
DXD Motif-Dependent and -Independent Effects of the Chlamydia trachomatis Cytotoxin CT166

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Inhibition of Ras-ERK Signaling in Ctr D/UW3-Infected HeLa Cells

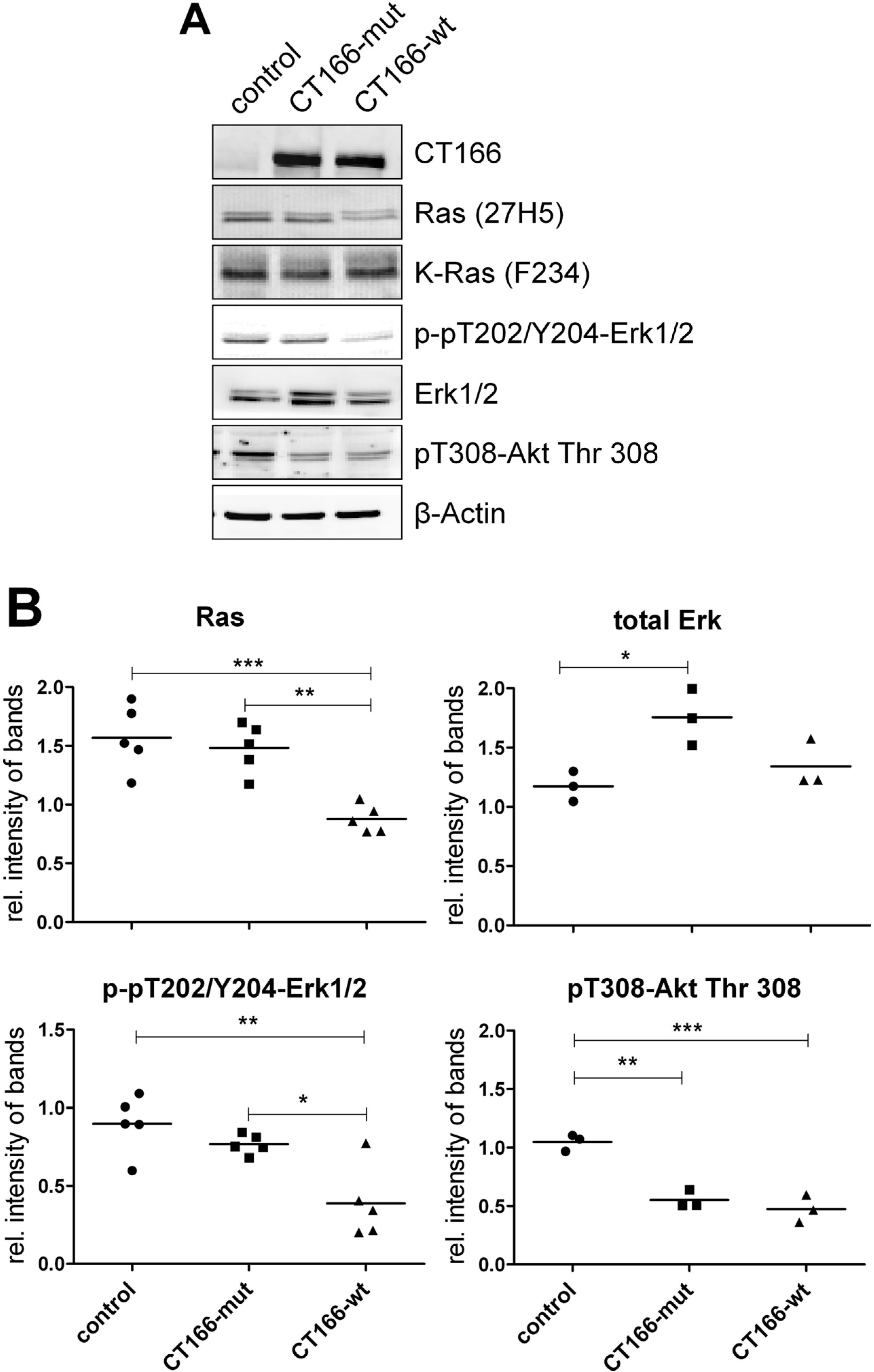
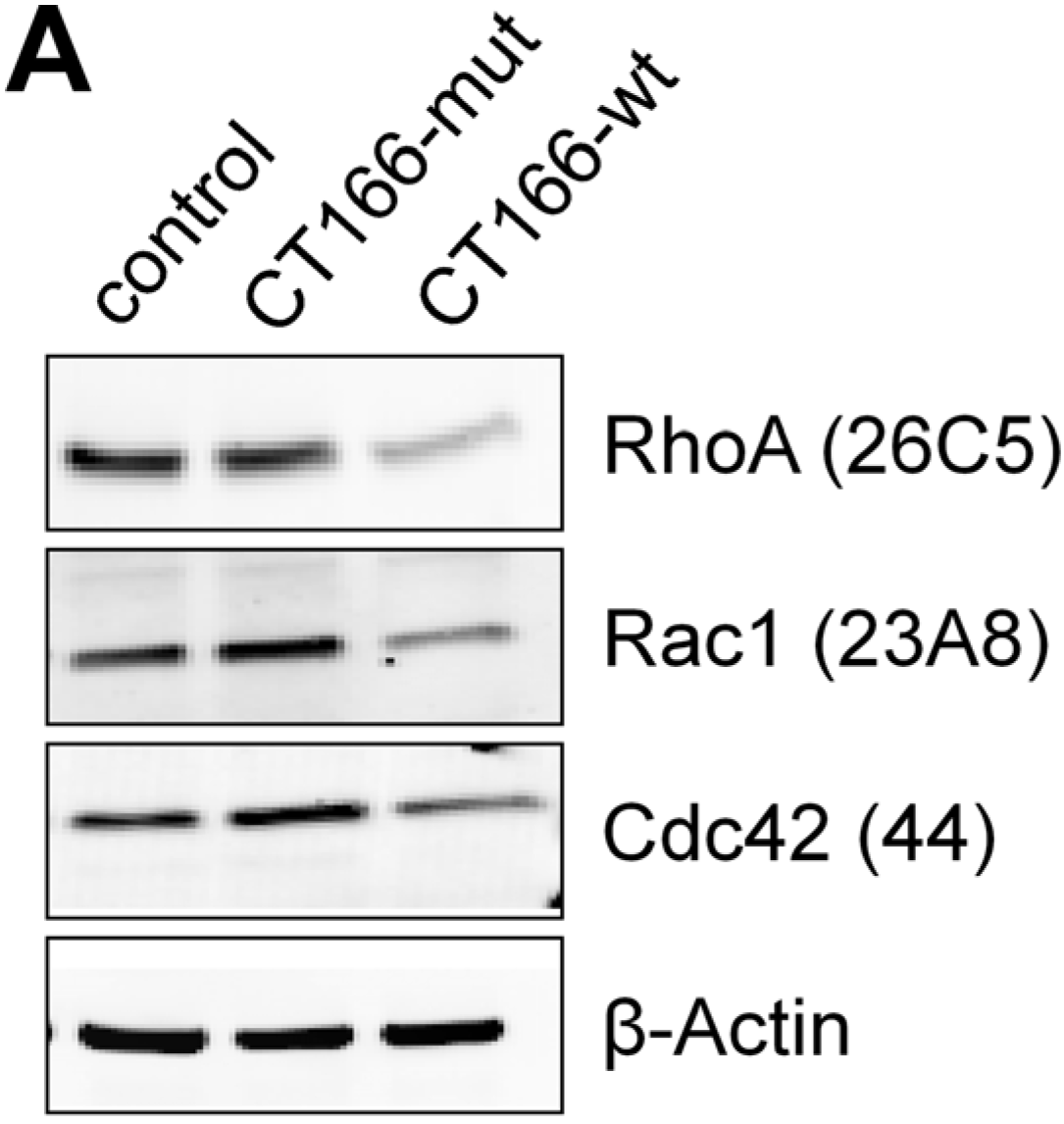
2.2. CT166-Mediated Inhibition of Ras-ERK and PI3K/Akt Signaling


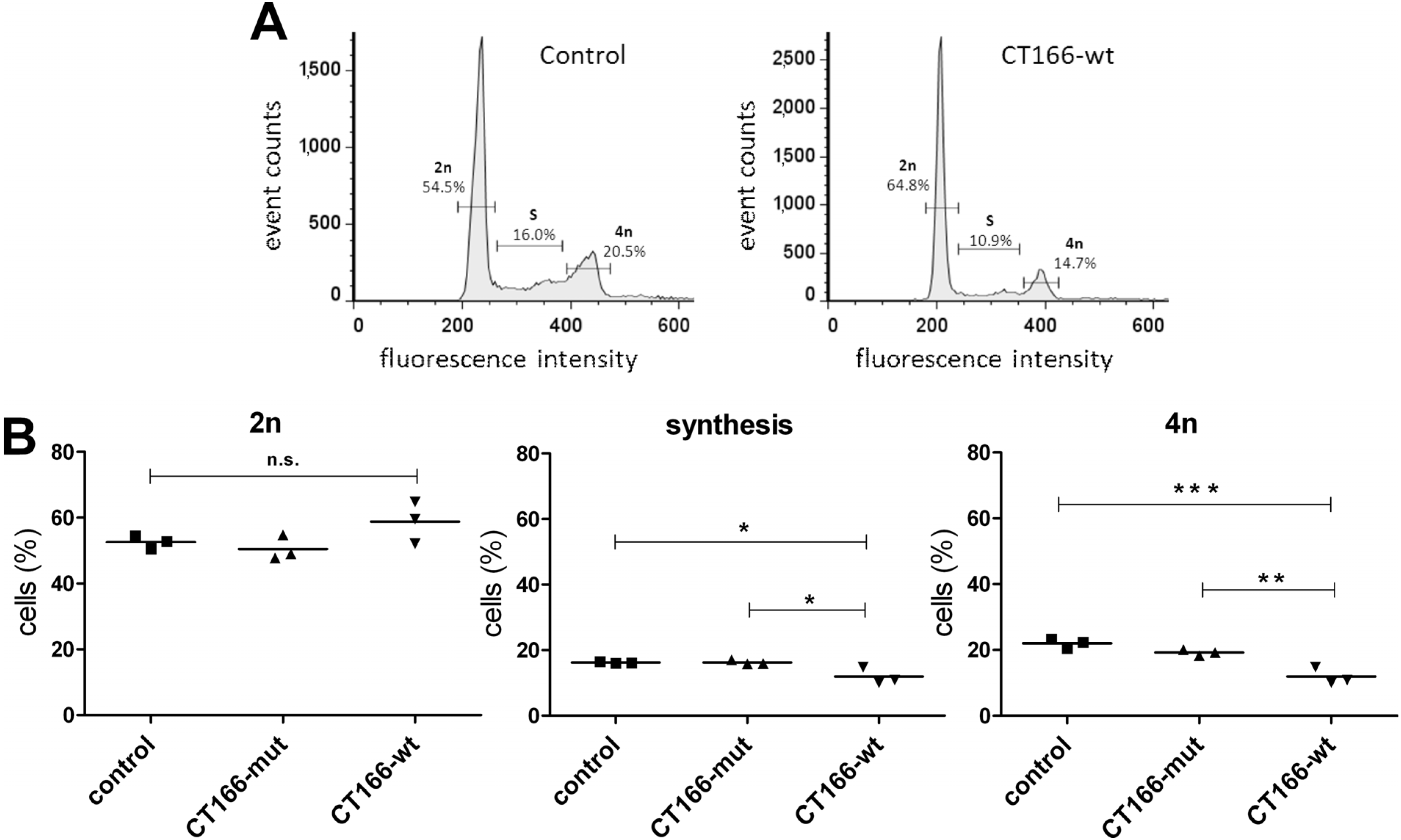
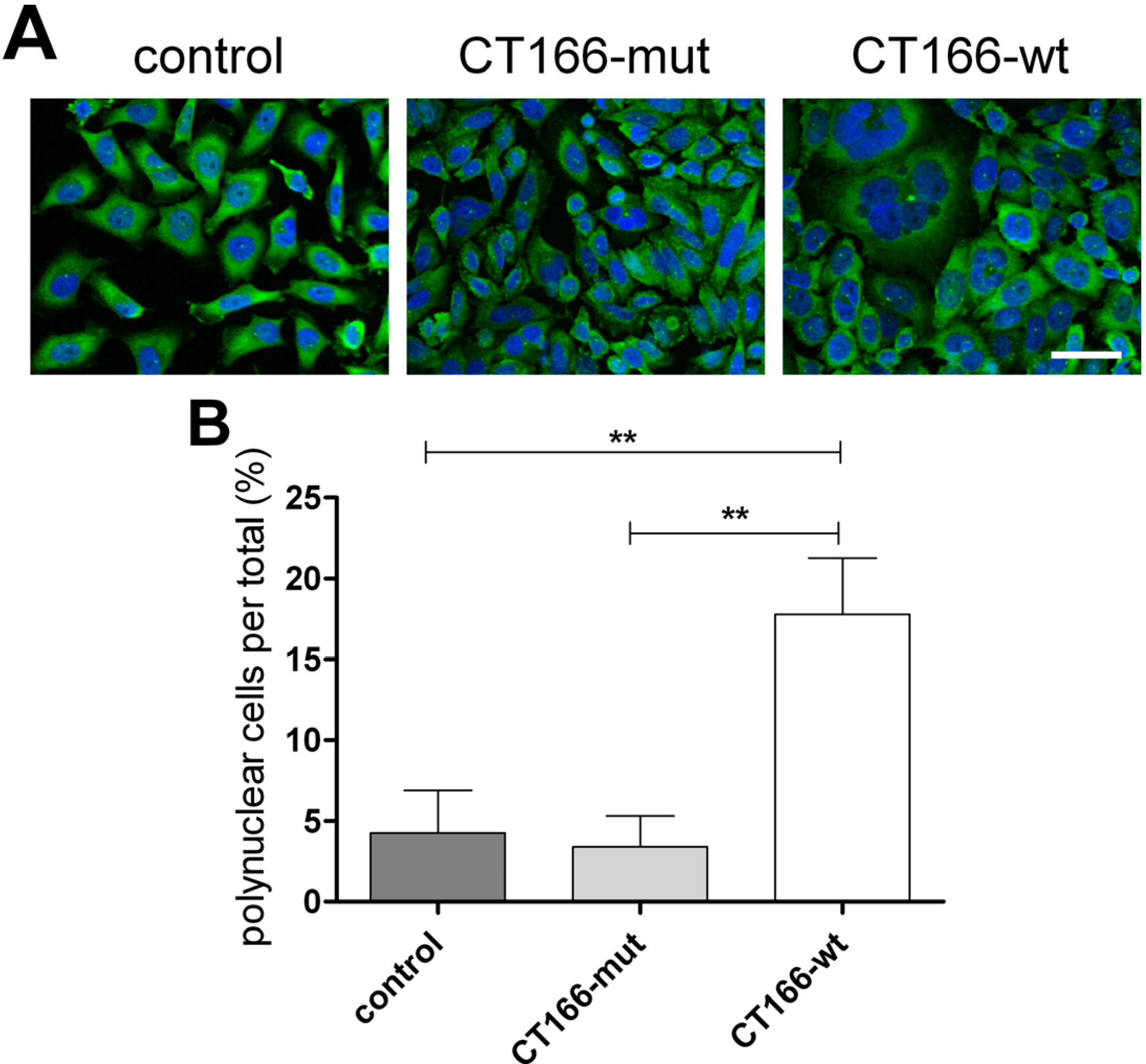
2.3. Formation of Multinucleated CT166-Expressing HeLa Cells



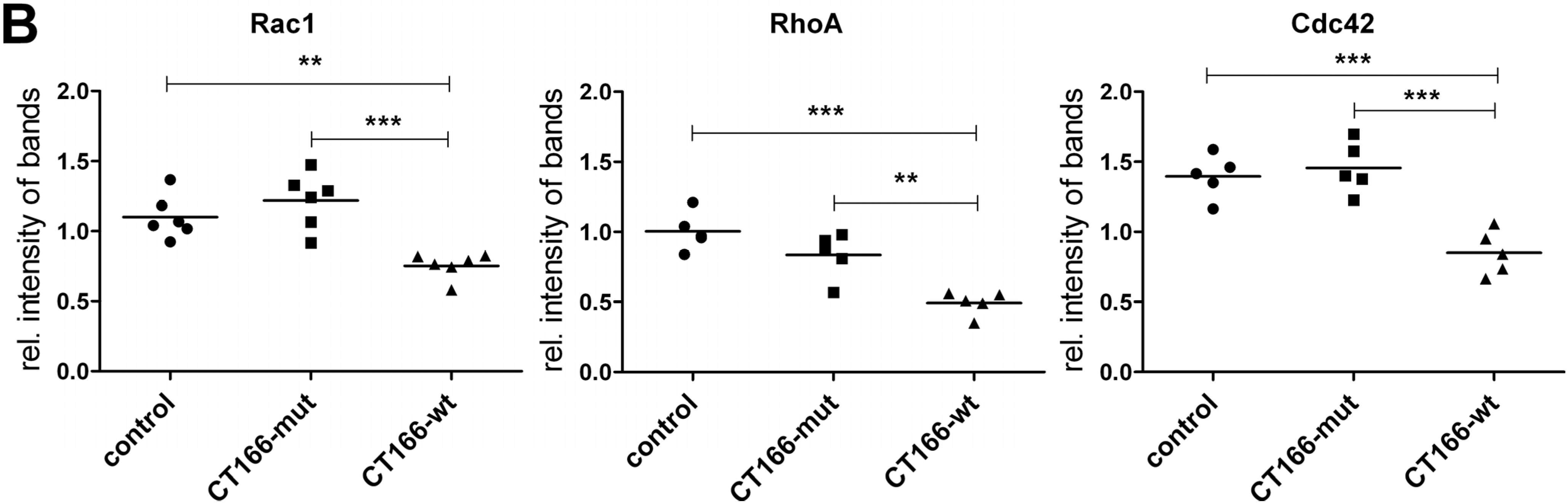
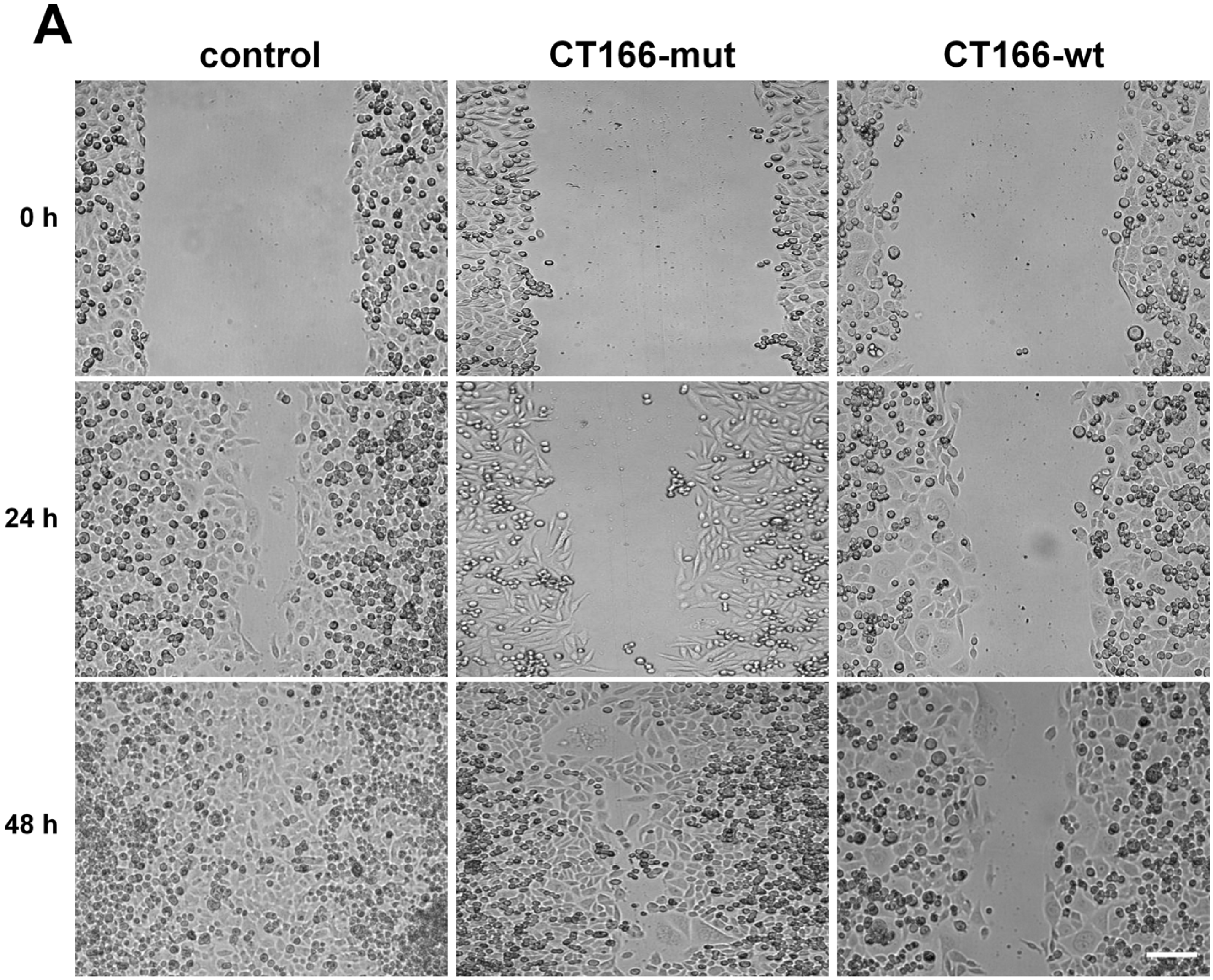
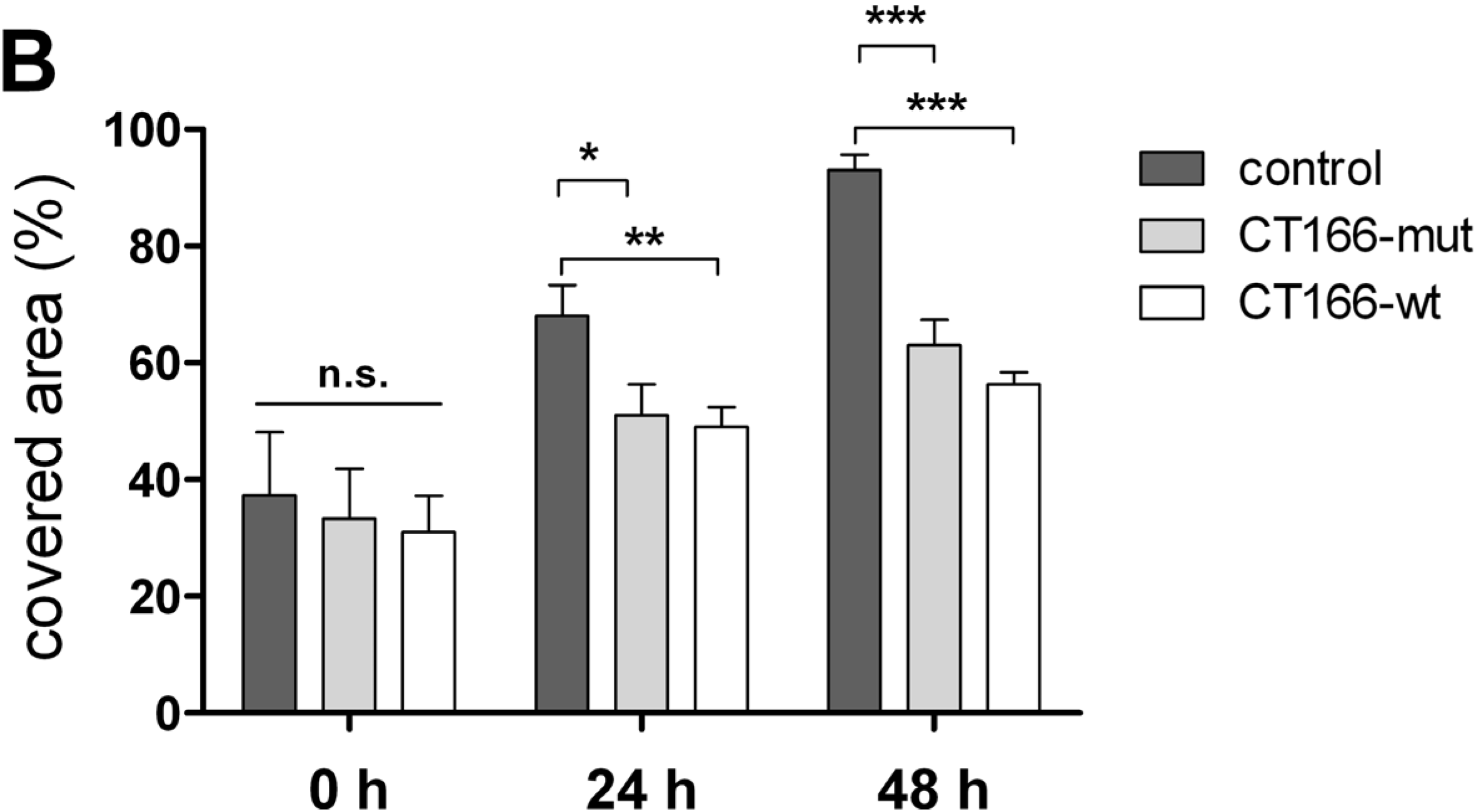
2.4. Reduced Migration of CT166-Expressing HeLa Cells


3. Experimental Section
3.1. Cell Cultures
3.2. Chlamydial Cultures
3.3. Infection Experiments of TRex™-HeLa Cells
3.4. Western Blotting
3.5. Analysis of Cell Cycle Progression
3.6. Cell Proliferation
3.7. Quantification of Multinuclear Cell Formation
3.8. Scratch Assay
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Breton, C.; Snajdrova, L.; Jeanneau, C.; Koca, J.; Imberty, A. Structures and mechanisms of glycosyltransferases. Glycobiology 2006, 16, 29R–37R. [Google Scholar] [CrossRef] [PubMed]
- Busch, C.; Aktories, K. Microbial toxins and the glycosylation of rho family GTPases. Curr. Opin. Struct. Biol. 2000, 10, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Dreger, S.C.; Huelsenbeck, J.; Just, I. Clostridium