Cytotoxic Necrotizing Factor 1 Contributes to Escherichia coli Meningitis

Abstract
:1. Introduction
2. E. coli Penetration of the Blood-Brain Barrier
3. Cytotoxic Necrotizing Factor 1 (CNF1) Contributes to E. coli Invasion of HBMEC and Penetration into the Brain

4. Identification of the Host Receptor for CNF1
5. Secretion of CNF1 across the Bacterial Inner and Outer Membrane
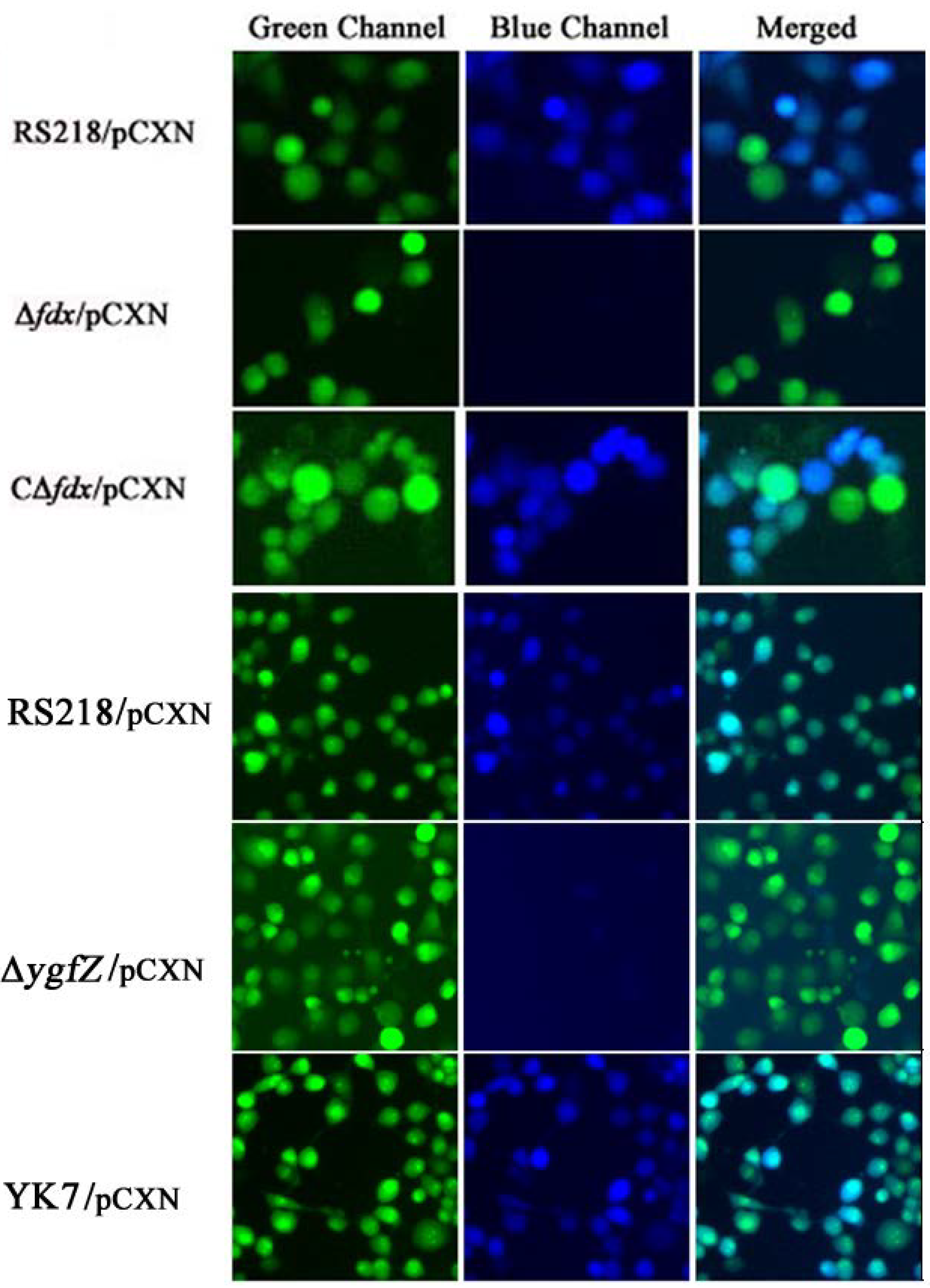
6. Identification of Tn5 Mutants Defective in CNF1 Secretion



{kind=link}
{kind=link}
{kind=link}
{kind=link}
7. Modulation of Host Receptor and Signaling Molecules for Prevention of E. coli Invasion of the Blood-Brain Barrier
8. Conclusion
Acknowledgements
Conflicts of Interest
References
- Gladstone, I.M.; Ehrenkranz, R.A.; Edberg, S.C.; Baltimore, R.S. A ten-year review of neonatal sepsis and comparison with the previous fifty-year experience. Pediatr. Infect. Dis. J. 1990, 9, 819–825. [Google Scholar] [CrossRef]
- Unhanand, M.; Musatafa, M.M.; McCracken, G.H.; Nelson, J.D. Gram-negative enteric bacillary meningitis: A twenty-one year experience. J. Pediatr. 1993, 122, 15–21. [Google Scholar] [CrossRef]
- Dawson, K.G.; Emerson, J.C.; Burns, J.L. Fifteen years of experience with bacterial meningitis. Pediatr. Infect. Dis. J. 1999, 18, 816–822. [Google Scholar] [CrossRef]
- Klinger, G.; Chin, C.-N.; Beyene, J.; Perlman, M. Predicting the outcome of neonatal bacterial meningitis. Pediatrics 2000, 106, 477–482. [Google Scholar] [CrossRef]
- Stevens, J.P.; Eames, M.; Kent, A.; Halket, S.; Holt, D.; Harvey, D. Long term outcome of neonatal meningitis. Arch. Dis. Child. Fetal Neonatal Ed. 2003, 88, F179–F184. [Google Scholar] [CrossRef]
- Kim, K.S. E. coli translocation at the blood-brain barrier. Infect. Immun. 2001, 69, 5217–5222. [Google Scholar] [CrossRef]
- Kim, K.S. Strategy of E. coli for crossing the blood-brain barrier. J. Infect. Dis. 2002, 186, S220–S224. [Google Scholar] [CrossRef]
- Kim, K.S. Neurological diseases: Pathogenesis of bacterial meningitis: From bacteremia to neuronal injury. Nat. Rev. Neurosci. 2003, 4, 376–385. [Google Scholar]
- Kim, K.S. Mechanisms of microbial traversal of the blood-brain barrier. Nat. Rev. Microbiol. 2008, 6, 625–634. [Google Scholar] [CrossRef]
- Kim, K.S. Acute bacterial meningitis in infants and children. Lancet Infect. Dis. 2010, 10, 32–42. [Google Scholar] [CrossRef]
- Kim, K.S. Current concepts on the pathogenesis of E. coli meningitis: Implications for prevention and therapy. Curr. Opin. Infect. Dis. 2012, 25, 273–278. [Google Scholar] [CrossRef]
- McCracken, G.H., Jr.; Threlkeld, N.; Mize, S.; Baker, C.J.; Kapal, S.L.; Fraingezicht, I.; Feldman, W.F.; Schad, U.; The Neonatal Meningitis Cooperative Study Group. Moxalactam therapy for neonatal meningitis due to gram-negative sepsis enteric bacilli. JAMA 1984, 252, 1427–1432. [Google Scholar] [CrossRef]
