Yessotoxin as a Tool to Study Induction of Multiple Cell Death Pathways

Abstract
:1. Introduction
2. Programmed Cell Death in Disease Models
3. Potential Medical Applications for Yessotoxin


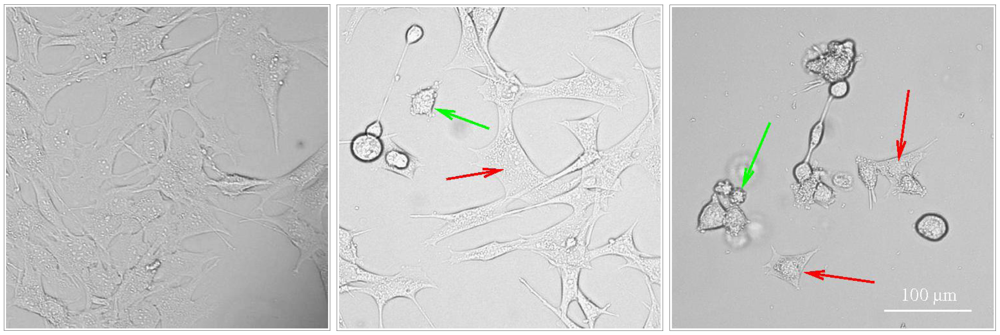
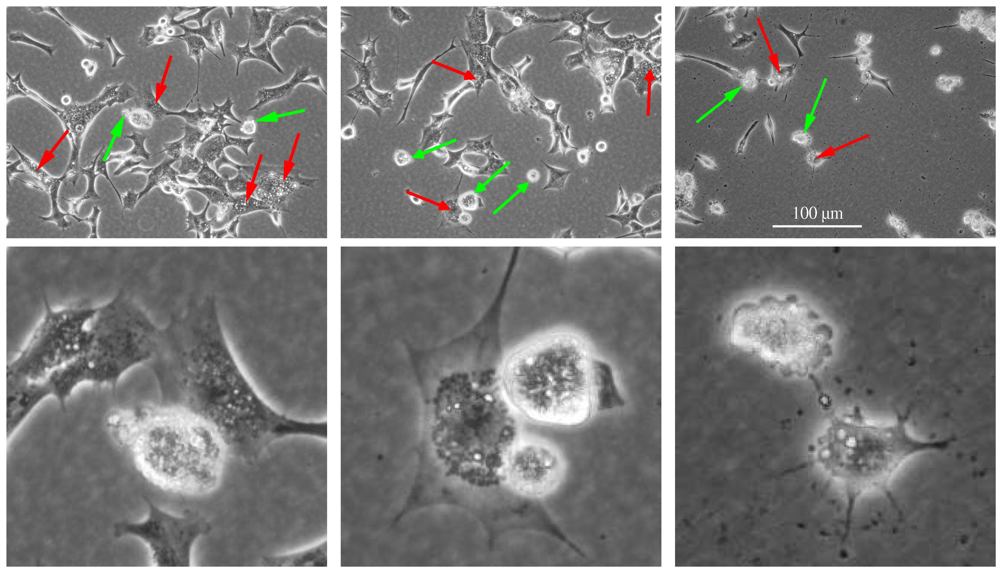
4. Other Examples of Complex Induction of Programmed Cell Death

{kind=link}
{kind=link}
| Reference | Cell type | Insult | Pathway | Mediated by |
|---|---|---|---|---|
| Sperandio et al. [24] | 293T cells, Apaf-1 null mouse embryonic fibroblast | Transfection with the human insulin-like growth factor I receptor (IGFIR) | apoptosis, paraptosis | caspase-9, Apaf-1 independent |
| Kanasaki et al. [51] | Rat Pituitary GH3 Cells | Bromocriptine | apoptosis | p38 |
| Palmeri et al. [52] | Experimental rat pituitary tumours | paraptosis | p38, ERK1/2, PKCδ | |
| Wang et al. [53] | HEK293, HeLa and 293T cells | Transfection with the TAJ/TROY (novel member of the TNFR family) | paraptosis-like | overexpression of programmed cell death 5 (PDCD5) |
| Samadder et al. [54] | MEFs (mouse embryonic fibroblasts) | Glycosylated anti-tumour ether lipids (GAELs) | paraptosis-like | mTOR-independent (mammalian target of rapamycin) |
| Li et al. [55] | HCT116 (colorectal cancer cells) | Ginsenoside RH2 | apoptosis, paraptosis-like | caspase-3 activation and p53 |
| Korsnes et al. [19] | BC3H1 myoblast cell lines | Yessotoxin | apoptosis | caspase-3 activation |
| Korsnes et al. [23] | paraptosis-like | caspase-9 activation, JNK/SAPK1 | ||
| Asare et al. [56] | Hepa1c1c7 cells | 1-Nitropyrene (1-NP) | apoptosis, paraptosis | ERK1/2, p38, JNK |
| Yoon et al. [57] | MDA-MB-231, MDA-MB-435S and Hs578T (breast cancer cells) | Curcumin | paraptosis-like | ERK, JNK |
| Zhang et al. [58] | human colon carcinoma SW620 cells | δ-tocotrienol | paraptosis-like | Suppression of the Wnt signalling pathway. |
5. Concluding Remarks
Acknowledgements
Conflict of Interests
References
- Broker, L.; Kruyt, F.; Giaccone, G. Cell death independent of caspases: A review. Clin. Cancer Res. 2005, 11, 3155–3162. [Google Scholar] [CrossRef]
- Zhivotovsky, B.; Orrenius, S. Cell death mechanisms: Cross-talk and role in disease. Exp. Cell Res. 2010, 316, 1374–1383. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 1, 107–120. [Google Scholar]
- Bredesen, D.E. Programmed cell death mechanisms in neurological disease. Curr. Mol. Med. 2008, 8, 173–86. [Google Scholar] [CrossRef]