difficile toxins: More than mere inhibitors of Rho proteins. Int. J. Biochem. Cell Biol. 2008, 40, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R.; Geny, B. Rho/Ras-GTPase-dependent and -independent activity of clostridial glucosylating toxins. J. Med. Microbiol. 2011, 60, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Pauillac, S.; Schelle, I.; Bouvet, P.; Bouchier, C.; Varela-Chavez, C.; Just, I.; Popoff, M.R. Haemorrhagic toxin and lethal toxin from Clostridium sordellii strain vpi9048: Molecular characterization and comparative analysis of substrate specificity of the large clostridial glucosylating toxins. Cell. Microbiol. 2014, 16, 1706–1721. [Google Scholar] [CrossRef] [PubMed]
- Dreger, S.C.; Schulz, F.; Huelsenbeck, J.; Gerhard, R.; Hofmann, F.; Just, I.; Genth, H. Killing of rat basophilic leukemia cells by lethal toxin from Clostridium sordellii: Critical role of phosphatidylinositide 3′-OH kinase/Akt signaling. Biochemistry 2009, 48, 1785–1792. [Google Scholar] [CrossRef] [PubMed]
- Nottrott, S.; Schoentaube, J.; Genth, H.; Just, I.; Gerhard, R. Clostridium difficile toxin A-induced apoptosis is p53-independent but depends on glucosylation of Rho GTPases. Apoptosis 2007, 12, 1443–1453. [Google Scholar] [CrossRef] [PubMed]
