Inhibitors of the Cellular Trafficking of Ricin





{kind=link}
{kind=link}
{kind=link}
Abstract
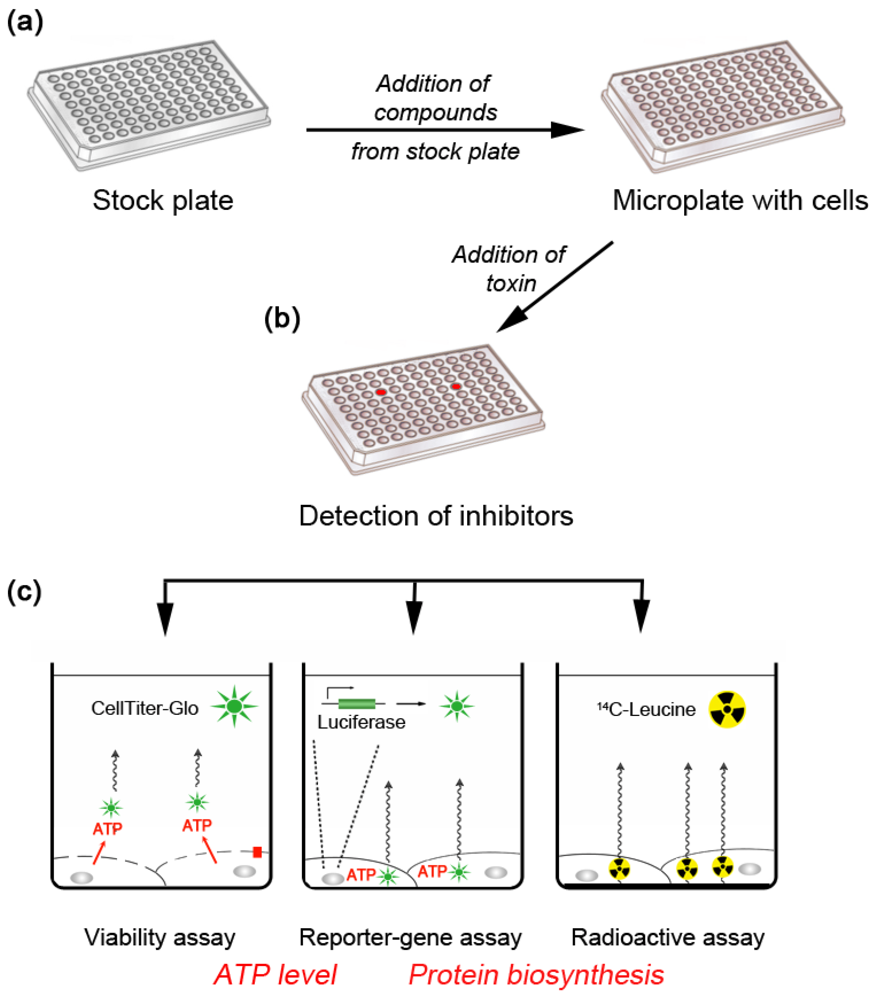
:1. Why Target Cellular Components? A Chemical Genetics Approach

2. Inhibitors of Retrograde Transport Toxins that Act Inside Cells
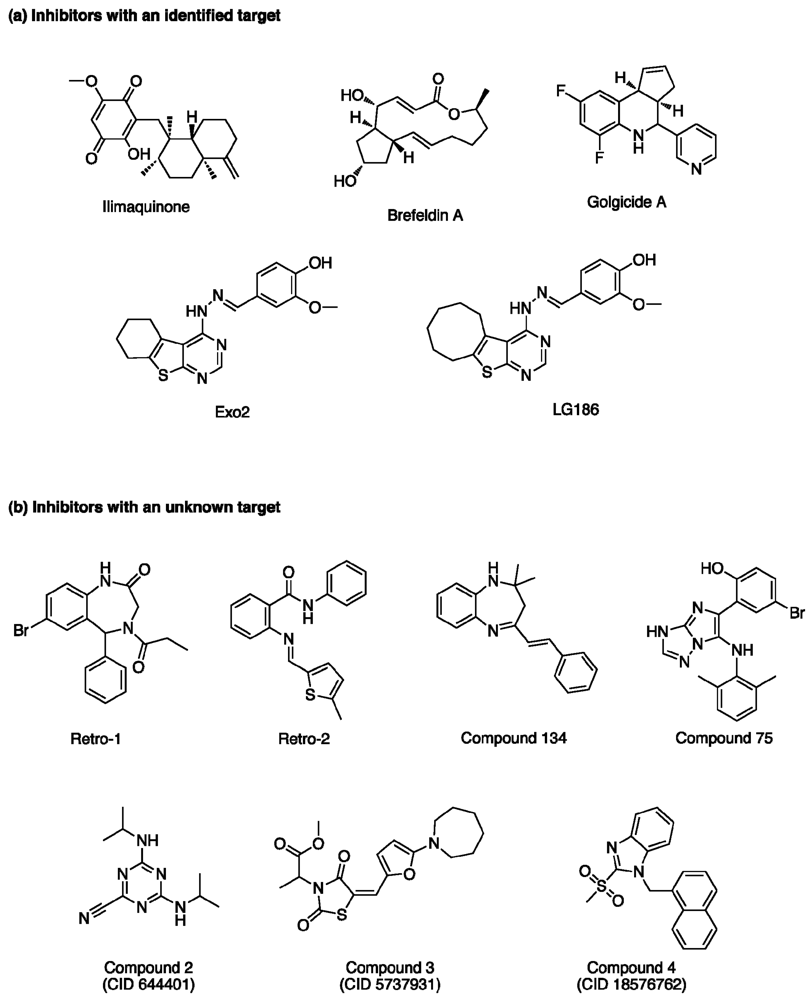
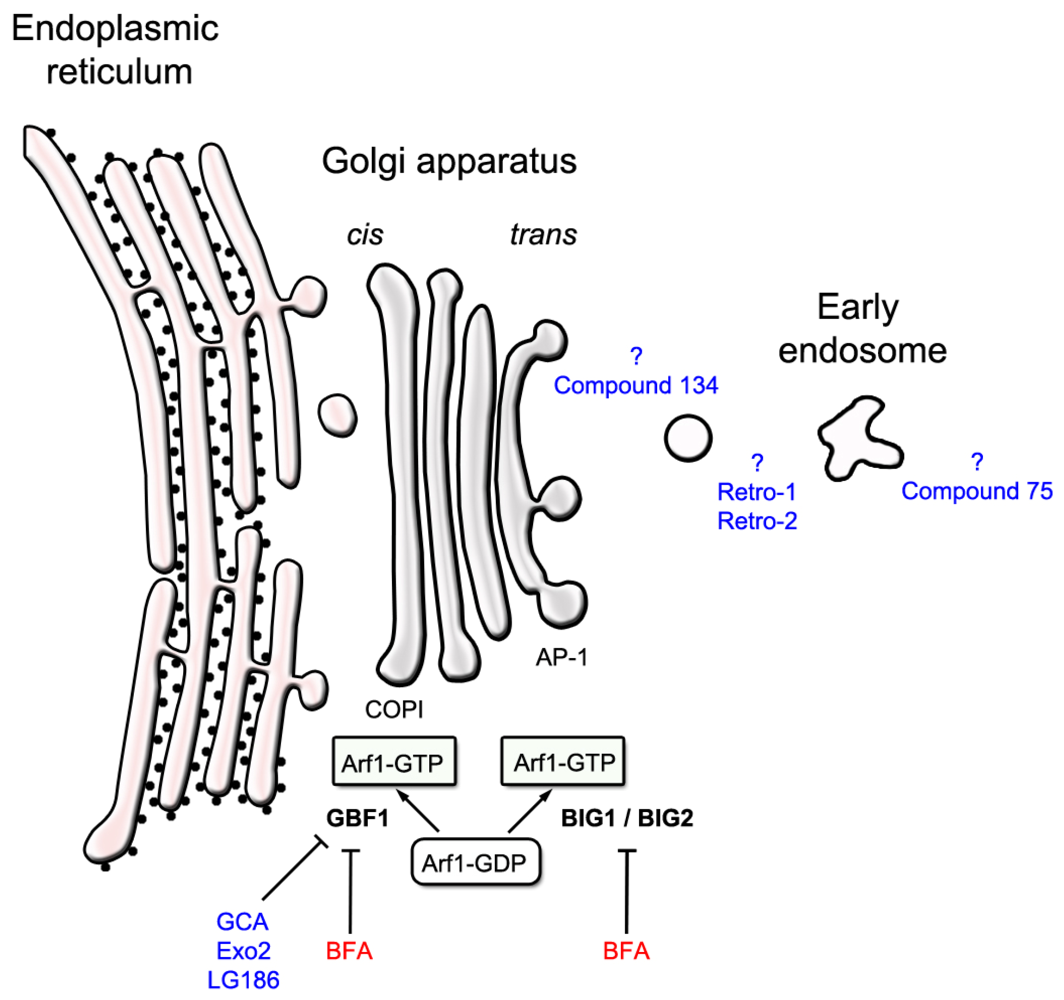
2.1. Compounds that Target an Identified Host Target

2.2. Compounds with Unknown Molecular Targets

3. Identifying Intracellular Protein-Binding Targets of Ricin Inhibitors
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Smallshaw, J.E.; Richardson, J.A.; Vitetta, E.S. RiVax, a recombinant ricin subunit vaccine, protects mice against ricin delivered by gavage or aerosol. Vaccine 2007, 25, 7459–7469. [Google Scholar] [CrossRef] [PubMed]
- Vitetta, E.S.; Smallshaw, J.E.; Coleman, E.; Jafri, H.; Foster, C.; Munford, R.; Schindler, J. A pilot clinical trial of a recombinant ricin vaccine in normal humans. Proc. Natl. Acad. Sci. USA 2006, 103, 2268–2273. [Google Scholar]
- Lemley, P.V.; Amanatides, P.; Wright, D.C. Identification and characterization of a monoclonal antibody that neutralizes ricin toxicity in vitro and in vivo. Hybridoma 1994, 13, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Prigent, J.; Panigai, L.; Lamourette, P.; Sauvaire, D.; Devilliers, K.; Plaisance, M.; Volland, H.; Creminon, C.; Simon, S. Neutralising antibodies against ricin toxin. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Schramm, V.L. Transition state analogues for enzymes of nucleic acid metabolism. Nucleic Acids Res. Suppl. 2003, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Monzingo, A.F.; Robertus, J.D. The X-ray structure of ricin A chain with a novel inhibitor. Arch. Biochem. Biophys. 2009, 483, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.J.; Ravikumar, K.; Shen, H.; Suh, J.K.; Kerwin, S.M.; Robertus, J.D. Structure-based design and characterization of novel platforms for ricin and shiga toxin inhibition. J. Med. Chem. 2002, 45, 90–98. [Google Scholar] [PubMed]