- Chipuk, J.; Green, D. Do inducers of apoptosis trigger caspase-independent cell death? Nat. Rev. Mol. Cell Biol. 2005, 6, 268–275. [Google Scholar] [CrossRef]
- Köhler, C.; Orrenius, S.; Zhivotovsky, B. Evaluation of caspase activity in apoptotic cells. J. Immunol. Methods 2002, 265, 97–110. [Google Scholar] [CrossRef]
- Dlamini, Z.; Mbita, Z.; Zungu, M. Genealogy, expression and molecular mechanisms in apoptosis. Pharmacol. Ther. 2004, 10, 1–15. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Y.; Liu, Y.; Cui, Y. Pathways to caspase activation. Cell Biol. Int. 2005, 29, 489–496. [Google Scholar] [CrossRef]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochim. Biophys. Acta 1757, 639–647. [Google Scholar]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Hu, W.; Kavanagh, J. Anticancer therapy targeting the apoptotic pathway. Lancet Oncol. 2003, 4, 721–729. [Google Scholar] [CrossRef]
- Lowe, M.; Lane, J.D.; Woodman, P.G.; Allan, V.J. Caspase-mediated cleavage of syntaxin 5 and giantin accompanies inhibition of secretory traffic during apoptosis. J. Cell. Sci. 2004, 117, 1139–1150. [Google Scholar] [CrossRef]
- Adams, J.; Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007, 26, 1324–1337. [Google Scholar] [CrossRef]
- De la Rosa, L.; Alfonso, A.; Vilarino, N.; Vieytes, M.R.; Yasumoto, T.; Botana, L.M. Maitotoxininduced calcium entry in human lymphocytes: Modulation by yessotoxin, Ca2+ channel blockers and kinases. Cell Signal. 2001, 13, 711–716. [Google Scholar] [CrossRef]
- De la Rosa, L.; Alfonso, A.; Vilarino, N.; Vieytes, M.R.; Yasumoto, T.; Botana, L.M. Modulation of cytosolic calcium levels if human lymphocytes by yessotoxin, a novel marine phycotoxin. Biochem. Pharmacol. 2001, 61, 827–833. [Google Scholar]
- Ronzitti, G.; Callegari, F.; Malagati, C.; Rossini, G.P. Selective disruption of the E-cadherin-catenin system by an algal toxin. Br. J. Cancer 2004, 90, 1100–1107. [Google Scholar] [CrossRef]
- Pérez-Gómez, A.; Ferrerro-Gutiérrez, A.; Novelli, A.; Franco, J.M.; Paz, B.; Fernández-Sánchez, M.T. Potent neurotoxic action of the shellfish biotoxin yessotoxin on cultured cerebellar neurons. Toxicol. Sci. 2006, 90, 168–177. [Google Scholar]
- Korsnes, M.S.; Hetland, D.L.; Espenes, A.; Tranulis, M.A.; Aune, T. Apoptotic events induced by yessotoxin in myoblast cell lines from rat and mouse. Toxicol. Vitro 2006, 20, 1077–1087. [Google Scholar] [CrossRef]
- Korsnes, M.S.; Hetland, D.L.; Espenes, A.; Aune, T. Induction of apoptosis by YTX in myoblast cell lines via mitochondrial signalling transduction pathway. Toxicol. Vitro 2006, 20, 1419–1426. [Google Scholar]
