Venom Peptides as a Rich Source of Cav2.2 Channel Blockers



Abstract
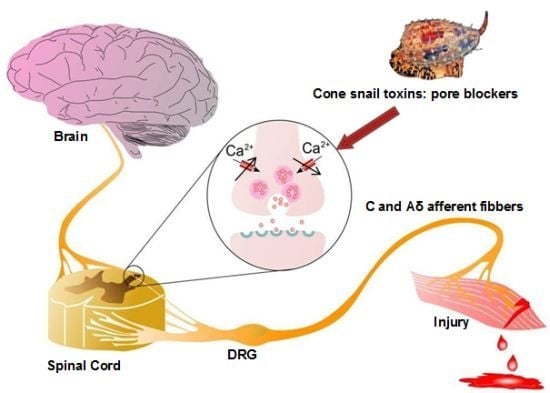
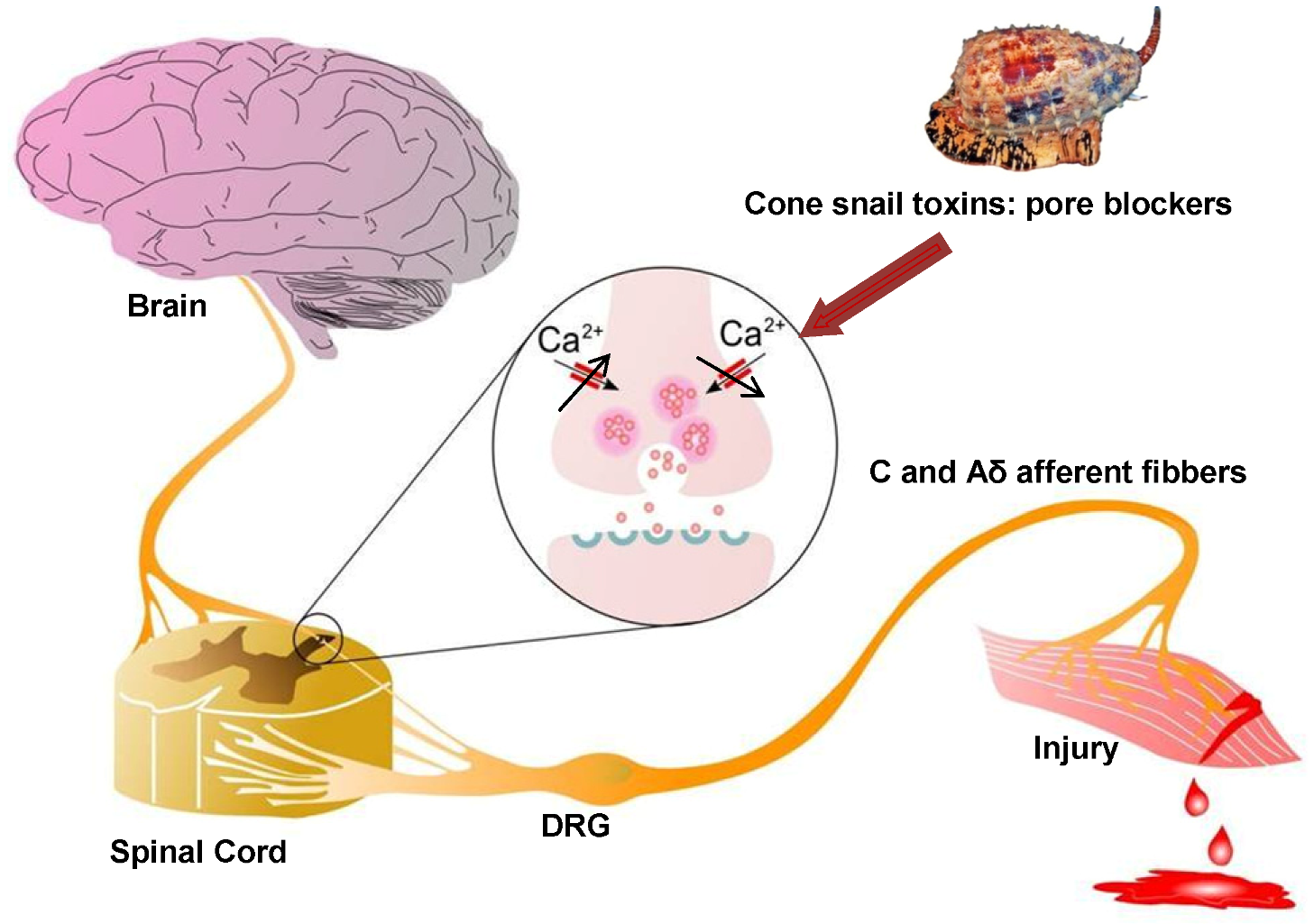
:
1. Introduction
2. Cav Channels

{kind=link}
{kind=link}
{kind=link}
{kind=link}
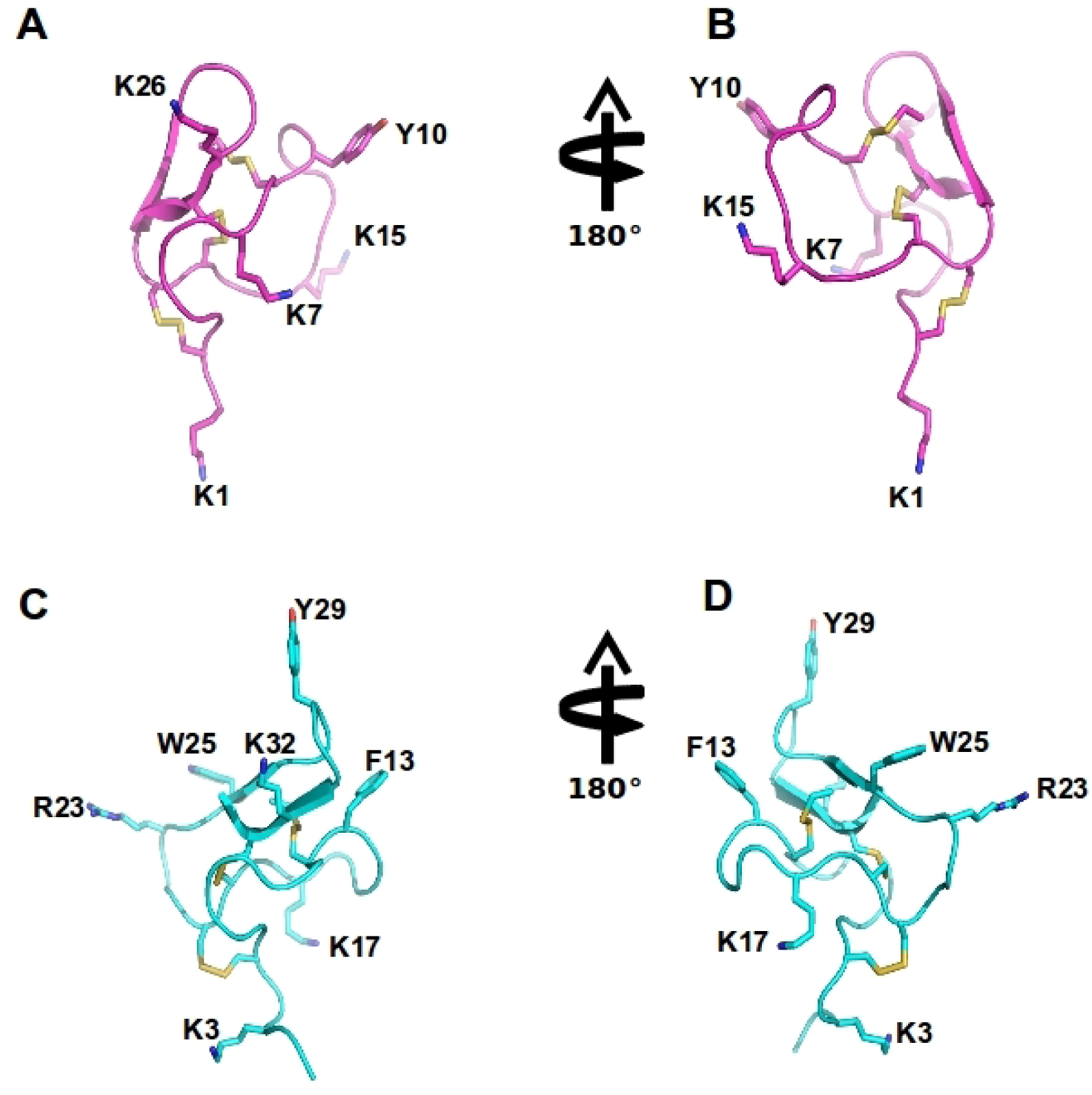{kind=link}
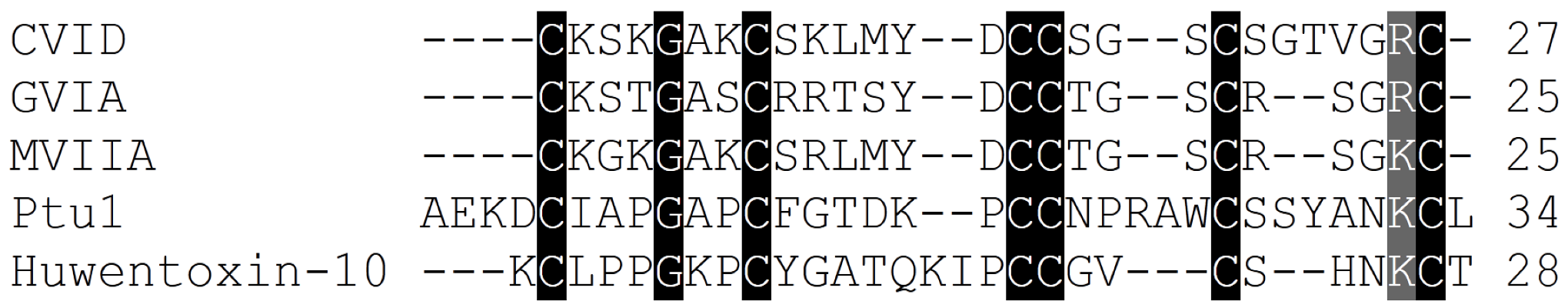{kind=link}
| Cav subtype | Current type | Localization | Antagonist class/Name | Physiological function |
|---|---|---|---|---|
| Cav1.1 | L | Skeletal muscle, transverse tubules | DHP, PHA, BTZ | Excitation-contraction coupling, gene regulation |
| Cav1.2 | L | Cardiac myocytes, smooth muscle myocytes, endocrine cells, neuronal cell bodies, proximal dendrites | DHP, PHA, BTZ | Excitation-contraction coupling, hormone secretion, gene regulation |
| Cav1.3 | L | Endocrine cells, neuronal cell bodies and dendrites, cardiac atrial myocytes and pacemarker cells, cochlear hair cells | DHP, PHA, BTZ | Hormone secretion, gene regulation, tonic transmitter release |
| Cav1.4 | L | Retinal rod and bipolar cells, spinal cord, adrenal gland, mast cells | DHP, PHA, BTZ | Tonic neurotransmitter release |
| Cav2.1 | P/Q | Nerve terminals and dendrites, neuroendocrine cells | ω-agatoxin IVA | Neurotransmitter release, dendritic Ca2+ transient currents |
| Cav2.2 | N | Nerve terminals and dendrites, neuroendocrine cells | ω-conotoxin CVID, GVIA MVIIA | Neurotransmitter release, Ca2+-dependent action potentials |
| Cav2.3 | R | Neuronal cell bodies and dendrites | ω-theraphotoxin-Hg1a(SNX-482) | NeurotransmitterRelease |
| Cav3.1 | T | Neuronal cell bodies and dendrites, cerebellum and thalamus, cardiac and smooth muscles | Pimozide, mibefradil,TTA-P2, Ni2+, Zn2+ | Pacemaking, repetitive firing |
| Cav3.2 | T | CNS: neuronal cell bodiesand dendrites, heart, liver, kidney, lung, skeletal muscle, pancreas | Kurtoxin, pimopzide, mibefradil, Z123212, TTA-P2, Ni2+, Zn2+ | Pacemaking, repetitive firing |
| Cav3.3 | T | CNS: neuronal cell bodies and dendrites | Pimozide, TTA-P2, Zn2+ Ni2+, mibefradil | Pacemaking, repetitive firing |

2.1. Cav2.2 Channels
2.2. Cav2.2 Splice Variants
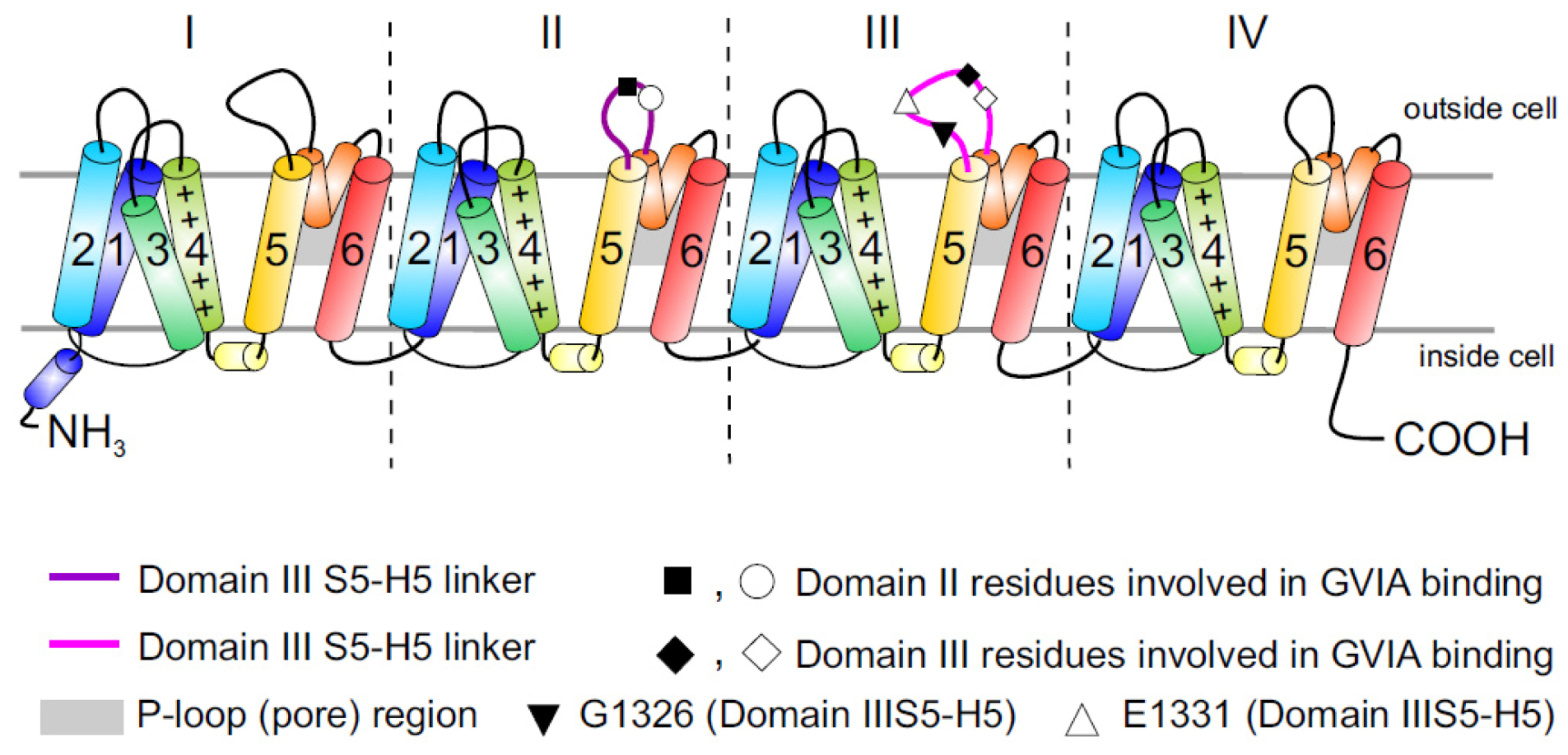
2.3. Cav2.2 Toxin Binding Sites
2.4. Auxiliary Subunits of Cav Channels
2.4.1. α2δ Subunit
2.4.2. β Subunit
2.4.3. γ Subunit
2.5. Cav2.2 Channels as Analgesic Target

