.; Cohen, M.; Doyle, T.; Fischer, M.; et al. Investigation of bioterrorism-related anthrax, United States, 2001: Epidemiologic findings. Emerg. Infect. Dis. 2002, 8, 1019–1028. [Google Scholar] [PubMed]
- Ramsay, C.N.; Stirling, A.; Smith, J.; Hawkins, G.; Brooks, T.; Hood, J.; Penrice, G.; Browning, L.M.; Ahmed, S. An outbreak of infection with Bacillus anthracis in injecting drug users in Scotland. Euro. Surveill. 2010, 15, 2. [Google Scholar] [PubMed]
- Ringertz, S.H.; Hoiby, E.A.; Jensenius, M.; Maehlen, J.; Caugant, D.A.; Myklebust, A.; Fossum, K. Injectional anthrax in a heroin skin-popper. Lancet 2000, 356, 1574–1575. [Google Scholar]
- Winters, B.D.; Eberlein, M.; Leung, J.; Needham, D.M.; Pronovost, P.J.; Sevransky, J.E. Long-term mortality and quality of life in sepsis: A systematic review. Crit. Care Med. 2010, 38, 1276–1283. [Google Scholar]
- Booth, M.G.; Hood, J.; Brooks, T.J.; Hart, A. Anthrax infection in drug users. Lancet 2010, 375, 1345–1346. [Google Scholar]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar]
- Scobie, H.M.; Rainey, G.J.; Bradley, K.A.; Young, J.A. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar]
- Sherer, K.; Li, Y.; Cui, X.; Eichacker, P.Q. Lethal and edema toxins in the pathogenesis of Bacillus anthracis septic shock: Implications for therapy. Am. J. Respir. Crit. Care Med. 2007, 175, 211–221. [Google Scholar]
- van der Goot, G.; Young, J.A. Receptors of anthrax toxin and cell entry. Mol. Aspects. Med. 2009, 30, 406–412. [Google Scholar]
- Bell, S.E.; Mavila, A.; Salazar, R.; Bayless, K.J.; Kanagala, S.; Maxwell, S.A.; Davis, G.E. Differential gene expression during capillary morphogenesis in 3D collagen matrices: Regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J. Cell Sci. 2001, 114, 2755–2773. [Google Scholar] [PubMed]
- Carson-Walter, E.B.; Watkins, D.N.; Nanda, A.; Vogelstein, B.; Kinzler, K.W.; St Croix, B. Cell surface tumor endothelial markers are conserved in mice and humans. Cancer Res. 2001, 61, 6649–6655. [Google Scholar]
- Cui, X.; Li, Y.; Li, X.; Laird, M.W.; Subramanian, M.; Moayeri, M.; Leppla, S.H.; Fitz, Y.; Su, J.; Sherer, K.; et al. Bacillus anthracis edema and lethal toxin have different hemodynamic effects but function together to worsen shock and outcome in a rat model. J. Infect. Dis. 2007, 195, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Klimpel, K.R.; Molloy, S.S.; Thomas, G.; Leppla, S.H. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl. Acad. Sci. USA 1992, 89, 10277–10281. [Google Scholar]
- Milne, J.C.; Furlong, D.; Hanna, P.C.; Wall, J.S.; Collier, R.J. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J. Biol. Chem. 1994, 269, 20607–20612. [Google Scholar]
- Mogridge, J.; Cunningham, K.; Collier, R.J. Stoichiometry of anthrax toxin complexes. Biochemistry 2002, 41, 1079–1082. [Google Scholar]
- Tonello, F.; Montecucco, C. The anthrax lethal factor and its MAPK kinase-specific metalloprotease activity. Mol. Aspects. Med. 2009, 30, 431–438. [Google Scholar]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar]
- Tang, W.J.; Guo, Q. The adenylyl cyclase activity of anthrax edema factor. Mol. Aspects. Med. 2009, 30, 423–430. [Google Scholar]
- Tippetts, M.T.; Robertson, D.L. Molecular cloning and expression of the Bacillus anthracis edema factor toxin gene: A calmodulin-dependent adenylate cyclase. J. Bacteriol. 1988, 170, 2263–2266. [Google Scholar]