- Busch, C.; Hofmann, F.; Selzer, J.; Munro, J.; Jeckel, D.; Aktories, K. A common motif of eukaryotic glycosyltransferases is essential for the enzyme activity of large clostridial cytotoxins. J. Biol. Chem. 1998, 273, 19566–19572. [Google Scholar] [CrossRef] [PubMed]
- Belyi, Y.; Aktories, K. Bacterial toxin and effector glycosyltransferases. Biochim. Biophys. Acta 2010, 1800, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Belyi, Y.; Jank, T.; Aktories, K. Cytotoxic glucosyltransferases of Legionella pneumophila. Curr. Top. Microbiol. Immunol. 2013, 376, 211–226. [Google Scholar] [PubMed]
- Byrne, G.I. Chlamydia trachomatis strains and virulence: Rethinking links to infection prevalence and disease severity. J. Infect. Dis. 2010, 201 (Suppl. 2), S126–S133. [Google Scholar] [CrossRef] [PubMed]
- Belland, R.J.; Scidmore, M.A.; Crane, D.D.; Hogan, D.M.; Whitmire, W.; McClarty, G.; Caldwell, H.D. Chlamydia trachomatis cytotoxicity associated with complete and partial cytotoxin genes. Proc. Natl. Acad. Sci. USA 2001, 98, 13984–13989. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, J.; Janik, K.; May, M.; Sommer, K.; Ebeling, J.; Hofmann, F.; Genth, H.; Klos, A. Actin re-organization induced by Chlamydia trachomatis serovar D—Evidence for a critical role of the effector protein CT166 targeting Rac. PLoS One 2010, 5, e9887. [Google Scholar] [CrossRef] [PubMed]
- Somboonna, N.; Wan, R.; Ojcius, D.M.; Pettengill, M.A.; Joseph, S.J.; Chang, A.; Hsu, R.; Read, T.D.; Dean, D. Hypervirulent Chlamydia trachomatis clinical strain is a recombinant between lymphogranuloma venereum (L(2)) and D lineages. MBio 2011, 2, e00045-11. [Google Scholar] [CrossRef] [PubMed]