2.6. Venoms as a Rich Source of Cav2.2 Channel Blockers
| ω-conotoxin name | ω-conotoxin Sequence | 125I-Ctx binding assays to rat brain IC50/Kd (nM) | Reference |
|---|---|---|---|
| CnVIIA | CKGKGAOCTRLMYDCCHGSCSSSKGRC* | 0.4 (2.2 > 2.1) | [82] |
| CVIA | CKSTGASCRRTSYDCCTGSCRSGRC | 0.6 (2.2 > 1.2) | [8] |
| CVIB | CKGKGASCRKTMYDCCRGSCRSGRC | 7.7 (2.2~2.1 > 2.3) | [8] |
| CVIC | CKGKGQSCSKLMYDCCTGSC-SRRGKC | 7.6 (2.1~2.2) | [8] |
| CVID | CKSKGAKCSKLMYDCCSGSCSGTVGRC | 0.07 (2.2 > 2.1) | [8] |
| CVIE | CKGKGASCRRTSYDCCTGSCRSGRC | 0.025 (2.2 > 2.1 > 1.2~1.3~2.3 | [16] |
| CVIF | CKGKGASCRRTSYDCCTGSCRLGRC | 0.098 (2.2 > 2.1 > 1.2~1.3~2.3) | [16] |
| FVIA | CKGTGKSCSRIAYNCCTGSCRSGKC | ND (2.2 > 2.1 > 3.2) | [83] |
| GVIA | CKSOGSSCSOTSYNCCRSCNOYTKRCY* | 0.04 (2.2 > 2.1) | [84,85,86,87,88] |
| GVIB | CKSOGSSCSOTSYNCCR-SCNOYTKRCYG* | ND | [88,89] |
| GVIIA | CKSOGTOCSRGMRDCCTSCLLYSNKCRRY* | 3.7 (ND) | [88,89,90] |
| GVIIB | CKSOGTOCSRGMRDCCTSCLSYSNKCRRY* | ND | [88] |
| MVIIA | CKGKGAKCSRLMYDCCTGSCRSGKC | 0.055 (2.2 > 2.1) | [13,81,87] |
| RVIA | CKPPGSPCRVSSYNCCSSCKSYNKKCG | 0.25 (2.2) | [10] |
| TVIA | CLSXGSSCSXTSYNCCRSCNXYSRKCR | ND (2.2 > 2.1) | [91,92] |

2.6.1. Cone Snail Venom Peptide Cav2.2 Inhibitors for the Treatment of Pain
2.6.2. Effect of Selectivity on Side Effect Profile
2.7. Cav2.2 Inhibitor Toxins from Spiders
| Toxin name/Synonym | Functional (IC50)/Binding (Kd) at Cav2.2 | Amino acid sequence | Reference |
|---|---|---|---|
| μ/ω-theraphotoxin-Hh1a/Huwentoxin-1 | 100 nM (ND) | ACKGVFGACTPGKNECCPNRVCSDKHKWCKWKL | [110,111] |
| μ/ω-theraphotoxin-Hh1b/Huwentoxin1a3 | (ND) | ACKGVFGACTPGKNECCPNRVCSDKHKWCKWKL | [112] |
| μ/ω-theraphotoxin Hh1c/Huwentoxin1a10 | (ND) | ACKGVFDACTPGKNECCSNRVCSDKHKWCKWKL | [112,113] |
| μ/ω-theraphotoxin-Hh1d/Huwentoxin-1a6 | (ND) | ACKGVFDACTPGKNECCPNRVCSDEHKWCKWKL | [112] |
| ω-agatoxin-Aa2a/ω-agatoxin IIA | 10 nM (Y) | GCIEIGGDCDGYQEKSYCQCCRNNGFCS | [114,115] |
| ω-agatoxin-Aa3a/ω-agatoxin IIIA | 1.4 nM/170 pM (N) | SCIDIGGDCDGEKDDCQCCRRNGYCSCYSLFGYLKSGCKCVVGTSAEFQGICRRKARQCYNSDPDKCESHNKPKRR | [116,133] |
| ω-agatoxin-Aa3b/ω-agatoxin IIIB | 140 nM/2.4 nM (N) | SCIDFGGDCDGEKDDCQCCRSNGYCSCYNLFGYLKSGCKCEVGTSAEFRRICRRKAKQCYNSDPDKCVSVYKPKRR | [116,117] |
| ω-agatoxin-Aa3d/ω-agatoxin IIID | 35 nM (N) | SCIKIGEDCDGDKDDCQCCRTNGYCSXYXLFGYLKSG | [116] |