- Moayeri, M.; Leppla, S.H. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol. Aspects. Med. 2009, 30, 439–455. [Google Scholar]
- Firoved, A.M.; Miller, G.F.; Moayeri, M.; Kakkar, R.; Shen, Y.; Wiggins, J.F.; McNally, E.M.; Tang, W.J.; Leppla, S.H. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am. J. Pathol. 2005, 167, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Hicks, C.W.; Li, Y.; Okugawa, S.; Solomon, S.B.; Moayeri, M.; Leppla, S.H.; Mohanty, A.; Subramanian, G.M.; Mignone, T.S.; Fitz, Y.; et al. Anthrax edema toxin has cAMP-mediated stimulatory effects and high-dose lethal toxin has depressant effects in an isolated perfused rat heart model. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1108–H1118. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, D.A.; Cui, X.; Solomon, S.B.; Vitberg, D.A.; Migone, T.S.; Scher, D.; Danner, R.L.; Natanson, C.; Subramanian, G.M.; Eichacker, P.Q. Anthrax lethal and edema toxins produce different patterns of cardiovascular and renal dysfunction and synergistically decrease survival in canines. J. Infect. Dis. 2010, 202, 1885–1896. [Google Scholar]
- Smith, H.; Keppie, J.; Stanley, J.L.; Harris-Smith, P.W. The chemical basis of the virulence of Bacillus anthracis. IV. Secondary shock as the major factor in death of guinea-pigs from anthrax. Br. J. Exp. Pathol. 1955, 36, 323–335. [Google Scholar] [PubMed]
- Stanley, J.L.; Smith, H. Purification of factor I and recognition of a third factor of the anthrax toxin. J. Gen. Microbiol. 1961, 26, 49–63. [Google Scholar]
- Beall, F.A.; Dalldorf, F.G. The pathogenesis of the lethal effect of anthrax toxin in the rat. J. Infect. Dis. 1966, 116, 377–389. [Google Scholar]
- Bonventre, P.F.; Eckert, N.J. Toxin production as a criterion for differentiating Bacillus cereus and Bacillus anthracis. J. Bacteriol. 1963, 85, 490–491. [Google Scholar] [PubMed]
- Fish, D.C.; Lincoln, R.E. In vivo-produced anthrax toxin. J. Bacteriol. 1968, 95, 919–924. [Google Scholar] [PubMed]
- Vick, J.A.; Lincoln, R.E.; Klein, F.; Mahlandt, B.G.; Walker, J.S.; Fish, D.C. Neurological and physiological responses of the primate to anthrax toxin. J. Infect. Dis. 1968, 118, 85–96. [Google Scholar]
- Moayeri, M.; Haines, D.; Young, H.A.; Leppla, S.H. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J. Clin. Invest. 2003, 112, 670–682. [Google Scholar] [PubMed]
- Cui, X.; Moayeri, M.; Li, Y.; Li, X.; Haley, M.; Fitz, Y.; Correa-Araujo, R.; Banks, S.M.; Leppla, S.H.; Eichacker, P.Q. Lethality during continuous anthrax lethal toxin infusion is associated with circulatory shock but not inflammatory cytokine or nitric oxide release in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R699–R709. [Google Scholar]
- Gozes, Y.; Moayeri, M.; Wiggins, J.F.; Leppla, S.H. Anthrax lethal toxin induces ketotifen-sensitive intradermal vascular leakage in certain inbred mice. Infect. Immun. 2006, 74, 1266–1272. [Google Scholar]
- Kirby, J.E. Anthrax lethal toxin induces human endothelial cell apoptosis. Infect. Immun. 2004, 72, 430–439. [Google Scholar]
- Rolando, M.; Munro, P.; Stefani, C.; Auberger, P.; Flatau, G.; Lemichez, E. Injection of Staphylococcus aureus EDIN by the Bacillus anthracis protective antigen machinery induces vascular permeability. Infect. Immun. 2009, 77, 3596–3601. [Google Scholar] [PubMed]