- Korsnes, M.S.; Hetland, D.L.; Espenes, A.; Aune, T. Cleavage of tensin during cytoskeleton disruption in YTX-induced apoptosis. Toxicol. Vitro 2007, 21, 9–15. [Google Scholar] [CrossRef]
- Korsnes, M.S.; Espenes, A. Yessotoxin as an apoptotic inducer. Toxicon 2011, 57, 947–958. [Google Scholar] [CrossRef]
- Korsnes, M.S.; Espenes, A.; Hetland, D.L.; Hermansen, L.C. Paraptosis-like cell death induced by yessotoxin. Toxicol. Vitro 2011, 25, 1764–1770. [Google Scholar] [CrossRef]
- Sperandio, S.; de Belle, I.; Bredesen, D. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 14376–14381. [Google Scholar]
- Okada, H.; Mak, T.W. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar] [CrossRef]
- Brown, J.; Attardi, L. The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 2005, 5, 231–237. [Google Scholar] [CrossRef]
- Ricci, M.; Zong, W. Chemotherapeutic approaches for targeting cell death pathways. Oncologist 2006, 11, 342–357. [Google Scholar] [CrossRef]
- Eskelinen, E.L. Doctor Jekyll and Mister Hyde: Autophagy can promote both cell survival and cell death. Cell Death Differ. 2005, 12, 1468–1472. [Google Scholar] [CrossRef]
- Krantic, S.; Mechawar, N.; Reix, S.; Quirion, R. Apoptosis-inducing factor: A matter of neuron life and death. Prog. Neurobiol. 2007, 81, 179–196. [Google Scholar] [CrossRef]
- Ünal-Çevik, I.; Kılınç, M.; Can, A.; Gürsoy-Özdemir, Y.; Dalkara, T. Apoptotic and necrotic death mechanisms are concomitantly activated in the same cell after cerebral ischemia. Stroke 2004, 35, 2189–2194. [Google Scholar] [CrossRef]
- Boujrad, H.; Gubkina, O.; Robert, N.; Krantic, S.; Susin, S.A. AIF-mediated programmed necrosis: A highly regulated way to die. Cell Cycle 2007, 6, 2612–2619. [Google Scholar] [CrossRef]
- Kitanaka, C.; Kuchino, Y. Caspase-independent programmed cell death with necrotic morphology. Cell Death Differ. 1999, 6, 508–515. [Google Scholar]
- McConkey, D. Biochemical determinants of apoptosis and necrosis. Toxicol. Lett. 1998, 99, 157–168. [Google Scholar] [CrossRef]
- Nicotera, P.; Leist, M.; Fava, E.; Berliocchi, L.; Volbracht, C. Energy requirement for caspase activation and neuronal cell death. Brain Pathol. 2000, 10, 276–282. [Google Scholar]
- Leist, M.; Jaattela, M. Four deaths and a funeral: From caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2001, 2, 589–598. [Google Scholar] [CrossRef]