- Read, T.D.; Brunham, R.C.; Shen, C.; Gill, S.R.; Heidelberg, J.F.; White, O.; Hickey, E.K.; Peterson, J.; Utterback, T.; Berry, K.; et al. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 2000, 28, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Brandes, V.; Schelle, I.; Brinkmann, S.; Schulz, F.; Schwarz, J.; Gerhard, R.; Genth, H. Protection from Clostridium difficile toxin B-catalysed Rac1/Cdc42 glucosylation by tauroursodeoxycholic acid-induced Rac1/Cdc42 phosphorylation. Biol. Chem. 2012, 393, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Huelsenbeck, J.; Hartmann, B.; Hofmann, F.; Just, I.; Gerhard, R. Cellular stability of Rho-GTPases glucosylated by Clostridium difficile toxin B. FEBS Lett. 2006, 580, 3565–3569. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, S.C.; Klose, I.; Reichenbach, M.; Huelsenbeck, J.; Genth, H. Distinct kinetics of (H/K/N)Ras glucosylation and Rac1 glucosylation catalysed by Clostridium sordellii lethal toxin. FEBS Lett. 2009, 583, 3133–3139. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Cheng, X.-L.; Zhou, M.; Li, Q. Chlamydia inhibit host cell apoptosis by inducing Bag-1 via the MAPK/ERK survival pathway. Apoptosis 2013, 18, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Zheng, Q.; Zhou, M.; Zhu, L.; Ai, B.; Zhou, L. Chlamydial antiapoptotic activity involves activation of the Raf/MEK/ERK survival pathway. Curr. Microbiol. 