- Kim, K.S. Comparison of cefotaxime, imipenem-cilastatin, ampicillin-gentamicin and ampicillin-chloramphenicol in the treatment of experimental E. coli bacteremia and meningitis. Antimicrob. Agents Chemother. 1985, 28, 433–436. [Google Scholar] [CrossRef]
- Blanco, J.; Mora, A.; Mamani, R.; Lopez, C.; Blanco, M.; Dabhi, G.; Herrea, A.; Blanco, J.E.; Alonso, M.P.; Garcia-Garrote, F.; et al. National survey of Escherichia coli causing extraintestinal infections reveal the spread of drug-resistant clonal groups O25b:H4-B2-ST131, O15:H5-D-ST393 and CGA-D-ST69 with high virulence gene content in Spain. J. Antimicrob. Chemother. 2011, 66, 2011–2012. [Google Scholar] [CrossRef]
- Moissenet, D.; Slauze, B.; Clermont, O.; Bingen, E.; Arlet, G.; Denamur, E.; Merens, A.; Mitanchez, D.; Vu-Thien, H. Meningitis caused by Escherichia coli producing TEM-52 extended-spectrum β-lactamase within an extensive outbreak in a neonatal ward: epidemiological investigation and characterization of the strain. J. Clin. Microbiol. 2011, 48, 2459–2463. [Google Scholar]
- Dietzman, D.E.; Fischer, G.W.; Schoenknecht, F.D. Neonatal Escherichia coli septicemia—Bacterial counts in blood. J. Pediatr. 1974, 85, 128–130. [Google Scholar] [CrossRef]
- Berman, P.H.; Banker, B.Q. Neonatal meningitis: A clinical and pathologic study of 29 cases. Pediatrics 1966, 38, 6–24. [Google Scholar]
- Kim, K.S.; Itabashi, H.; Gemski, P.; Sadoff, J.; Warren, R.L.; Cross, A.S. The K1 capsule is the critical determinant in the development of Escherichia coli meningitis in the rat. J. Clin. Invest. 1992, 90, 897–905. [Google Scholar] [CrossRef]
- Stins, M.F.; Gilles, F.; Kim, K.S. Selective expression of adhesion molecules on human brain microvascular endothelial cells. J. Neuroimmunol. 1997, 76, 81–90. [Google Scholar] [CrossRef]
- Stins, M.F.; Badger, J.L.; Kim, K.S. Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells. Microb. Pathog. 2001, 30, 19–28. [Google Scholar] [CrossRef]
- Kim, K.S.; Wass, C.A.; Cross, A.S. Blood-brain barrier permeability during the development of experimental bacterial meningitis in the rat. Exp. Neurol. 1997, 145, 253–257. [Google Scholar] [CrossRef]
- Boquet, P. The cytotoxic necrotizing factor 1 (CNF1) from Escherichia coli. Toxicon 2001, 39, 1673–1680. [Google Scholar] [CrossRef]
- Badger, J.; Wass, C.; Weissman, S.; Kim, K.S. Application of signature-tagged mutagenesis for the identification of E. coli K1 genes that contribute to invasion of the blood-brain barrier. Infect. Immun. 2000, 68, 5056–5061. [Google Scholar] [CrossRef]