- Yan, X.; Hollis, T.; Svinth, M.; Day, P.; Monzingo, A.F.; Milne, G.W.; Robertus, J.D. Structure-based identification of a ricin inhibitor. J. Mol. Biol. 1997, 266, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Sturm, M.B.; Roday, S.; Schramm, V.L. Circular DNA and DNA/RNA hybrid molecules as scaffolds for ricin inhibitor design. J. Am. Chem. Soc. 2007, 129, 5544–5550. [Google Scholar] [PubMed]
- Fan, S.; Wu, F.; Martiniuk, F.; Hale, M.L.; Ellington, A.D.; Tchou-Wong, K.M. Protective effects of anti-ricin A-chain RNA aptamer against ricin toxicity. World J. Gastroenterol. 2008, 14, 6360–6365. [Google Scholar] [PubMed]
- Hesselberth, J.R.; Miller, D.; Robertus, J.; Ellington, A.D. In vitro selection of RNA molecules that inhibit the activity of ricin A-chain. J. Biol. Chem. 2000, 275, 4937–4942. [Google Scholar] [PubMed]
- Robertus, J.D.; Yan, X.; Ernst, S.; Monzingo, A.; Worley, S.; Day, P.; Hollis, T.; Svinth, M. Structural analysis of ricin and implications for inhibitor design. Toxicon 1996, 34, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Watt, B.; Wahome, P.G.; Mantis, N.J.; Robertus, J.D. Identification of new classes of ricin toxin inhibitors by virtual screening. Toxicon 2011, 56, 526–534. [Google Scholar]
- Wahome, P.G.; Bai, Y.; Neal, L.M.; Robertus, J.D.; Mantis, N.J. Identification of small-molecule inhibitors of ricin and shiga toxin using a cell-based high-throughput screen. Toxicon 2010, 56, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Stechmann, B.; Bai, S.K.; Gobbo, E.; Lopez, R.; Merer, G.; Pinchard, S.; Panigai, L.; Tenza, D.; Raposo, G.; Beaumelle, B.; et al. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 2010, 141, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Saenz, J.B.; Doggett, T.A.; Haslam, D.B. Identification and characterization of small molecules that inhibit intracellular toxin transport. Infect. Immun. 2007, 75, 4552–4561. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Popoff, V. Tracing the retrograde route in protein trafficking. Cell 2008, 135, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.; Spooner, R.A. Toxin entry and trafficking in mammalian cells. Adv. Drug Deliv. Rev. 2006, 58, 1581–1596. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Spilsberg, B.; Lauvrak, S.U.; Torgersen, M.L.; Iversen, T.G.; van Deurs, B. Pathways followed by protein toxins into cells. Int. J. Med. Microbiol. 2004, 293, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.M.; Roberts, L.M.; Lencer, W.I. Entry of protein toxins into mammalian cells by crossing the endoplasmic reticulum membrane: Co-opting basic mechanisms of endoplasmic reticulum-associated degradation. Curr. Top. Microbiol. Immunol. 2005, 300, 149–168. [Google Scholar] [PubMed]
- Zhao, L.; Haslam, D.B. A quantitative and highly sensitive luciferase-based assay for bacterial toxins that inhibit protein synthesis. J. Med. Microbiol. 2005, 54, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, M.P.; Wu, H.C. Ilimaquinone inhibits the cytotoxicities of ricin, diphtheria toxin, and other protein toxins in Vero cells. Exp. Cell Res. 1995, 219, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, P.A.; Yucel, J.K.; Veit, B.; Faulkner, D.J.; Deerinck, T.; Soto, G.; Ellisman, M.; Malhotra, V. Complete vesiculation of Golgi membranes and inhibition of protein transport by a novel sea sponge metabolite, ilimaquinone. Cell 1993, 73, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.H.; Chueh, S.C.; Kung, F.L.; Pan, S.L.; Shen, Y.C.; Guh, J.H. Ilimaquinone, a marine sponge metabolite, displays anticancer activity via GADD153-mediated pathway. Eur. J. Pharmacol. 2007, 556, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Radeke, H.S.; Digits, C.A.; Casaubon, R.L.; Snapper, M.L. Interactions of (-)-ilimaquinone with methylation enzymes: Implications for vesicular-mediated secretion. Chem. Biol. 1999, 6, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Chen, C.C.; Zhang, M.S.; Wu, H.C. Disruption of the Golgi apparatus by brefeldin A inhibits the cytotoxicity of ricin, modeccin, and Pseudomonas toxin. Exp. Cell Res. 1991, 192, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.L.; Scovill, J.P.; Pace, J.G. Drugs that show protective effects from ricin toxicity in in vitro protein synthesis assays. Nat. Toxins 1995, 3, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Donta, S.T.; Tomicic, T.K.; Donohue-Rolfe, A. Inhibition of Shiga-like toxins by brefeldin A. J. Infect. Dis. 1995, 171, 721–724. [Google Scholar] [PubMed]