- Yamashima, T. Implication of cysteine proteases calpain, cathepsin and caspase in ischemic neuronal death of primates. Prog. Neurobiol. 2000, 62, 273–295. [Google Scholar] [CrossRef]
- Foghsgaard, L.; Wissing, D.; Mauch, D.; Lademann, U.; Bastholm, L.; Boes, M.; Elling, F.; Leist, M.; Jäättelä, M. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis attel¨induced by tumor necrosis factor. J. Cell Biol. 2001, 153, 999–1010. [Google Scholar] [CrossRef]
- Hoa, N.; Myers, M.; Douglass, T.; Zhang, J.; Delgado, C.; Driggers, L.; Callahan, L.; VanDeusen, G.; Pham, J.; Bhakta, N.; et al. Molecular mechanisms of paraptosis induction: Implications for a non-genetically modified tumor vaccine. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12, 1509–1518. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Kundu, M.; Thompson, C.B. Macroautophagy versus mitochondrial autophagy: A question of fate? Cell Death Differ. 2005, 12, 1484–1489. [Google Scholar] [CrossRef]
- Pehar, M.; O’Riordan, K.J.; Burns-Cusato, M.; Andrzejewski, M.E.; del Alcazar, C.G.; Burger, C.; Scrable, H.; Puglielli, L. Altered longevity-assurance activity of p53:p44 in the mouse causes memory loss, neurodegeneration and premature death. Aging Cell 2010, 9, 174–190. [Google Scholar] [CrossRef]
- Ankarcrona, M.; Dypbukt, J.; Bonfoco, E.; Zhivotovsky, B.; Orrenius, S.; Lipton, S.; Nicotera, P. Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron 1995, 15, 961–973. [Google Scholar] [CrossRef]
- Portera-Cailliau, C.; Price, D.; Martin, L. Non-NMDA and NMDA receptor-mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: Further evidence for an apoptosis-necrosis continuum. J. Comp. Neurol. 1997, 378, 88–104. [Google Scholar] [CrossRef]
- Leist, M.; Fava, E.; Montecucco, C.; Nicotera, P. Peroxynitrite and nitric oxide donors induce neuronal apoptosis by eliciting autocrine excitotoxicity. Eur. J. Neurosci. 1997, 9, 1488–1498. [Google Scholar] [CrossRef]
- Melino, G.; Bernassola, F.; Knight, R.; Corasaniti, M.; Nistic, G.; Finazzi-Agr, A. S-nitrosylation regulates apoptosis. Nature 1997, 388, 432–433. [Google Scholar]
- Nicotera, P.; Ankarcrona, M.; Bonfoco, E.; Orrenius, S.; Lipton, S.A. Neuronal necrosis and apoptosis: Two distinct events induced by exposure to glutamate or oxidative stress. Adv. Neurol. 1997, 72, 95–101. [Google Scholar]
- López, L.M.B.; Rancano, A.A.; Vieytes, M.R.; Garcia, M.I.L. Therapeutic Use of Yessotoxins as Human Tumor Cell Growth Inhibitors. EPO Patent EP1875906.