2011, 63, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Chumduri, C.; Gurumurthy, R.K.; Zadora, P.K.; Mi, Y.; Meyer, T.F. Chlamydia infection promotes host DNA damage and proliferation but impairs the DNA damage response. Cell Host Microbe 2013, 13, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Just, I.; Genth, H. Prevention of Clostridium sordellii lethal toxin-induced apoptotic cell death by tauroursodeoxycholic acid. Biochemistry 2009, 48, 9002–9010. [Google Scholar] [CrossRef] [PubMed]
- Guttenberg, G.; Hornei, S.; Jank, T.; Schwan, C.; Lu, W.; Einsle, O.; Papatheodorou, P.; Aktories, K. Molecular characteristics of Clostridium perfringens TpeL toxin and consequences of mono-O-GlcNAcylation of Ras in living cells. J. Biol. Chem. 2012, 287, 24929–24940. [Google Scholar] [CrossRef] [PubMed]
- Chambard, J.C.; Lefloch, R.; Pouyssegur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.; Jiao, H.; Liao, J.; Xu, X. Cytokinesis and cancer: Polo loves ROCK‘n’ Rho(A). J. Genet. Genomics 2010, 37, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Moorman, J.P.; Bobak, D.A.; Hahn, C.S. Inactivation of the small GTP binding protein Rho induces multinucleate cell formation and apoptosis in murine T lymphoma EL4. J. Immunol. 1996, 156, 4146–4153. [Google Scholar] [PubMed]