| ω-agatoxin-Aa3f/ω-agatoxin IIIA (58T) | 1.4 nM (N) | SCIDIGGDCDGEKDDCQCCRRNGYCSCYSLFGYLKSGCKCVVGTSAEFQGICRRKARTCYNSDPDKCESHNKPKRR | [116] |
| ω-agatoxin-Aa3g/ω-agatoxin IIIB (35R) | 2.4 nM (N) | SCIDFGGDCDGEKDDCQCCRSNGYCSCYNLFGYLRSGCKCEVGTSAEFRRICRRKAKQCYNSDPDKCVSVYKPKRR | [116] |
| ω-agatoxin-Aa3h/ω-agatoxin IIIB (29S) | 2.4 nM (N) | SCIDFGGDCDGEKDDCQCCRSNGYCSCYSLFGYLKSGCKCEVGTSAEFRRICRRKAKQCYNSDPDKCVSVYKPKRR | [116] |
| ω-ctenitoxin-Pn2a/Neurotoxin Tx3–3 | >320nM/50 pM (N) | GCANAYKSCNGPHTCCWGYNGYKKACICSGXNWK | [7,118,119] |
| ω-ctenitoxin-Pn3a/Neurotoxin Tx3–4 | 50 pM (N) | SCINVGDFCDGKKDDCQCCRDNAFCSCSVIFGYKTNCRCEVGTTATSYGICMAKHKCGRQTTCTKPCLSKRCKKNH | [119] |
| ω-ctenitoxin-Pn4a/Neurotoxin Tx3–6 PnTx3–6/Phα1β | 122 nM (N) | ACIPRGEICTDDCECCGCDNQCYCPPGSSLGIFKCSCAHANKYFCNRKKEKCKKA | [119,120] |
| ω-ctenitoxin-Pr1a/Neurotoxin PRTx3–7 | >1000 nM (N) | ACAGLYKKCGKGVNTCCENRPCKCDLAMGNCICKKKFVEFFGG | [121,122] |
| ω-segestritoxin-Sf1a/SNX-325 | ~10 nM (Y) | GSCIESGKSCTHSRSMKNGLCCPKSRCNCRQIQHRHDYLGKRKYSCRCS | [123] |
| ω-theraphotoxin-Hh1a/Huwentoxin-10 | 40 nM (Y) | KCLPPGKPCYGATQKIPCCGVCSHNKCT | [113] |


2.7.1. ω-Agatoxin-Aa2a
2.7.2. ω-Theraphotoxin-Hh1a
2.7.3. ω-Segestritoxin-Sf1a
2.8. Spider Venom Cav2.2 Inhibitors in Pain
3. Cav2.2 Inhibitors from Other Venomous Animals
3.1. Scorpion Venom Peptides
3.2. Snake Venom Peptides
3.3. Centipede Venom Peptides
3.4. Assassin Bug Toxins
4. High Throughput Assays for Novel Cav2.2 Channel Inhibitors
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2003, 2, 790–802. [Google Scholar]
- Gomez, M.V.; Kalapothakis, E.; Guatimosim, C.; Prado, M.A. Phoneutria nigriventer venom: A cocktail of toxins that affect ion channels. Cell Mol. Neurobiol. 2002, 22, 579–588. [Google Scholar]
- Klint, J.K.; Senff, S.; Rupasinghe, D.B.; Er, S.Y.; Herzig, V.; Nicholson, G.M.; King, G.F. Spider-venom peptides that target voltage-gated sodium channels: Pharmacological tools and potential therapeutic leads. Toxicon 2012, 60, 478–491. [Google Scholar] [CrossRef]
- Herzig, V.; Wood, D.L.; Newell, F.; Chaumeil, P.A.; Kaas, Q.; Binford, G.J.; Nicholson, G.M.; Gorse, D.; King, G.F. Arachnoserver 2.0, an updated online resource for spider toxin sequences and structures. Nucleic Acids Res. 2011, 39, D653–D657. [Google Scholar] [CrossRef]
- Arachnoserver 2.0. Available online: www.arachnoserver.org (accessed on 12 December 2012).