- Klausner, R.D.; Donaldson, J.G.; Lippincott-Schwartz, J. Brefeldin A: Insights into the control of membrane traffic and organelle structure. J. Cell Biol. 1992, 116, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Prydz, K.; Hansen, S.H.; van Deurs, B. Ricin transport in brefeldin A-treated cells: Correlation between Golgi structure and toxic effect. J. Cell Biol. 1991, 115, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, J.G.; Finazzi, D.; Klausner, R.D. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature 1992, 360, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Peyroche, A.; Antonny, B.; Robineau, S.; Acker, J.; Cherfils, J.; Jackson, C.L. Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex: Involvement of specific residues of the Sec7 domain. Mol. Cell 1999, 3, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Mossessova, E.; Corpina, R.A.; Goldberg, J. Crystal structure of ARF1*Sec7 complexed with Brefeldin A and its implications for the guanine nucleotide exchange mechanism. Mol. Cell 2003, 12, 1403–1411. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, J.G.; Honda, A. Localization and function of Arf family GTPases. Biochem. Soc. Trans. 2005, 33, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, J.G.; Honda, A.; Weigert, R. Multiple activities for Arf1 at the Golgi complex. Biochim. Biophys. Acta 2005, 1744, 364–373. [Google Scholar]
- Bonifacino, J.S.; Glick, B.S. The mechanisms of vesicle budding and fusion. Cell 2004, 116, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.S. Adaptable adaptors for coated vesicles. Trends Cell Biol. 2004, 14, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Niu, T.K.; Pfeifer, A.C.; Lippincott-Schwartz, J.; Jackson, C.L. Dynamics of GBF1, a Brefeldin A-sensitive Arf1 exchange factor at the Golgi. Mol. Biol. Cell 2005, 16, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Saenz, J.B.; Sun, W.J.; Chang, J.W.; Li, J.; Bursulaya, B.; Gray, N.S.; Haslam, D.B. Golgicide A reveals essential roles for GBF1 in Golgi assembly and function. Nat. Chem. Biol. 2009, 5, 157–165. [Google Scholar] [PubMed]