- Warfel, J.M.; Steele, A.D.; D’Agnillo, F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am. J. Pathol. 2005, 166, 1871–1881. [Google Scholar]
- Guichard, A.; McGillivray, S.M.; Cruz-Moreno, B.; van Sorge, N.M.; Nizet, V.; Bier, E. Anthrax toxins cooperatively inhibit endocytic recycling by the Rab11/Sec15 exocyst. Nature 2010, 467, 854–858. [Google Scholar]
- Bolcome, R.E., III.; Chan, J. Constitutive MEK1 activation rescues anthrax lethal toxin-induced vascular effects in vivo. Infect. Immun. 2010, 78, 5043–5053. [Google Scholar] [CrossRef] [PubMed]
- Bolcome, R.E., III.; Sullivan, S.E.; Zeller, R.; Barker, A.P.; Collier, R.J.; Chan, J. Anthrax lethal toxin induces cell death-independent permeability in zebrafish vasculature. Proc. Natl. Acad. Sci. USA 2008, 105, 2439–2444. [Google Scholar]
- Watson, L.E.; Kuo, S.R.; Katki, K.; Dang, T.; Park, S.K.; Dostal, D.E.; Tang, W.J.; Leppla, S.H.; Frankel, A.E. Anthrax toxins induce shock in rats by depressed cardiac ventricular function. PLoS One 2007, 2. [Google Scholar]
- Watson, L.E.; Mock, J.; Lal, H.; Lu, G.; Bourdeau, R.W.; Tang, W.J.; Leppla, S.H.; Dostal, D.E.; Frankel, A.E. Lethal and edema toxins of anthrax induce distinct hemodynamic dysfunction. Front. Biosci. 2007, 12, 4670–4675. [Google Scholar]
- Golden, H.B.; Watson, L.E.; Lal, H.; Verma, S.K.; Foster, D.M.; Kuo, S.R.; Sharma, A.; Frankel, A.; Dostal, D.E. Anthrax toxin: Pathologic effects on the cardiovascular system. Front. Biosci. 2009, 14, 2335–2357. [Google Scholar]
- Moayeri, M.; Crown, D.; Dorward, D.W.; Gardner, D.; Ward, J.M.; Li, Y.; Cui, X.; Eichacker, P.; Leppla, S.H. The heart is an early target of anthrax lethal toxin in mice: A protective role for neuronal nitric oxide synthase (nNOS). PLoS Pathog. 2009, 5. [Google Scholar]
- Lips, D.J.; Bueno, O.F.; Wilkins, B.J.; Purcell, N.H.; Kaiser, R.A.; Lorenz, J.N.; Voisin, L.; Saba-El-Leil, M.K.; Meloche, S.; et al. MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation 2004, 109, 1938–1941. [Google Scholar] [PubMed]
- Purcell, N.H.; Wilkins, B.J.; York, A.; Saba-El-Leil, M.K.; Meloche, S.; Robbins, J.; Molkentin, J.D. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 14074–14079. [Google Scholar]
- Yamaguchi, O.; Watanabe, T.; Nishida, K.; Kashiwase, K.; Higuchi, Y.; Takeda, T.; Hikoso, S.; Hirotani, S.; Asahi, M.; Taniike, M.; et al. Cardiac-specific disruption of the c-raf-1 gene induces cardiac dysfunction and apoptosis. J. Clin. Invest. 2004, 114, 937–943. [Google Scholar] [PubMed]
- Kandadi, M.R.; Hua, Y.; Ma, H.; Li, Q.; Kuo, S.R.; Frankel, A.E.; Ren, J. Anthrax lethal toxin suppresses murine cardiomyocyte contractile function and intracellular Ca2+ handling via a NADPH oxidase-dependent mechanism. PLoS One 2010, 5, e13335. [Google Scholar]
- Cui, X.; Li, Y.; Li, X.; Haley, M.; Moayeri, M.; Fitz, Y.; Leppla, S.H.; Eichacker, P.Q. Sublethal doses of Bacillus anthracis lethal toxin inhibit inflammation with lipopolysaccharide and Escherichia coli challenge but have opposite effects on survival. J. Infect. Dis. 2006, 193, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Hanna, P.C.; Acosta, D.; Collier, R.J. On the role of macrophages in anthrax. Proc. Natl. Acad. Sci. USA 1993, 90, 10198–10201. [Google Scholar]