- Adams, M.E.; Bindokas, V.P.; Hasegawa, L.; Venema, V.J. Omega-agatoxins: Novel calcium channel antagonists of two subtypes from funnel web spider (agelenopsis aperta) venom. J. Biol. Chem. 1990, 265, 861–867. [Google Scholar]
- Dos Santos, R.G.; Van Renterghem, C.; Martin-Moutot, N.; Mansuelle, P.; Cordeiro, M.N.; Diniz, C.R.; Mori, Y.; De Lima, M.E.; Seagar, M. Phoneutria nigriventer ω-phonetoxin iia blocks the cav2 family of calcium channels and interacts with ω-conotoxin-binding sites. J. Biol. Chem.. 2002, 277, 13856–13862. [Google Scholar]
- Lewis, R.J.; Nielsen, K.J.; Craik, D.J.; Loughnan, M.L.; Adams, D.A.; Sharpe, I.A.; Luchian, T.; Adams, D.J.; Bond, T.; Thomas, L. Novel ω-conotoxins from conus catus discriminate among neuronal calcium channel subtypes. J. Biol. Chem. 2000, 275, 35335–35344. [Google Scholar]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef]
- Abbott, J.R.; Litzinger, M.J. Different ω-conotoxins mark the development of swiss webster mouse cortex suggesting N-type voltage sensitive calcium channel subtypes. Int. J. Dev. Neurosci. 1994, 12, 43–47. [Google Scholar] [CrossRef]
- Ellinor, P.T.; Zhang, J.F.; Horne, W.A.; Tsien, R.W. Structural determinants of the blockade of N-type calcium channels by a peptide neurotoxin. Nature 1994, 372, 272–275. [Google Scholar] [CrossRef]
- Wagner, J.; Snowman, A.; Biswas, A.; Olivera, B.; Snyder, S. Ω-conotoxin gvia binding to a high affinity receptor in brain: Characterization, calcium sensitivity and solubilization. J. Neurosci. 1988, 9, 3354–3359. [Google Scholar]
- Olivera, B.M.; Cruz, L.J.; de Santos, V.; LeCheminant, G.W.; Griffin, D.; Zeikus, R.; McIntosh, J.M.; Galyean, R.; Varga, J.; Gray, W.R. Neuronal calcium channel antagonists. Discrimination between calcium channel subtypes using ω-conotoxin from conus magus venom. Biochemistry 1987, 26, 2086–2090. [Google Scholar]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Miljanich, G.P. Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef]
- Berecki, G.; Motin, L.; Haythornthwaite, A.; Vink, S.; Bansal, P.; Drinkwater, R.; Wang, C.I.; Moretta, M.; Lewis, R.J.; Alewood, P.F.; et al. Analgesic ω-conotoxins cvie and cvif selectively and voltage-dependently block recombinant and native N-type calcium channels. Mol. Pharmacol. 2010, 77, 139–148. [Google Scholar]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef]
- Wermeling, D.P. Ziconotide, an intrathecally administered N-type calcium channel antagonist for the treatment of chronic pain. Pharmacotherapy 2005, 25, 1084–1094. [Google Scholar] [CrossRef]
- Alicino, I.; Giglio, M.; Manca, F.; Bruno, F.; Puntillo, F. Intrathecal combination of ziconotide and morphine for refractory cancer pain: A rapidly acting and effective choice. Pain 2012, 153, 245–249. [Google Scholar] [CrossRef]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International union of pharmacology. Xlviii. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef]
- Williams, M.E.; Brust, P.F.; Feldman, D.H.; Patthi, S.; Simerson, S.; Maroufi, A.; McCue, A.F.; Velicelebi, G.; Ellis, S.B.; Harpold, M.M. Structure and functional expression of an ω-conotoxin-sensitive human N-type calcium channel. Science 1992, 257, 389–395. [Google Scholar]
- Catterall, W.A. Structure and regulation of voltage-gated ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef]
- Yu, F.H.; Catterall, W.A. The vgl-chanome: A protein superfamily specialized for electrical signaling and ionic homeostasis. Sci. STKE 2004, 2004. [Google Scholar] [CrossRef]
- Payandeh, J.; Scheuer, T.; Zheng, N.; Catterall, W.A. The crystal structure of a voltage-gated sodium channel. Nature 2011, 475, 353–358. [Google Scholar]
- Payandeh, J.; Gamal El-Din, T.M.; Scheuer, T.; Zheng, N.; Catterall, W.A. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 2012, 486, 135–139. [Google Scholar]
- Lipscombe, D.; Pan, J.Q.; Gray, A.C. Functional diversity in neuronal voltage-gated calcium channels by alternative splicing of ca(v)α1. Mol. Neurobiol. 2002, 26, 21–44. [Google Scholar] [CrossRef]
- Lin, Z.; Haus, S.; Edgerton, J.; Lipscombe, D. Identification of functionally distinct isoforms of the N-type ca2+ channel in rat sympathetic ganglia and brain. Neuron 1997, 18, 153–166. [Google Scholar] [CrossRef]
- Bell, T.J.; Thaler, C.; Castiglioni, A.J.; Helton, T.D.; Lipscombe, D. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron 2004, 41, 127–138. [Google Scholar]
- Lipscombe, D.; Raingo, J. Alternative splicing matters: N-type calcium channels in nociceptors. Channels (Austin) 2007, 1, 225–227. [Google Scholar]
- Zamponi, G.W.; McCleskey, E.W. Splicing it up: A variant of the N-type calcium channel specific for pain. Neuron 2004, 41, 3–4. [Google Scholar] [CrossRef]
- Kaneko, S.; Cooper, C.B.; Nishioka, N.; Yamasaki, H.; Suzuki, A.; Jarvis, S.E.; Akaike, A.; Satoh, M.; Zamponi, G.W. Identification and characterization of novel human ca(v)2.2 (α1b) calcium channel variants lacking the synaptic protein interaction site. J. Neurosci. 2002, 22, 82–92. [Google Scholar]
- McDonough, S.I.; Boland, L.M.; Mintz, I.M.; Bean, B.P. Interactions among toxins that inhibit N-type and P-type calcium channels. J. Gen. Physiol. 2002, 119, 313–328. [Google Scholar] [CrossRef]
- Stocker, J.W.; Nadasdi, L.; Aldrich, R.W.; Tsien, R.W. Preferential interaction of ω-conotoxins with inactivated N-type Ca2+ channels. J. Neurosci. 1997, 17, 3002–3013. [Google Scholar]
- Feng, Z.P.; Hamid, J.; Doering, C.; Bosey, G.M.; Snutch, T.P.; Zamponi, G.W. Residue gly1326 of the N-type calcium channel α1b subunit controls reversibility of ω-conotoxin gvia and mviia block. J. Biol. Chem. 2001, 276, 15728–15735. [Google Scholar]
- Mould, J.; Yasuda, T.; Schroeder, C.I.; Beedle, A.M.; Doering, C.J.; Zamponi, G.W.; Adams, D.J.; Lewis, R.J. The α2δ auxiliary subunit reduces affinity of ω-conotoxins for recombinant N-type (Cav2.2) calcium channels. J. Biol. Chem. 2004, 279, 34705–34714. [Google Scholar]
- Dolphin, A.; Wyatt, C.; Richards, J.; Beattie, R.; Craig, P.; Lee, J.H.; Cribbs, L.; Volsen, S.; Perez‐Reyes, E. The effect of α2δ and other accessory subunits on expression and properties of the calcium channel α1g. J. Physiol. 1999, 519, 35–45. [Google Scholar]
- Birnbaumer, L.; Qin, N.; Olcese, R.; Tareilus, E.; Platano, D.; Costantin, J.; Stefani, E. Structures and functions of calcium channel β subunits. J. Bioenerg. Biomembr. 1998, 30, 357–375. [Google Scholar] [CrossRef]
- Takahashi, M.; Seagar, M.J.; Jones, J.F.; Reber, B.F.; Catterall, W.A. Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proc. Natl. Acad. Sci. USA 1987, 84, 5478–5482. [Google Scholar] [CrossRef]
- Arikkath, J.; Campbell, K.P. Auxiliary subunits: Essential components of the voltage-gated calcium channel complex. Curr. Opin. Neurobiol. 2003, 13, 298–307. [Google Scholar] [CrossRef]