- Feng, Y.; Yu, S.; Lasell, T.K.; Jadhav, A.P.; Macia, E.; Chardin, P.; Melancon, P.; Roth, M.; Mitchison, T.; Kirchhausen, T. Exo1: A new chemical inhibitor of the exocytic pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 6469–6474. [Google Scholar]
- Yarrow, J.C.; Feng, Y.; Perlman, Z.E.; Kirchhausen, T.; Mitchison, T.J. Phenotypic screening of small molecule libraries by high throughput cell imaging. Comb. Chem. High. Throughput Screen. 2003, 6, 279–286. [Google Scholar] [PubMed]
- Feng, Y.; Jadhav, A.P.; Rodighiero, C.; Fujinaga, Y.; Kirchhausen, T.; Lencer, W.I. Retrograde transport of cholera toxin from the plasma membrane to the endoplasmic reticulum requires the trans-Golgi network but not the Golgi apparatus in Exo2-treated cells. EMBO Rep. 2004, 5, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.A.; Watson, P.; Smith, D.C.; Boal, F.; Amessou, M.; Johannes, L.; Clarkson, G.J.; Lord, J.M.; Stephens, D.J.; Roberts, L.M. The secretion inhibitor Exo2 perturbs trafficking of Shiga toxin between endosomes and the trans-Golgi network. Biochem. J. 2008, 414, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Boal, F.; Guetzoyan, L.; Sessions, R.B.; Zeghouf, M.; Spooner, R.A.; Lord, J.M.; Cherfils, J.; Clarkson, G.J.; Roberts, L.M.; Stephens, D.J. LG186: An inhibitor of GBF1 function that causes Golgi disassembly in human and canine cells. Traffic 2010, 11, 1537–1551. [Google Scholar] [CrossRef] [PubMed]
- Guetzoyan, L.J.; Spooner, R.A.; Boal, F.; Stephens, D.J.; Lord, J.M.; Roberts, L.M.; Clarkson, G.J. Fine tuning Exo2, a small molecule inhibitor of secretion and retrograde trafficking pathways in mammalian cells. Mol. Biosyst. 2010, 6, 2030–2038. [Google Scholar] [CrossRef] [PubMed]
- Guetzoyan, L.J.; Spooner, R.A.; Lord, J.M.; Roberts, L.M.; Clarkson, G.J. Simple oxidation of pyrimidinylhydrazones to triazolopyrimidines and their inhibition of Shiga toxin trafficking. Eur. J. Med. Chem. 2010, 45, 275–283. [Google Scholar] [PubMed]
- Mallard, F.; Johannes, L. Shiga toxin B-subunit as a tool to study retrograde transport. Methods Mol. Med. 2003, 73, 209–220. [Google Scholar] [PubMed]