- López, L.M.B.; López, E.A.; Gonzalez, C.V. Use of Yessotoxin and Analogues and Derivatives Thereof for Treating and/or Preserving Neurodegenerative Diseases Linked to Tau and Beta Amyloid. European Patent Application PCT/ES2011/070078, 2011. [Google Scholar]
- Nicotera, P.; Leist, M.; Manzo, L. Neuronal cell death: A demise with different shapes. Trends Pharmacol. Sci. 1999, 20, 46–51. [Google Scholar] [CrossRef]
- Kanasaki, H.; Fukunaga, K.; Takahashi, K.; Miyazaki, K.; Miyamoto, E. Involvement of p38 mitogen-activated protein kinase activation in bromocriptine-induced apoptosis in rat pituitary GH3 cells. Biol. Reprod. 2000, 62, 1486–1494. [Google Scholar] [CrossRef]
- Palmeri, C.; Petiti, J.; Sosa, L.D.V.; Guti´errez, S.; Paul, A.D.; Mukdsi, J.; Torres, A. Bromocriptine induces parapoptosis as the main type of cell death responsible for experimental pituitary tumor shrinkage. Toxicol. Appl. Pharmacol. 2009, 240, 55–65. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, N.; Wang, L.; Ding, P.; Zhang, D.; Han, W.; Ma, D. An alternative form of paraptosis-like cell death, triggered by TAJ/TROY and enhanced by PDCD5 overexpression. J. Cell. Sci. 2004, 117, 1525–1532. [Google Scholar] [CrossRef]
- Samadder, P.; Bittman, R.; Byun, H.; Arthur, G. A glycosylated antitumor ether lipid kills cells via paraptosis-like cell death. Biochem. Cell. Biol. 2009, 87, 401–414. [Google Scholar] [CrossRef]
- Li, B.; Zhao, J.; Wang, C.Z.; Searle, J.; He, T.C.; Yuan, C.S.; Du, W. Ginsenoside Rh2 induces apoptosis and paraptosis-like cell death in colorectal cancer cells through activation of p53. Cancer Lett. 2011, 301, 185–192. [Google Scholar] [CrossRef]
- Asare, N.; Landvik, N.; Lagadic-Gossmann, D.; Rissel, M.; Tekpli, X.; Ask, K.; Lag, M.; Holme, J. 1-Nitropyrene (1-NP) induces apoptosis and apparently a non-apoptotic programmed cell death (paraptosis) in Hepa1c1c7 cells. Toxicol. Appl. Pharmacol. 2008, 230, 175–186. [Google Scholar] [CrossRef]
- Yoon, M.J.; Kim, E.H.; Lim, J.H.; Kwon, T.K.; Choi, K.S. Superoxide anion and proteasomal dysfunction contribute to curcumin-induced paraptosis of malignant breast cancer cells. Free Radic. Biol. Medi. 2010, 48, 713–726. [Google Scholar] [CrossRef]
- Zhang, J.S.; Li, D.M.; He, N.; Liu, Y.H.; Wang, C.H.; Jiang, S.Q.; Chen, B.Q.; Liu, J.R. A paraptosis-like cell death induced by Î-tocotrienol in human colon carcinoma SW620 cells is associated with the suppression of the Wnt signaling pathway. Toxicology 2011, 285, 8–17. [Google Scholar] [CrossRef]
- Bursch, W.; Hochegger, K.; Torok, L.; Marian, B.; Ellinger, A.; Hermann, R. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J. Cell. Sci. 2000, 113, 1189–1198. [Google Scholar]
- Chi, S.; Kitanaka, C.; Noguchi, K.; Mochizuki, T.; Nagashima, Y.; Shirouzu, M.; Fujita, H.; Yoshida, M.; Chen, W.; Asai, A.; et al. Oncogenic Ras triggers cell suicide through the activation of a caspase-independent cell death program in human cancer cells. Oncogene 1999, 18, 2281–2290. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Korsnes, M.S. Yessotoxin as a Tool to Study Induction of Multiple Cell Death Pathways. Toxins 2012, 4, 568-579. https://doi.org/10.3390/toxins4070568
Korsnes MS. Yessotoxin as a Tool to Study Induction of Multiple Cell Death Pathways. Toxins. 2012; 4(7):568-579. https://doi.org/10.3390/toxins4070568
Chicago/Turabian StyleKorsnes, Mónica Suárez. 2012. "Yessotoxin as a Tool to Study Induction of Multiple Cell Death Pathways" Toxins 4, no. 7: 568-579. https://doi.org/10.3390/toxins4070568
APA StyleKorsnes, M. S. (2012). Yessotoxin as a Tool to Study Induction of Multiple Cell Death Pathways. Toxins, 4(7), 568-579. https://doi.org/10.3390/toxins4070568