- Tournier, J.N.; Rossi Paccani, S.; Quesnel-Hellmann, A.; Baldari, C.T. Anthrax toxins: A weapon to systematically dismantle the host immune defenses. Mol. Aspects. Med. 2009, 30, 456–466. [Google Scholar]
- Pezard, C.; Berche, P.; Mock, M. Contribution of individual toxin components to virulence of Bacillus anthracis. Infect. Immun. 1991, 59, 3472–3477. [Google Scholar] [PubMed]
- Molin, F.D.; Fasanella, A.; Simonato, M.; Garofolo, G.; Montecucco, C.; Tonello, F. Ratio of lethal and edema factors in rabbit systemic anthrax. Toxicon 2008, 52, 824–828. [Google Scholar]
- Quinn, C.P.; Shone, C.C.; Turnbull, P.C.; Melling, J. Purification of anthrax-toxin components by high-performance anion-exchange, gel-filtration and hydrophobic-interaction chromatography. Biochem. J. 1988, 252, 753–758. [Google Scholar]
- Cooksey, B.A.; Sampey, G.C.; Pierre, J.L.; Zhang, X.; Karwoski, J.D.; Choi, G.H.; Laird, M.W. Production of biologically active Bacillus anthracis edema factor in Escherichia coli. Biotechnol. Prog. 2004, 20, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, J.A.; Stephens, D.S.; Ashford, D.A.; Omenaca, C.; Topiel, M.S.; Galbraith, M.; Tapper, M.; Fisk, T.L.; Zaki, S.; Popovic, T.; et al. Bioterrorism-related inhalational anthrax: The first 10 cases reported in the United States. Emerg. Infect. Dis. 2001, 7, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.M.; Taylor, H.J. Cyclic AMP mediates EDHF-type relaxations of rabbit jugular vein. Biochem. Biophys. Res. Commun. 1999, 263, 52–57. [Google Scholar]
- Stehlik, J.; Movsesian, M.A. Inhibitors of cyclic nucleotide phosphodiesterase 3 and 5 as therapeutic agents in heart failure. Expert. Opin. Investig. Drugs 2006, 15, 733–742. [Google Scholar]
- Borer, J.S. Drug insight: If inhibitors as specific heart-rate-reducing agents. Nat. Clin. Pract. Cardiovasc. Med. 2004, 1, 103–109. [Google Scholar]
- Katz, A.M. Physilogy of the Heart, 3rd ed; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; pp. 255–286. [Google Scholar]
- Shen, Y.; Zhukovskaya, N.L.; Zimmer, M.I.; Soelaiman, S.; Bergson, P.; Wang, C.R.; Gibbs, C.S.; Tang, W.J. Selective inhibition of anthrax edema factor by adefovir, a drug for chronic hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 3242–3247. [Google Scholar]
- Friedler, R.M.; Kurokawa, K.; Coburn, J.W.; Massry, S.G. Renal action of cholera toxin: I. Effects on urinary excretion of electrolytes and cyclic AMP. Kidney Int. 1975, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, K.; Friedler, R.M.; Massry, S.G. Renal action of cholera toxin: II. Effects on adenylate cyclase-cyclic AMP system. Kidney Int. 1975, 7, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Pierce, N.F.; Graybill, J.R.; Kaplan, M.M.; Bouwman, D.L. Systemic effects of parenteral cholera enterotoxin in dogs. J. Lab. Clin. Med. 1972, 79, 145–156. [Google Scholar]
- Kuehnert, M.J.; Doyle, T.J.; Hill, H.A.; Bridges, C.B.; Jernigan, J.A.; Dull, P.M.; Reissman, D.B.; Ashford, D.A.; Jernigan, D.B. Clinical features that discriminate inhalational anthrax from other acute respiratory illnesses. Clin. Infect. Dis. 2003, 36, 328–336. [Google Scholar]
- Firoved, A.M.; Moayeri, M.; Wiggins, J.F.; Shen, Y.; Tang, W.J.; Leppla, S.H. Anthrax edema toxin sensitizes DBA/2J mice to lethal toxin. Infect. Immun. 2007, 75, 2120–2125. [Google Scholar]