- Ong, S.E.; Schenone, M.; Margolin, A.A.; Li, X.; Do, K.; Doud, M.K.; Mani, D.R.; Kuai, L.; Wang, X.; Wood, J.L.; et al. Identifying the proteins to which small-molecule probes and drugs bind in cells. Proc. Natl. Acad. Sci. USA 2009, 106, 4617–4622. [Google Scholar]
- Kley, N. Chemical dimerizers and three-hybrid systems: Scanning the proteome for targets of organic small molecules. Chem. Biol. 2004, 11, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Becker, F.; Murthi, K.; Smith, C.; Come, J.; Costa-Roldan, N.; Kaufmann, C.; Hanke, U.; Degenhart, C.; Baumann, S.; Wallner, W.; et al. A three-hybrid approach to scanning the proteome for targets of small molecule kinase inhibitors. Chem. Biol. 2004, 11, 211–223. [Google Scholar] [PubMed]
- Lomenick, B.; Olsen, R.W.; Huang, J. Identification of direct protein targets of small molecules. ACS Chem. Biol. 2011, 6, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Maly, D.J.; Choong, I.C.; Ellman, J.A. Combinatorial target-guided ligand assembly: Identification of potent subtype-selective c-Src inhibitors. Proc. Natl. Acad. Sci. USA 2000, 97, 2419–2424. [Google Scholar]
- Sem, D.S.; Bertolaet, B.; Baker, B.; Chang, E.; Costache, A.D.; Coutts, S.; Dong, Q.; Hansen, M.; Hong, V.; Huang, X.; et al. Systems-based design of bi-ligand inhibitors of oxidoreductases: Filling the chemical proteomic toolbox. Chem. Biol. 2004, 11, 185–194. [Google Scholar] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Barbier, J.; Bouclier, C.; Johannes, L.; Gillet, D. Inhibitors of the Cellular Trafficking of Ricin. Toxins 2012, 4, 15-27. https://doi.org/10.3390/toxins4010015
Barbier J, Bouclier C, Johannes L, Gillet D. Inhibitors of the Cellular Trafficking of Ricin. Toxins. 2012; 4(1):15-27. https://doi.org/10.3390/toxins4010015
Chicago/Turabian StyleBarbier, Julien, Céline Bouclier, Ludger Johannes, and Daniel Gillet. 2012. "Inhibitors of the Cellular Trafficking of Ricin" Toxins 4, no. 1: 15-27. https://doi.org/10.3390/toxins4010015
APA StyleBarbier, J., Bouclier, C., Johannes, L., & Gillet, D. (2012). Inhibitors of the Cellular Trafficking of Ricin. Toxins, 4(1), 15-27. https://doi.org/10.3390/toxins4010015