- Dolphin, A.C. Calcium channel diversity: Multiple roles of calcium channel subunits. Curr. Opin. Neurobiol. 2009, 19, 237–244. [Google Scholar] [CrossRef]
- Davies, A.; Hendrich, J.; Van Minh, A.T.; Wratten, J.; Douglas, L.; Dolphin, A.C. Functional biology of the α2δ subunits of voltage-gated calcium channels. Trends. Pharmacol. Sci. 2007, 28, 220–228. [Google Scholar] [CrossRef]
- Cole, R.L.; Lechner, S.M.; Williams, M.E.; Prodanovich, P.; Bleicher, L.; Varney, M.A.; Gu, G. Differential distribution of voltage-gated calcium channel α2δ delta (alpha2delta) subunit mrna-containing cells in the rat central nervous system and the dorsal root ganglia. J. Comp. Neurol. 2005, 491, 246–269. [Google Scholar] [CrossRef]
- Luo, Z.D.; Chaplan, S.R.; Higuera, E.S.; Sorkin, L.S.; Stauderman, K.A.; Williams, M.E.; Yaksh, T.L. Upregulation of dorsal root ganglion α2δ calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J. Neurosci. 2001, 21, 1868–1875. [Google Scholar]
- Luo, Z.; Calcutt, N.; Higuera, E.; Valder, C.; Song, Y.H.; Svensson, C.; Myers, R. Injury type-specific calcium channel α2δ/1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J. Pharmacol. Exp. Ther. 2002, 303, 1199–1205. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Sindrup, S.H.; Jensen, T.S. Chronic neuropathic pain: Mechanisms, drug targets and measurement. Fundam. Clin. Pharmacol. 2007, 21, 129–136. [Google Scholar] [CrossRef]
- Hendrich, J.; Van Minh, A.T.; Heblich, F.; Nieto-Rostro, M.; Watschinger, K.; Striessnig, J.; Wratten, J.; Davies, A.; Dolphin, A.C. Pharmacological disruption of calcium channel trafficking by the α2δ ligand gabapentin. Proc. Natl. Acad. Sci. USA 2008, 105, 3628–3633. [Google Scholar]
- Dolphin, A.C. Calcium Channel α2δ Subunits in Epilepsy and as Targets for Antiepileptic Drugs. In Jasper's Basic Mechanisms Epilepsies, 4th; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; Oxford University Press: Bethesda, Maryland, MD, USA, 2012. [Google Scholar]
- Taylor, C.P.; Angelotti, T.; Fauman, E. Pharmacology and mechanism of action of pregabalin: The calcium channel α2/δ (alpha2-delta) subunit as a target for antiepileptic drug discovery. Epilepsy. Res. 2007, 73, 137–150. [Google Scholar]
- Eroglu, C.; Allen, N.J.; Susman, M.W.; O'Rourke, N.A.; Park, C.Y.; Ozkan, E.; Chakraborty, C.; Mulinyawe, S.B.; Annis, D.S.; Huberman, A.D.; et al. Gabapentin receptor α2δ/1 is a neuronal thrombospondin receptor responsible for excitatory cns synaptogenesis. Cell 2009, 139, 380–392. [Google Scholar]
- Pragnell, M.; De Waard, M.; Mori, Y.; Tanabe, T.; Snutch, T.P.; Campbell, K.P. Calcium channel β-subunit binds to a conserved motif in the i-ii cytoplasmic linker of the α1-subunit. Nature 1994, 368, 67–70. [Google Scholar] [CrossRef]
- De Waard, M.; Pragnell, M.; Campbell, K.P. Ca2+ channel regulation by a conserved β subunit domain. Neuron 1994, 13, 495–503. [Google Scholar] [CrossRef]
- Bichet, D.; Cornet, V.; Geib, S.; Carlier, E.; Volsen, S.; Hoshi, T.; Mori, Y.; De Waard, M. The i-ii loop of the ca2+ channel alpha1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron 2000, 25, 177–190. [Google Scholar] [CrossRef]
- Buraei, Z.; Yang, J. The ss subunit of voltage-gated Ca2+ channels. Physiol. Rev. 2010, 90, 1461–1506. [Google Scholar] [CrossRef]
- Arias, J.M.; Murbartian, J.; Vitko, I.; Lee, J.H.; Perez-Reyes, E. Transfer of beta subunit regulation from high to low voltage-gated Ca2+ channels. FEBS Lett. 2005, 579, 3907–3912. [Google Scholar]
- Altier, C.; Garcia-Caballero, A.; Simms, B.; Walcher, J.; Tedford, H.; Hermosilla, G.; Zamponi, G. The Cav β subunit prevents RFT2-mediated ubiquitination and proteasomal degradation l-type calcium channels via the derlin-1/p97 erad protein complex. Nat. Neurosci. 2011, 14, 173–180. [Google Scholar] [CrossRef]
- Cornet, V.; Bichet, D.; Sandoz, G.; Marty, I.; Brocard, J.; Bourinet, E.; Mori, Y.; Villaz, M.; De Waard, M. Multiple determinants in voltage-dependent p/q calcium channels control their retention in the endoplasmic reticulum. Eur. J. Neurosci. 2002, 16, 883–895. [Google Scholar] [CrossRef]
- Flucher, B.E.; Kasielke, N.; Grabner, M. The triad targeting signal of the skeletal muscle calcium channel is localized in the cooh terminus of the alpha(1s) subunit. J. Cell Biol. 2000, 151, 467–478. [Google Scholar]
- Kobrinsky, E.; Tiwari, S.; Maltsev, V.A.; Harry, J.B.; Lakatta, E.; Abernethy, D.R.; Soldatov, N.M. Differential role of the alpha1c subunit tails in regulation of the cav1.2 channel by membrane potential, beta subunits, and Ca2+ ions. J. Biol. Chem. 2005, 280, 12474–12485. [Google Scholar]
- Waithe, D.; Ferron, L.; Page, K.M.; Chaggar, K.; Dolphin, A.C. Beta-subunits promote the expression of Cav2.2 channels by reducing their proteasomal degradation. J. Biol. Chem. 2011, 286, 9598–9611. [Google Scholar]
- Letts, V.A.; Felix, R.; Biddlecome, G.H.; Arikkath, J.; Mahaffey, C.L.; Valenzuela, A.; Bartlett, F.S., II; Mori, Y.; Campbell, K.P.; Frankel, W.N. The mouse stargazer gene encodes a neuronal Ca2+-channel γ subunit. Nat. Genet. 1998, 19, 340–347. [Google Scholar] [CrossRef]
- Kang, M.G.; Chen, C.C.; Felix, R.; Letts, V.A.; Frankel, W.N.; Mori, Y.; Campbell, K.P. Biochemical and biophysical evidence for γ2 subunit association with neuronal voltage-activated Ca2+ channels. J. Biol. Chem. 2001, 276, 32917–32924. [Google Scholar]
- Yang, L.; Katchman, A.; Morrow, J.P.; Doshi, D.; Marx, S.O. Cardiac l-type calcium channel (Cav1.2) associates with γ subunits. FASEB J. 2011, 25, 928–936. [Google Scholar] [CrossRef]
- Hansen, J.P.; Chen, R.S.; Larsen, J.K.; Chu, P.J.; Janes, D.M.; Weis, K.E.; Best, P.M. Calcium channel γ6 subunits are unique modulators of low voltage-activated (Cav3.1) calcium current. J. Mol. Cell. Cardiol. 2004, 37, 1147–1158. [Google Scholar] [CrossRef]
- Sandoval, A.; Arikkath, J.; Monjaraz, E.; Campbell, K.P.; Felix, R. Γ1-dependent down-regulation of recombinant voltage-gated Ca2+ channels. Cell. Mol. Neurobiol. 2007, 27, 901–908. [Google Scholar]
- Wallace, M.S.; Rauck, R.L.; Deer, T. Ziconotide combination intrathecal therapy: Rationale and evidence. Clin. J. Pain 2010, 26, 635–644. [Google Scholar] [CrossRef]
- Cizkova, D.; Marsala, J.; Lukacova, N.; Marsala, M.; Jergova, S.; Orendacova, J.; Yaksh, T.L. Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Exp. Brain Res. 2002, 147, 456–463. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Lewis, R.J.; Todorovic, S.M.; Arneric, S.P.; Snutch, T.P. Role of voltage-gated calcium channels in ascending pain pathways. Brain Res. Rev. 2009, 60, 84–89. [Google Scholar] [CrossRef]
- Krarup, C. An update on electrophysiological studies in neuropathy. Curr. Opin. Neurol. 2003, 16, 603–612. [Google Scholar] [CrossRef]