- Maldonado-Arocho, F.J.; Fulcher, J.A.; Lee, B.; Bradley, K.A. Anthrax oedema toxin induces anthrax toxin receptor expression in monocyte-derived cells. Mol. Microbiol. 2006, 61, 324–337. [Google Scholar]
- Paccani, S.R.; Tonello, F.; Ghittoni, R.; Natale, M.; Muraro, L.; D’Elios, M.M.; Tang, W.J.; Montecucco, C.; Baldari, C.T. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 2005, 201, 325–331. [Google Scholar]
- Tournier, J.N.; Quesnel-Hellmann, A.; Mathieu, J.; Montecucco, C.; Tang, W.J.; Mock, M.; Vidal, D.R.; Goossens, P.L. Anthrax edema toxin cooperates with lethal toxin to impair cytokine secretion during infection of dendritic cells. J. Immunol. 2005, 174, 4934–4941. [Google Scholar]
- Chakrabarty, K.; Wu, W.; Booth, J.L.; Duggan, E.S.; Nagle, N.N.; Coggeshall, K.M.; Metcalf, J.P. Human lung innate immune response to Bacillus anthracis spore infection. Infect. Immun. 2007, 75, 3729–3738. [Google Scholar] [PubMed]
- Heninger, S.; Drysdale, M.; Lovchik, J.; Hutt, J.; Lipscomb, M.F.; Koehler, T.M.; Lyons, C.R. Toxin-deficient mutants of Bacillus anthracis are lethal in a murine model for pulmonary anthrax. Infect. Immun. 2006, 74, 6067–6074. [Google Scholar] [PubMed]
- Pickering, A.K.; Osorio, M.; Lee, G.M.; Grippe, V.K.; Bray, M.; Merkel, T.J. Cytokine response to infection with Bacillus anthracis spores. Infect. Immun. 2004, 72, 6382–6389. [Google Scholar] [PubMed]
- Stearns-Kurosawa, D.J.; Lupu, F.; Taylor, F.B., Jr.; Kinasewitz, G.; Kurosawa, S. Sepsis and pathophysiology of anthrax in a nonhuman primate model. Am. J. Pathol. 2006, 169, 433–444. [Google Scholar]
- Popov, S.G.; Villasmil, R.; Bernardi, J.; Grene, E.; Cardwell, J.; Popova, T.; Wu, A.; Alibek, D.; Bailey, C.; Alibek, K. Effect of Bacillus anthracis lethal toxin on human peripheral blood mononuclear cells. FEBS Lett. 2002, 527, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, M.; Uddin, A.; Maher, S.; Charalambous, N.; Hamm, T.S.; Alsumaiti, A.; Triantafilou, K. Anthrax toxin evades Toll-like receptor recognition, whereas its cell wall components trigger activation via TLR2/6 heterodimers. Cell Microbiol. 2007, 9, 2880–2892. [Google Scholar]
- Hsu, L.C.; Ali, S.R.; McGillivray, S.; Tseng, P.H.; Mariathasan, S.; Humke, E.W.; Eckmann, L.; Powell, J.J.; Nizet, V.; Dixit, V.M.; et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc. Natl. Acad. Sci. USA 2008, 105, 7803–7808. [Google Scholar]
- Cui, X.; Su, J.; Li, Y.; Shiloach, J.; Solomon, S.; Kaufman, J.B.; Mani, H.; Fitz, Y.; Weng, J.; Altaweel, L.; et al. Bacillus anthracis cell wall produces injurious inflammation but paradoxically decreases the lethality of anthrax lethal toxin in a rat model. Intensiv. Care Med. 2010, 36, 148–156. [Google Scholar] [CrossRef]
- Iyer, J.K.; Khurana, T.; Langer, M.; West, C.M.; Ballard, J.D.; Metcalf, J.P.; Merkel, T.J.; Coggeshall, K.M. Inflammatory cytokine response to Bacillus anthracis peptidoglycan requires phagocytosis and lysosomal trafficking. Infect. Immun. 2010, 78, 2418–2428. [Google Scholar] [CrossRef] [PubMed]
- Langer, M.; Malykhin, A.; Maeda, K.; Chakrabarty, K.; Williamson, K.S.; Feasley, C.L.; West, C.M.; Metcalf, J.P.; Coggeshall, K.M. Bacillus anthracis peptidoglycan stimulates an inflammatory response in monocytes through the p38 mitogen-activated protein kinase pathway. PLoS One 2008, 3, e3706. [Google Scholar] [PubMed]