- Khan, N.A.; Wang, Y.; Kim, K.J.; Chung, J.W.; Wass, C.A.; Kim, K.S. Cytotoxic necrotizing factor 1 contributes to Escherichia coli K1 invasion of the central nervous system. J. Biol. Chem. 2002, 277, 15607–15612. [Google Scholar]
- Khan, N.A.; Shin, S.; Chung, J.W.; Kim, K.J.; Elliot, S.; Wang, Y.; Kim, K.S. Outer membrane protein A and cytotoxic necrotizing factor-1 use diverse signaling mechanisms for Escherichia coli K1 invasion of human brain microvascular endothelial cells. Microb. Pathog. 2003, 35, 35–42. [Google Scholar] [CrossRef]
- Fiorentini, C.; Fabbri, A.; Flatau, G.; Donelli, G.; Matarrese, P.; Lemichez, E.; Falzano, L.; Boquet, P. Escherichia coli cytotoxic necrotizing factor 1 (CNF1), a toxin that activates the Rho GTPase. J. Biol. Chem. 1997, 272, 19532–19537. [Google Scholar] [CrossRef]
- Fabbri, A.; Falzano, L.; Travaglione, S.; Stringaro, A.; Malorni, W.; Fais, S.; Fiorentini, C. Rho-activating Escherichia coli cytotoxic necrotizing factor 1: Macropinocytosis of apoptotic bodies in human epithelial cells. Int. J. Med. Microbiol. 2002, 291, 551–554. [Google Scholar]
- Doye, A.; Mettouchi, A.; Bossis, G.; Clément, R.; Buisson-Touati, C.; Flatau, G.; Gagnoux, L.; Piechaczyk, M.; Boquet, P.; Lemichez, E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 2002, 111, 553–564. [Google Scholar]
- Visvikis, O.; Boyer, L.; Torrino, S.; Doye, A.; Lemonnier, M.; Lorès, P.; Rolando, M.; Flatau, G.; Mettouchi, A.; Bouvard, D.; et al. Escherichia coli producing CNF1 toxin hijacks Tollip to trigger Rac1-dependent cell invasion. Traffic 2011, 12, 579–590. [Google Scholar] [CrossRef]
- Maruvada, R.; Kim, K.S. IbeA and OmpA of Escherichia coli K1 exploit Rac1 activation for invasion of human brain microvascular endothelial cells. Infect. Immun. 2012, 80, 2035–2041. [Google Scholar] [CrossRef]
- Fabbri, A.; Travaglione, S.; Ballan, G.; Loizzo, S.; Fiorentini, C. The cytotoxic necrotizing factor 1 from E. coli: A janus toxin playing with cancer regulators. Toxins 2013, 5, 1462–1474. [Google Scholar] [CrossRef]
- Stallmach, A.; Orzechowski, H.D.; Feldmann, P.; Riecken, E.O.; Zeitz, M.; Herbst, H. 32/67-kD laminin receptor expression in human colonic neoplasia: elevated transcript levels correlate with the degree of epithelial dysplasia. Am. J. Gastroenterol. 1999, 94, 3341–3347. [Google Scholar] [CrossRef]
- Chung, J.W.; Hong, S.J.; Kim, K.J.; Goti, D.; Stins, M.F.; Shin, S.; Dawson, V.L.; Dawson, T.M.; Kim, K.S. 37 kDa laminin receptor precursor modulates cytotoxic necrotizing factor 1-mediated RhoA activation and bacterial uptake. J. Biol. Chem. 2003, 278, 16857–16862. [Google Scholar] [CrossRef]