- Saegusa, H.; Kurihara, T.; Zong, S.; Kazuno, A.; Matsuda, Y.; Nonaka, T.; Han, W.; Toriyama, H.; Tanabe, T. Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. EMBO J. 2001, 20, 2349–2356. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Wakamori, M.; Ino, M.; Miyamoto, N.; Takahashi, E.; Yoshinaga, T.; Sawada, K.; Imoto, K.; Tanaka, I.; Yoshizawa, T.; et al. Differential nociceptive responses in mice lacking the α1b subunit of N-type Ca(2+) channels. Neuroreport 2001, 12, 2423–2427. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Snutch, T.P. Modulation of voltage-dependent calcium channels by g proteins. Curr. Opin. Neurobiol. 1998, 8, 351–356. [Google Scholar] [CrossRef]
- Li, C.Y.; Song, Y.H.; Higuera, E.S.; Luo, Z.D. Spinal dorsal horn calcium channel α2δ/1 subunit upregulation contributes to peripheral nerve injury-induced tactile allodynia. J. Neurosci. 2004, 24, 8494–8499. [Google Scholar] [CrossRef]
- Li, C.Y.; Zhang, X.L.; Matthews, E.A.; Li, K.W.; Kurwa, A.; Boroujerdi, A.; Gross, J.; Gold, M.S.; Dickenson, A.H.; Feng, G.; et al. Calcium channel α2δ1 subunit mediates spinal hyperexcitability in pain modulation. Pain 2006, 125, 20–34. [Google Scholar] [CrossRef]
- Jain, K.K. An evaluation of intrathecal ziconotide for the treatment of chronic pain. Expert. Opin. Investig. Drugs 2000, 9, 2403–2410. [Google Scholar] [CrossRef]
- Staats, P.S.; Yearwood, T.; Charapata, S.G.; Presley, R.W.; Wallace, M.S.; Byas-Smith, M.; Fisher, R.; Bryce, D.A.; Mangieri, E.A.; Luther, R.R.; et al. Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or aids: A randomized controlled trial. JAMA 2004, 291, 63–70. [Google Scholar]
- Olivera, B.M.; Gray, W.R.; Zeikus, R.; McIntosh, J.M.; Varga, J.; Rivier, J.; de Santos, V.; Cruz, L.J. Peptide neurotoxins from fish-hunting cone snails. Science 1985, 230, 1338–1343. [Google Scholar]
- Skalicky, J.J.; Metzler, W.J.; Ciesla, D.J.; Galdes, A.; Pardi, A. Solution structure of the calcium channel antagonist omega-conotoxin gvia. Protein. Sci. 1993, 2, 1591–1603. [Google Scholar]
- Norton, R.S.; Pallaghy, P.K. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon 1998, 36, 1573–1583. [Google Scholar]
- Olivera, B.M. Conus peptides: Biodiversity-based discovery and exogenomics. J. Biol. Chem. 2006, 281, 31173–31177. [Google Scholar] [CrossRef]
- Jakubowski, J.A.; Kelley, W.P.; Sweedler, J.V. Screening for post-translational modifications in conotoxins using liquid chromatography/mass spectrometry: An important component of conotoxin discovery. Toxicon 2006, 47, 688–699. [Google Scholar] [CrossRef]
- Nielsen, K.J.; Adams, D.; Thomas, L.; Bond, T.; Alewood, P.F.; Craik, D.J.; Lewis, R.J. Structure-activity relationships of ω-conotoxins mviia, mviic and 14 loop splice hybrids at n and p/q-type calcium channels. J. Mol. Biol. 1999, 289, 1405–1421. [Google Scholar] [CrossRef]
- Favreau, P.; Gilles, N.; Lamthanh, H.; Bournaud, R.; Shimahara, T.; Bouet, F.; Laboute, P.; Letourneux, Y.; Menez, A.; Molgo, J.; et al. A new ω-conotoxin that targets N-type voltage-sensitive calcium channels with unusual specificity. Biochemistry 2001, 40, 14567–14575. [Google Scholar]
- Lee, S.; Kim, Y.; Back, S.K.; Choi, H.W.; Lee, J.Y.; Jung, H.H.; Ryu, J.H.; Suh, H.W.; Na, H.S.; Kim, H.J.; et al. Analgesic effect of highly reversible ω-conotoxin fvia on n type Ca2+ channels. Mol. Pain 2010, 6, 97. [Google Scholar] [CrossRef]
- Olivera, B.M.; McIntosh, J.M.; Cruz, L.J.; Luque, F.A.; Gray, W.R. Purification and sequence of a presynaptic peptide toxin from conus geographus venom. Biochemistry 1984, 23, 5087–5090. [Google Scholar]
- Sato, K.; Park, N.G.; Kohno, T.; Maeda, T.; Kim, J.I.; Kato, R.; Takahashi, M. Role of basic residues for the binding of ω-conotoxin gvia to N-type calcium channels. Biochem. Biophys. Res. Commun. 1993, 194, 1292–1296. [Google Scholar] [CrossRef]
- Kim, J.I.; Takahashi, M.; Ogura, A.; Kohno, T.; Kudo, Y.; Sato, K. Hydroxyl group of tyr13 is essential for the activity of ω-conotoxin gvia, a peptide toxin for N-type calcium channel. J. Biol. Chem. 1994, 269, 23876–23878. [Google Scholar]
- Kim, J.I.; Takahashi, M.; Ohtake, A.; Wakamiya, A.; Sato, K. Tyr13 is essential for the activity of ω-conotoxin mviia and gvia, specific N-type calcium channel blockers. Biochem. Biophys. Res. Commun. 1995, 206, 449–454. [Google Scholar] [CrossRef]
- Olivera, B.M.; Gray, W.R.; Zeikus, R.; McIntosh, J.M.; Varga, J.; Rivier, J.; Santos, V.D.; Cruz, L.J. Peptide neurotoxins from fish-hunting cone snails. Science 1985, 230, 1338–1343. [Google Scholar]
- Bingham, J.-P.; Baker, M.R.; Chun, J.B. Analysis of a cone snail's killer cocktail - the milked venom of conus geographus. Toxicon 2012, 60, 1166–1170. [Google Scholar]
- Nielsen, K.J.; Schroeder, T.; Lewis, R. Structure-activity relationships of ω-conotoxins at N-type voltage-sensitive calcium channels. J. Mol. Recognit. 2000, 13, 55–70. [Google Scholar] [CrossRef]
- Wang, Y.X.; Bezprozvannaya, S.; Bowersox, S.S.; Nadasdi, L.; Miljanich, G.; Mezo, G.; Silva, D.; Tarczy-Hornoch, K.; Luther, R.R. Peripheral versus central potencies of N-type voltage-sensitive calcium channel blockers. Naunyn. Schmiedebergs. Arch. Pharmacol. 1998, 357, 159–168. [Google Scholar] [CrossRef]
- Chung, D.; Gaur, S.; Bell, J.R.; Ramachandran, J.; Nadasdi, L. Determination of disulfide bridge pattern in ω-conopeptides. Int. J. Pept. Protein. Res. 1995, 46, 320–325. [Google Scholar]
- Lew, M.J.; Flinn, J.P.; Pallaghy, P.K.; Murphy, R.; Whorlow, S.L.; Wright, C.E.; Norton, R.S.; Angus, J.A. Structure-function relationships of ω-conotoxin gvia. Synthesis, structure, calcium channel binding, and functional assay of alanine-substituted analogues. J. Biol. Chem. 1997, 272, 12014–12023. [Google Scholar]
- Flinn, J.P.; Pallaghy, P.K.; Lew, M.J.; Murphy, R.; Angus, J.A.; Norton, R.S. Roles of key functional groups in ω-conotoxin gvia synthesis, structure and functional assay of selected peptide analogues. Eur. J. Biochem. 1999, 262, 447–455. [Google Scholar] [CrossRef]
- Motin, L.; Yasuda, T.; Schroeder, C.I.; Lewis, R.J.; Adams, D.J. Ω-conotoxin inhibition of excitatory synaptic transmission evoked by dorsal root stimulation in rat superficial dorsal horn-conotoxin cvib differentially inhibits native and recombinant n- and p/q-type calcium channels. Eur. J. Neurosci. 2007, 25, 435–444. [Google Scholar] [CrossRef]
- Motin, L.; Adams, D.J. Ω-conotoxin inhibition of excitatory synaptic transmission evoked by dorsal root stimulation in rat superficial dorsal horn. Neuropharmacology 2008, 55, 860–864. [Google Scholar] [CrossRef]
- Schroeder, K.; Neagle, B.; Trezise, D.J.; Worley, J. Ionworks ht: A new high-throughput electrophysiology measurement platform. J. Biomol. Screen 2003, 8, 50–64. [Google Scholar] [CrossRef]
- Swensen, A.M.; Niforatos, W.; Vortherms, T.A.; Perner, R.J.; Li, T.; Schrimpf, M.R.; Scott, V.E.; Lee, L.; Jarvis, M.F.; McGaraughty, S. An automated electrophysiological assay for differentiating Cav2. 2 inhibitors based on state dependence and kinetics. Assay Drug Develop. Technol. 2012, 10, 542–550. [Google Scholar] [CrossRef]