- Lica, M.; Schulz, F.; Schelle, I.; May, M.; Just, I.; Genth, H. Difference in the biological effects of Clostridium difficile toxin B in proliferating and non-proliferating cells. N–S Arch. Pharmacol. 2011, 383, 275–283. [Google Scholar] [CrossRef]
- Shoshan, M.C.; Aman, P.; Skog, S.; Florin, I.; Thelestam, M. Microfilament-disrupting Clostridium difficile toxin B causes multinucleation of transformed cells but does not block capping of membrane Ig. Eur. J. Cell Biol. 1990, 53, 357–363. [Google Scholar] [PubMed]
- Huelsenbeck, S.C.; May, M.; Schmidt, G.; Genth, H. Inhibition of cytokinesis by Clostridium difficile toxin B and cytotoxic necrotizing factors—Reinforcing the critical role of RhoA in cytokinesis. Cell Motil. Cytoskelet. 2009, 66, 967–975. [Google Scholar] [CrossRef]
- Hoffmann, C.; Pop, M.; Leemhuis, J.; Schirmer, J.; Aktories, K.; Schmidt, G. The Yersinia pseudotuberculosis cytotoxic necrotizing factor (CNFY) selectively activates RhoA. J. Biol. Chem. 2004, 279, 16026–16032. [Google Scholar] [CrossRef] [PubMed]
- Greene, W.; Zhong, G. Inhibition of host cell cytokinesis by Chlamydia trachomatis infection. J. Infect. 2003, 47, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Alzhanov, D.T.; Weeks, S.K.; Burnett, J.R.; Rockey, D.D. Cytokinesis is blocked in mammalian cells transfected with Chlamydia trachomatis gene CT223. BMC. Microbiol. 