- Flatau, G.; Lemichez, E.; Gauthier, M.; Chardin, P.; Paris, S.; Fiorentini, C.; Boquet, P. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature 1997, 387, 729–733. [Google Scholar] [CrossRef]
- Schmidt, G.; Sehr, P.; Wilm, M.; Selzer, J.; Mann, M.; Aktories, K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-I. Nature 1997, 387, 725–729. [Google Scholar] [CrossRef]
- Massia, S.P.; Rao, S.S.; Hubbell, J.A. Covalently immobilized laminin peptide Tyr-Ile-Gly-Ser-Arg (YIGSR) supports cell spreading and co-localization of the 67-kilodalton laminin receptor with alpha-actinin and vinculin. J. Biol. Chem. 1993, 268, 8053–8059. [Google Scholar]
- Kim, K.J.; Chung, J.W.; Kim, K.S. 67-kDa Laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J. Biol. Chem. 2005, 280, 1360–1368. [Google Scholar]
- Davis, J.M.; Carvalho, H.M.; Rasmussen, S.B.; O’Brien, A.D. Cytotoxic necrotizing factor type 1 delivered by outer membrane vesicles of uropathogenic Escherichia coli attenuates polymorphonuclear leukocyte antimicrobial activity and chemotaxis. Infect. Immun. 2006, 74, 4401–4408. [Google Scholar] [CrossRef]
- Kouokam, J.C.; Wai, S.N.; Fällman, M.; Dobrindt, U.; Hacker, J.; Uhlin, B.E. Active cytotoxic necrotizing factor 1 associated with outer membrane vesicles from uropathogenic Escherichia coli. Infect. Immun. 2006, 74, 2022–2030. [Google Scholar] [CrossRef]
- Yu, H.; Kim, K.S. Ferredoxin is involved in secretion of cytotoxic necrotizing factor 1 across the cytoplasmic membrane in Escherichia coli K1. Infect. Immun. 2010, 78, 838–844. [Google Scholar] [CrossRef]
- Yu, H.; Kim, K.S. YgfZ contributes to secretion of cytotoxic necrotizing factor 1 into outer membrane vesicles in Escherichia coli. Microbiology 2012, 158, 612–621. [Google Scholar] [CrossRef]
- Zhu, L.; Pearce, D.; Kim, K.S. Prevention of E. coli K1 penetration of the blood-brain barrier by counteracting host cell receptor and signaling molecule involved in E. coli invasion of human brain microvascular endothelial cells. Infect. Immun. 2010, 78, 3554–3559. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, M.-H.; Kim, K.S. Cytotoxic Necrotizing Factor 1 Contributes to Escherichia coli Meningitis. Toxins 2013, 5, 2270-2280. https://doi.org/10.3390/toxins5112270
Wang M-H, Kim KS. Cytotoxic Necrotizing Factor 1 Contributes to Escherichia coli Meningitis. Toxins. 2013; 5(11):2270-2280. https://doi.org/10.3390/toxins5112270
Chicago/Turabian StyleWang, Ming-Hsien, and Kwang Sik Kim. 2013. "Cytotoxic Necrotizing Factor 1 Contributes to Escherichia coli Meningitis" Toxins 5, no. 11: 2270-2280. https://doi.org/10.3390/toxins5112270
APA StyleWang, M. -H., & Kim, K. S. (2013). Cytotoxic Necrotizing Factor 1 Contributes to Escherichia coli Meningitis. Toxins, 5(11), 2270-2280. https://doi.org/10.3390/toxins5112270