- Popov, S.G.; Popova, T.G.; Hopkins, S.; Weinstein, R.S.; MacAfee, R.; Fryxell, K.J.; Chandhoke, V.; Bailey, C.; Alibek, K. Effective antiprotease-antibiotic treatment of experimental anthrax. BMC Infect. Dis. 2005, 5. [Google Scholar]
- Adekoya, O.A.; Sylte, I. The thermolysin family (M4) of enzymes: Therapeutic and biotechnological potential. Chem. Biol. Drug Des. 2009, 73, 7–16. [Google Scholar]
- Fisher, J.F.; Mobashery, S. Mechanism-based profiling of MMPs. Methods Mol. Biol. 2010, 622, 471–487. [Google Scholar]
- Holty, J.E.; Bravata, D.M.; Liu, H.; Olshen, R.A.; McDonald, K.M.; Owens, D.K. Systematic review: A century of inhalational anthrax cases from 1900 to 2005. Ann. Intern. Med. 2006, 144, 270–280. [Google Scholar]
- Migone, T.S.; Subramanian, G.M.; Zhong, J.; Healey, L.M.; Corey, A.; Devalaraja, M.; Lo, L.; Ullrich, S.; Zimmerman, J.; Chen, A.; et al. Raxibacumab for the treatment of inhalational anthrax. N. Engl. J. Med. 2009, 361, 135–144. [Google Scholar] [PubMed]
- Centers for Disease Control and Prevention, E-IND Protocol: One Time Emergency Use of Liquid 5% Anthrax Immune Globulin for Treatment of Severe Anthrax.
- Walsh, J.J.; Pesik, N.; Quinn, C.P.; Urdaneta, V.; Dykewicz, C.A.; Boyer, A.E.; Guarner, J.; Wilkins, P.; Norville, K.J.; Barr, J.R.; et al. A case of naturally acquired inhalation anthrax: Clinical care and analyses of anti-protective antigen immunoglobulin G and lethal factor. Clin. Infect. Dis. 2007, 44, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, G.M.; Cronin, P.W.; Poley, G.; Weinstein, A.; Stoughton, S.M.; Zhong, J.; Ou, Y.; Zmuda, J.F.; Osborn, B.L.; Freimuth, W.W. A phase 1 study of PAmAb, a fully human monoclonal antibody against Bacillus anthracis protective antigen, in healthy volunteers. Clin. Infect. Dis. 2005, 41, 12–20. [Google Scholar] [PubMed]
- Altaweel, L.; Chen, Z.; Moayeri, M.; Cui, X.; Li, Y.; Su, J.; Fitz, Y.; Johnson, S.; Leppla, S.H.; Purcell, R.; et al. Delayed treatment with W1-mAb, a chimpanzee-derived monoclonal antibody against protective antigen, reduces mortality from challenges with anthrax edema or lethal toxin in rats and with anthrax spores in mice. Crit. Care Med. 2011, 39, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hicks, C.W.; Cui, X.; Sweeney, D.A.; Li, Y.; Barochia, A.; Eichacker, P.Q. The Potential Contributions of Lethal and Edema Toxins to the Pathogenesis of Anthrax Associated Shock. Toxins 2011, 3, 1185-1202. https://doi.org/10.3390/toxins3091185
Hicks CW, Cui X, Sweeney DA, Li Y, Barochia A, Eichacker PQ. The Potential Contributions of Lethal and Edema Toxins to the Pathogenesis of Anthrax Associated Shock. Toxins. 2011; 3(9):1185-1202. https://doi.org/10.3390/toxins3091185
Chicago/Turabian StyleHicks, Caitlin W., Xizhong Cui, Daniel A. Sweeney, Yan Li, Amisha Barochia, and Peter Q. Eichacker. 2011. "The Potential Contributions of Lethal and Edema Toxins to the Pathogenesis of Anthrax Associated Shock" Toxins 3, no. 9: 1185-1202. https://doi.org/10.3390/toxins3091185
APA StyleHicks, C. W., Cui, X., Sweeney, D. A., Li, Y., Barochia, A., & Eichacker, P. Q. (2011). The Potential Contributions of Lethal and Edema Toxins to the Pathogenesis of Anthrax Associated Shock. Toxins, 3(9), 1185-1202. https://doi.org/10.3390/toxins3091185