- Bauer, C.S.; Rahman, W.; Tran-van-Minh, A.; Lujan, R.; Dickenson, A.H.; Dolphin, A.C. The anti-allodynic α2δ ligand pregabalin inhibits the trafficking of the calcium channel α1b subunit to presynaptic terminals in vivo. Biochem. Soc. Trans. 2010, 38, 525–528. [Google Scholar] [CrossRef]
- Perret, D.; Luo, Z.D. Targeting voltage-gated calcium channels for neuropathic pain management. Neurotherapeutics 2009, 6, 679–692. [Google Scholar] [CrossRef]
- Heinke, B.; Balzer, E.; Sandkuhler, J. Pre- and post-synaptic contributions of voltage-dependent Ca2+ channels to nociceptive transmission in rat spinal lamina i neurons. Eur. J. Neurosci. 2004, 19, 103–111. [Google Scholar] [CrossRef]
- Rycroft, B.K.; Vikman, K.S.; Christie, M.J. Inflammation reduces the contribution of N-type calcium channels to primary afferent synaptic transmission onto nk1 receptor-positive lamina i neurons in the rat dorsal horn. J. Physiol. 2007, 580, 883–894. [Google Scholar] [CrossRef]
- Hockerman, G.H.; Johnson, B.D.; Abbott, M.R.; Scheuer, T.; Catterall, W.A. Molecular determinants of high affinity phenylalkylamine block of l-type calcium channels in transmembrane segment iiis6 and the pore region of the α1 subunit. J. Biol. Chem. 1997, 272, 18759–18765. [Google Scholar]
- McGuire, D.; Bowersox, S.; Fellmann, J.D.; Luther, R.R. Sympatholysis after neuron-specific, N-type, voltage-sensitive calcium channel blockade: First demonstration of n-channel function in humans. J. Cardiovasc. Pharmacol. 1997, 30, 400–403. [Google Scholar] [CrossRef]
- Escoubas, P.; Sollod, B.; King, G.F. Venom landscapes: Mining the complexity of spider venoms via a combined cdna and mass spectrometric approach. Toxicon 2006, 47, 650–663. [Google Scholar] [CrossRef]
- Estrada, G.; Villegas, E.; Corzo, G. Spider venoms: A rich source of acylpolyamines and peptides as new leads for cns drugs. Nat. Product Rep. 2007, 24, 145–161. [Google Scholar] [CrossRef]
- Norton, R.S.; McDonough, S.I. Peptides targeting voltage-gated calcium channels. Curr. Pharm. Des. 2008, 14, 2480–2491. [Google Scholar] [CrossRef]
- Mintz, I.M.; Venema, V.J.; Swiderek, K.M.; Lee, T.D.; Bean, B.P.; Adams, M.E. P-type calcium channels blocked by the spider toxin omega-aga-iva. Nature 1992, 355, 827–829. [Google Scholar] [CrossRef]
- Newcomb, R.; Szoke, B.; Palma, A.; Wang, G.; Chen, X.; Hopkins, W.; Cong, R.; Miller, J.; Urge, L.; Tarczy-Hornoch, K.; et al. Selective peptide antagonist of the class e calcium channel from the venom of the tarantula hysterocrates gigas. Biochemistry 1998, 37, 15353–15362. [Google Scholar]
- Wang, M.; Guan, X.; Liang, S. The cross channel activities of spider neurotoxin huwentoxin-i on rat dorsal root ganglion neurons. Biochem. Biophys. Res. Commun. 2007, 357, 579–583. [Google Scholar] [CrossRef]
- Liang, S.P.; Zhang, D.Y.; Pan, X.; Chen, Q.; Zhou, P.A. Properties and amino acid sequence of huwentoxin-i, a neurotoxin purified from the venom of the chinese bird spider selenocosmia huwena. Toxicon 1993, 31, 969–978. [Google Scholar]
- Jiang, L.; Peng, L.; Chen, J.; Zhang, Y.; Xiong, X.; Liang, S. Molecular diversification based on analysis of expressed sequence tags from the venom glands of the chinese bird spider ornithoctonus huwena. Toxicon 2008, 51, 1479–1489. [Google Scholar]
- Liu, Z.; Dai, J.; Dai, L.; Deng, M.; Hu, Z.; Hu, W.; Liang, S. Function and solution structure of huwentoxin-x, a specific blocker of N-type calcium channels, from the chinese bird spider ornithoctonus huwena. J. Biol. Chem. 2006, 281, 8628–8635. [Google Scholar]
- Bindokas, V.P.; Adams, M.E. Ω-aga-i: A presynaptic calcium channel antagonist from venom of the funnel web spider, agelenopsis aperta. J. Neurobiol. 1989, 20, 171–188. [Google Scholar] [CrossRef]
- Adams, M.E.; Bindokas, V.P.; Hasegawa, L.; Venema, V.J. Ω-agatoxins: Novel calcium channel antagonists of two subtypes from funnel web spider (agelenopsis aperta) venom. J. Biol. Chem. 1990, 265, 861–867. [Google Scholar]
- Ertel, E.A.; Warren, V.A.; Adams, M.E.; Griffin, P.R.; Cohen, C.J.; Smith, M.M. Ω-agatoxins: A family of probes for similar binding sites on l- and N-type calcium channels. Biochemistry 1994, 33, 5098–5108. [Google Scholar]
- Yan, L.; Adams, M.E. The spider toxin ω-aga iiia defines a high affinity site on neuronal high voltage-activated calcium channels. J. Biol. Chem. 2000, 275, 21309–21316. [Google Scholar] [CrossRef]
- Leao, R.M.; Cruz, J.S.; Diniz, C.R.; Cordeiro, M.N.; Beirao, P.S. Inhibition of neuronal high-voltage activated calcium channels by the omega-phoneutria nigriventer tx3–3peptide toxin. Neuropharmacology 2000, 39, 1756–1767. [Google Scholar]
- Cordeiro Mdo, N.; de Figueiredo, S.G.; Valentim Ado, C.; Diniz, C.R.; von Eickstedt, V.R.; Gilroy, J.; Richardson, M. Purification and amino acid sequences of six tx3 type neurotoxins from the venom of the brazilian 'armed' spider phoneutria nigriventer (keys). Toxicon 1993, 31, 35–42. [Google Scholar] [CrossRef]
- Vieira, L.B.; Kushmerick, C.; Hildebrand, M.E.; Garcia, E.; Stea, A.; Cordeiro, M.N.; Richardson, M.; Gomez, M.V.; Snutch, T.P. Inhibition of high voltage-activated calcium channels by spider toxin pntx3-6. J. Pharmacol. Exp. Ther. 2005, 314, 1370–1377. [Google Scholar] [CrossRef]
- Vieira, L.B.; Pimenta, A.M.; Richardson, M.; Bemquerer, M.P.; Reis, H.J.; Cruz, J.S.; Gomez, M.V.; Santoro, M.M.; Ferreira-de-Oliveira, R.; Figueiredo, S.G.; et al. Leftward shift in the voltage-dependence for Ca2+ currents activation induced by a new toxin from phoneutria reidyi (aranae, ctenidae) venom. Cell. Mol. Neurobiol. 2007, 27, 129–146. [Google Scholar] [CrossRef]
- Richardson, M.; Pimenta, A.M.; Bemquerer, M.P.; Santoro, M.M.; Beirao, P.S.; Lima, M.E.; Figueiredo, S.G.; Bloch, C., Jr.; Vasconcelos, E.A.; Campos, F.A.; et al. Comparison of the partial proteomes of the venoms of brazilian spiders of the genus phoneutria. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2006, 142, 173–187. [Google Scholar]
- Newcomb, R.; Palma, A.; Fox, J.; Gaur, S.; Lau, K.; Chung, D.; Cong, R.; Bell, J.R.; Horne, B.; Nadasdi, L.; et al. Snx-325, a novel calcium antagonist from the spider segestria florentina. Biochemistry 1995, 34, 8341–8347. [Google Scholar]
- Benjamin, E.R.; Pruthi, F.; Olanrewaju, S.; Shan, S.; Hanway, D.; Liu, X.; Cerne, R.; Lavery, D.; Valenzano, K.J.; Woodward, R.M.; et al. Pharmacological characterization of recombinant N-type calcium channel (Cav2.2) mediated Calcium mobilization using flipr. Biochem. Pharmacol. 2006, 72, 770–782. [Google Scholar] [CrossRef]
- Adams, M.E.; Mintz, I.M.; Reily, M.D.; Thanabal, V.; Bean, B.P. Structure and properties of ω-agatoxin ivb, a new antagonist of p-type calcium channels. Mol. Pharmacol. 1993, 44, 681–688. [Google Scholar]
- Cardoso, F.C.; Pacifico, L.G.; Carvalho, D.C.; Victoria, J.M.; Neves, A.L.; Chavez-Olortegui, C.; Gomez, M.V.; Kalapothakis, E. Molecular cloning and characterization of phoneutria nigriventer toxins active on calcium channels. Toxicon 2003, 41, 755–763. [Google Scholar] [CrossRef]