2009, 9, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Doye, A.; Mettouchi, A.; Bossis, G.; Clement, R.; Buisson-Touati, C.; Flatau, G.; Gagnoux, L.; Piechaczyk, M.; Boquet, P.; Lemichez, E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 2002, 111, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Rohrbeck, A.; Kolbe, T.; Hagemann, S.; Genth, H.; Just, I. Distinct biological activities of C3 and ADP-ribosyltransferase-deficient C3-E174Q. FEBS J. 2012, 279, 2657–2671. [Google Scholar] [CrossRef] [PubMed]
- Heuer, D.; Rejman, L.A.; Machuy, N.; Karlas, A.; Wehrens, A.; Siedler, F.; Brinkmann, V.; Meyer, T.F. Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature 2009, 457, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Heymann, J.; Rejman, L.A.; Bauer, B.; Meyer, T.F.; Heuer, D. Chlamydia trachomatis infection prevents front-rear polarity of migrating HeLa cells. Cell. Microbiol. 2013, 15, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.; Dreger, S.; Gerhard, R.; Barth, H.; Just, I.; Genth, H. Difference in the cytotoxic effects of toxin B from Clostridium difficile strain VPI 10463 and toxin B from variant Clostridium difficile strain 1470. Infect. Immun. 2007, 75, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Reinert, D.J.; Jank, T.; Aktories, K.; Schulz, G.E. Structural basis for the function of Clostridium difficile toxin B. J. Mol. Biol. 2005, 351, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Teichert, M.; Tatge, H.; Schoentaube, J.; Just, I.; Gerhard, R. Application of mutated Clostridium difficile toxin A for determination of glucosyltransferase-dependent effects. Infect. Immun. 2006, 74, 6006–6010. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Geback, T.; Schulz, M.M.; Koumoutsakos, P.; Detmar, M. TScratch: A novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques 2009, 46, 265–274. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bothe, M.; Dutow, P.; Pich, A.; Genth, H.; Klos, A. DXD Motif-Dependent and -Independent Effects of the Chlamydia trachomatis Cytotoxin CT166. Toxins 2015, 7, 621-637. https://doi.org/10.3390/toxins7020621
Bothe M, Dutow P, Pich A, Genth H, Klos A. DXD Motif-Dependent and -Independent Effects of the Chlamydia trachomatis Cytotoxin CT166. Toxins. 2015; 7(2):621-637. https://doi.org/10.3390/toxins7020621
Chicago/Turabian StyleBothe, Miriam, Pavel Dutow, Andreas Pich, Harald Genth, and Andreas Klos. 2015. "DXD Motif-Dependent and -Independent Effects of the Chlamydia trachomatis Cytotoxin CT166" Toxins 7, no. 2: 621-637. https://doi.org/10.3390/toxins7020621
APA StyleBothe, M., Dutow, P., Pich, A., Genth, H., & Klos, A. (2015). DXD Motif-Dependent and -Independent Effects of the Chlamydia trachomatis Cytotoxin CT166. Toxins, 7(2), 621-637. https://doi.org/10.3390/toxins7020621