- Vieira, L.B.; Kushmerick, C.; Reis, H.J.; Diniz, C.R.; Cordeiro, M.N.; Prado, M.A.; Kalapothakis, E.; Romano-Silva, M.A.; Gomez, M.V. Pntx3–6a spider neurotoxin inhibits K+-evoked increase in [Ca2+](i) and Ca2+-dependent glutamate release in synaptosomes. Neurochem. Int. 2003, 42, 277–282. [Google Scholar] [CrossRef]
- Souza, A.H.; Ferreira, J.; Cordeiro Mdo, N.; Vieira, L.B.; De Castro, C.J.; Trevisan, G.; Reis, H.; Souza, I.A.; Richardson, M.; Prado, M.A.; et al. Analgesic effect in rodents of native and recombinant ph α1β toxin, a high-voltage-activated calcium channel blocker isolated from armed spider venom. Pain 2008, 140, 115–126. [Google Scholar] [CrossRef]
- de Souza, A.H.; Lima, M.C.; Drewes, C.C.; da Silva, J.F.; Torres, K.C.; Pereira, E.M.; de Castro Junior, C.J.; Vieira, L.B.; Cordeiro, M.N.; Richardson, M.; et al. Antiallodynic effect and side effects of pα1β, a neurotoxin from the spider phoneutria nigriventer: Comparison with ω-conotoxin mviia and morphine. Toxicon 2011, 58, 626–633. [Google Scholar] [CrossRef]
- Dalmolin, G.D.; Silva, C.R.; Rigo, F.K.; Gomes, G.M.; Cordeiro Mdo, N.; Richardson, M.; Silva, M.A.; Prado, M.A.; Gomez, M.V.; Ferreira, J. Antinociceptive effect of brazilian armed spider venom toxin tx3–3in animal models of neuropathic pain. Pain 2011, 152, 2224–2232. [Google Scholar] [CrossRef]
- Fukumoto, N.; Obama, Y.; Kitamura, N.; Niimi, K.; Takahashi, E.; Itakura, C.; Shibuya, I. Hypoalgesic behaviors of p/q-type voltage-gated Ca2+ channel mutant mouse, rolling mouse nagoya. Neuroscience 2009, 160, 165–173. [Google Scholar]
- Todorovic, S.M.; Jevtovic-Todorovic, V. T-type voltage-gated calcium channels as targets for the development of novel pain therapies. Br. J. Pharmacol. 2011, 163, 484–495. [Google Scholar] [CrossRef]
- Olivera, B.M.; Miljanich, G.P.; Ramachandran, J.; Adams, M.E. Calcium channel diversity and neurotransmitter release: The omega-conotoxins and omega-agatoxins. Annu. Rev. Biochem. 1994, 63, 823–867. [Google Scholar] [CrossRef]
- Stotz, S.C.; Spaetgens, R.L.; Zamponi, G.W. Block of voltage-dependent calcium channel by the green mamba toxin calcicludine. J. Membr. Biol. 2000, 174, 157–165. [Google Scholar] [CrossRef]
- Sidach, S.S.; Mintz, I.M. Kurtoxin, a gating modifier of neuronal high- and low-threshold Ca2+ channels. J. Neurosci. 2002, 22, 2023–2034. [Google Scholar]
- Meunier, F.A.; Feng, Z.P.; Molgo, J.; Zamponi, G.W.; Schiavo, G. Glycerotoxin from glycera convoluta stimulates neurosecretion by up-regulating N-type Ca2+ channel activity. EMBO J. 2002, 21, 6733–6743. [Google Scholar] [CrossRef]
- Lee, C.W.; Eu, Y.J.; Min, H.J.; Cho, E.M.; Lee, J.H.; Kim, H.H.; Nah, S.Y.; Swartz, K.J.; Kim, J.I. Expression and characterization of recombinant kurtoxin, an inhibitor of t-type voltage-gated calcium channels. Biochem. Biophys. Res. Commun. 2011, 416, 277–282. [Google Scholar]
- de Weille, J.R.; Schweitz, H.; Maes, P.; Tartar, A.; Lazdunski, M. Calciseptine, a peptide isolated from black mamba venom, is a specific blocker of the l-type calcium channel. Proc. Natl. Acad. Sci. USA 1991, 88, 2437–2440. [Google Scholar] [CrossRef]
- Schweitz, H.; Heurteaux, C.; Bois, P.; Moinier, D.; Romey, G.; Lazdunski, M. Calcicludine, a venom peptide of the kunitz-type protease inhibitor family, is a potent blocker of high-threshold Ca2+ channels with a high affinity for l-type channels in cerebellar granule neurons. Proc. Natl. Acad. Sci. USA 1994, 91, 878–882. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Z.; Xiao, Y.; Li, Y.; Rong, M.; Liang, S.; Zhang, Z.; Yu, H.; King, G.F.; Lai, R. Chemical punch packed in venoms makes centipedes excellent predators. Mol. Cell. Proteom. 2012, 11, 640–650. [Google Scholar] [CrossRef]
- Corzo, G.; Adachi-Akahane, S.; Nagao, T.; Kusui, Y.; Nakajima, T. Novel peptides from assassin bugs (hemiptera: Reduviidae): Isolation, chemical and biological characterization. FEBS Lett. 2001, 499, 256–261. [Google Scholar] [CrossRef]
- Bernard, C.; Corzo, G.; Mosbah, A.; Nakajima, T.; Darbon, H. Solution structure of ptu1, a toxin from the assassin bug peirates turpis that blocks the voltage-sensitive calcium channel N-type. Biochemistry 2001, 40, 12795–12800. [Google Scholar] [CrossRef]
- Vetter, I.; Davis, J.L.; Rash, L.D.; Anangi, R.; Mobli, M.; Alewood, P.F.; Lewis, R.J.; King, G.F. Venomics: A new paradigm for natural products-based drug discovery. Amino Acids 2011, 40, 15–28. [Google Scholar]
- Finley, M.F.; Lubin, M.L.; Neeper, M.P.; Beck, E.; Liu, Y.; Flores, C.M.; Qin, N. An integrated multiassay approach to the discovery of small-molecule N-type voltage-gated calcium channel antagonists. Assay Drug Dev. Technol. 2010, 8, 685–694. [Google Scholar] [CrossRef]
- Dai, G.; Haedo, R.J.; Warren, V.A.; Ratliff, K.S.; Bugianesi, R.M.; Rush, A.; Williams, M.E.; Herrington, J.; Smith, M.M.; McManus, O.B.; et al. A high-throughput assay for evaluating state dependence and subtype selectivity of Cav2 calcium channel inhibitors. Assay Drug Dev. Technol. 2008, 6, 195–212. [Google Scholar] [CrossRef]
- Dunlop, J.; Bowlby, M.; Peri, R.; Tawa, G.; LaRocque, J.; Soloveva, V.; Morin, J. Ion channel screening. Comb. Chem. High Throughput Screen. 2008, 11, 514–522. [Google Scholar] [CrossRef]
- Lubin, M.L.; Reitz, T.L.; Todd, M.J.; Flores, C.M.; Qin, N.; Xin, H. A nonadherent cell-based hts assay for N-type calcium channel using calcium 3 dye. Assay Drug Dev. Technol. 2006, 4, 689–694. [Google Scholar]
- Winquist, R.J.; Pan, J.Q.; Gribkoff, V.K. Use-dependent blockade of Cav2.2 voltage-gated calcium channels for neuropathic pain. Biochem. Pharmacol. 2005, 70, 489–499. [Google Scholar] [CrossRef]
- Sousa, S.R.; Ragnarsson, L.; Vetter, I.; Herzig, V.; King, G.F.; Lewis, R.J. Development of High Throughput Calcium Channel Assays to Accelerate the Discovery of Novel Toxins Targeting Human Cav2.2 Channels. In Proceedings of 17th World Congress the International Society on Toxinology & Venom Week 2012, Honolulu, Hawaii, USA, 8–13 July 2012; Volume 60, p. 111.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sousa, S.R.; Vetter, I.; Lewis, R.J. Venom Peptides as a Rich Source of Cav2.2 Channel Blockers. Toxins 2013, 5, 286-314. https://doi.org/10.3390/toxins5020286
Sousa SR, Vetter I, Lewis RJ. Venom Peptides as a Rich Source of Cav2.2 Channel Blockers. Toxins. 2013; 5(2):286-314. https://doi.org/10.3390/toxins5020286
Chicago/Turabian StyleSousa, Silmara R., Irina Vetter, and Richard J. Lewis. 2013. "Venom Peptides as a Rich Source of Cav2.2 Channel Blockers" Toxins 5, no. 2: 286-314. https://doi.org/10.3390/toxins5020286
APA StyleSousa, S. R., Vetter, I., & Lewis, R. J. (2013). Venom Peptides as a Rich Source of Cav2.2 Channel Blockers. Toxins, 5(2), 286-314. https://doi.org/10.3390/toxins5020286