
Citreoviridin Induces Autophagy-Dependent Apoptosis through Lysosomal-Mitochondrial Axis in Human Liver HepG2 Cells

Abstract
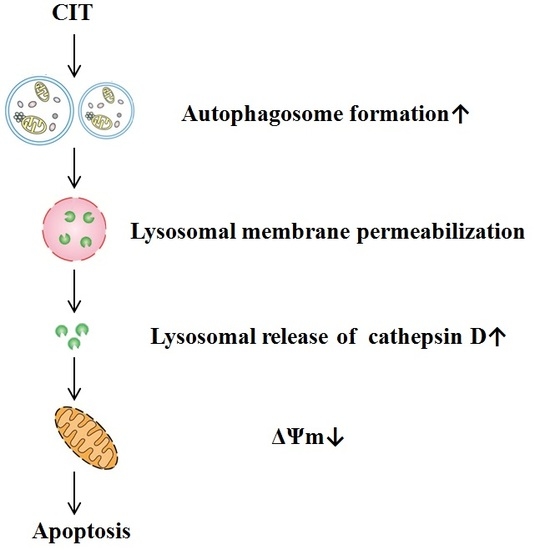
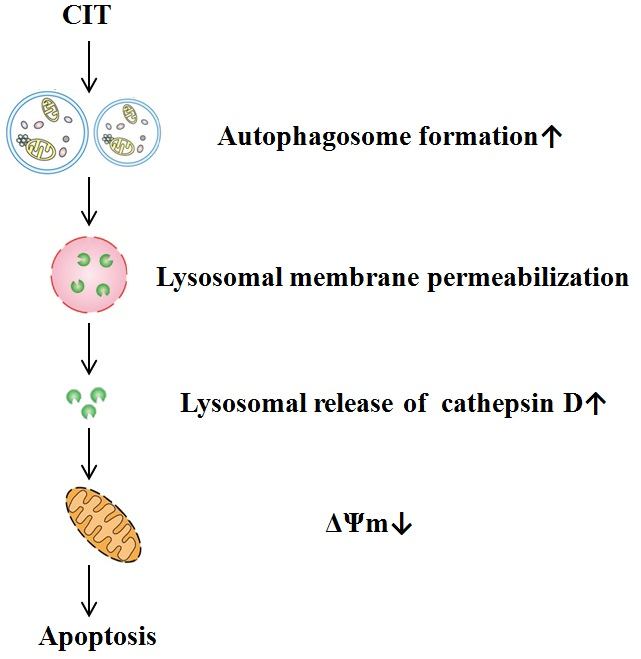
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
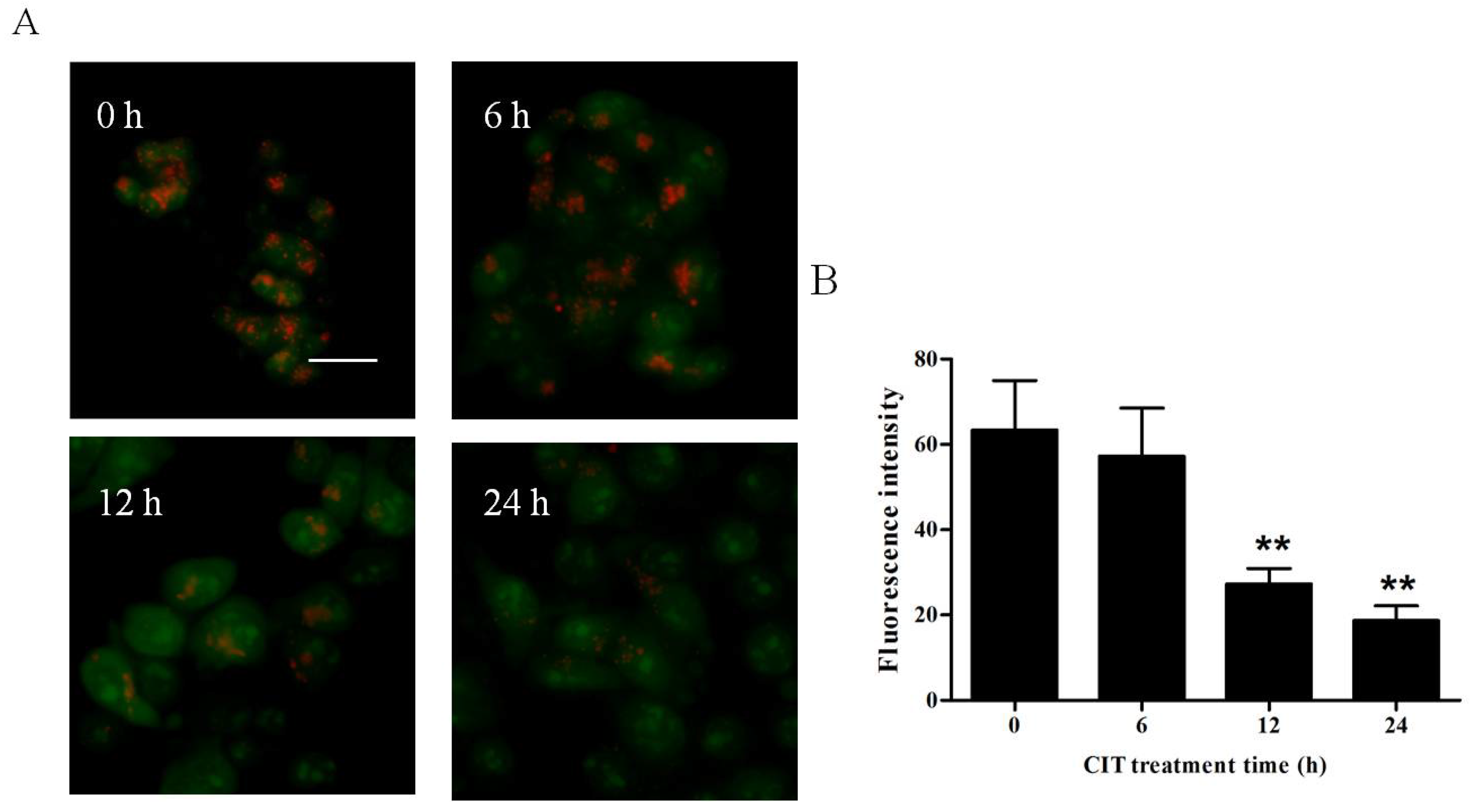
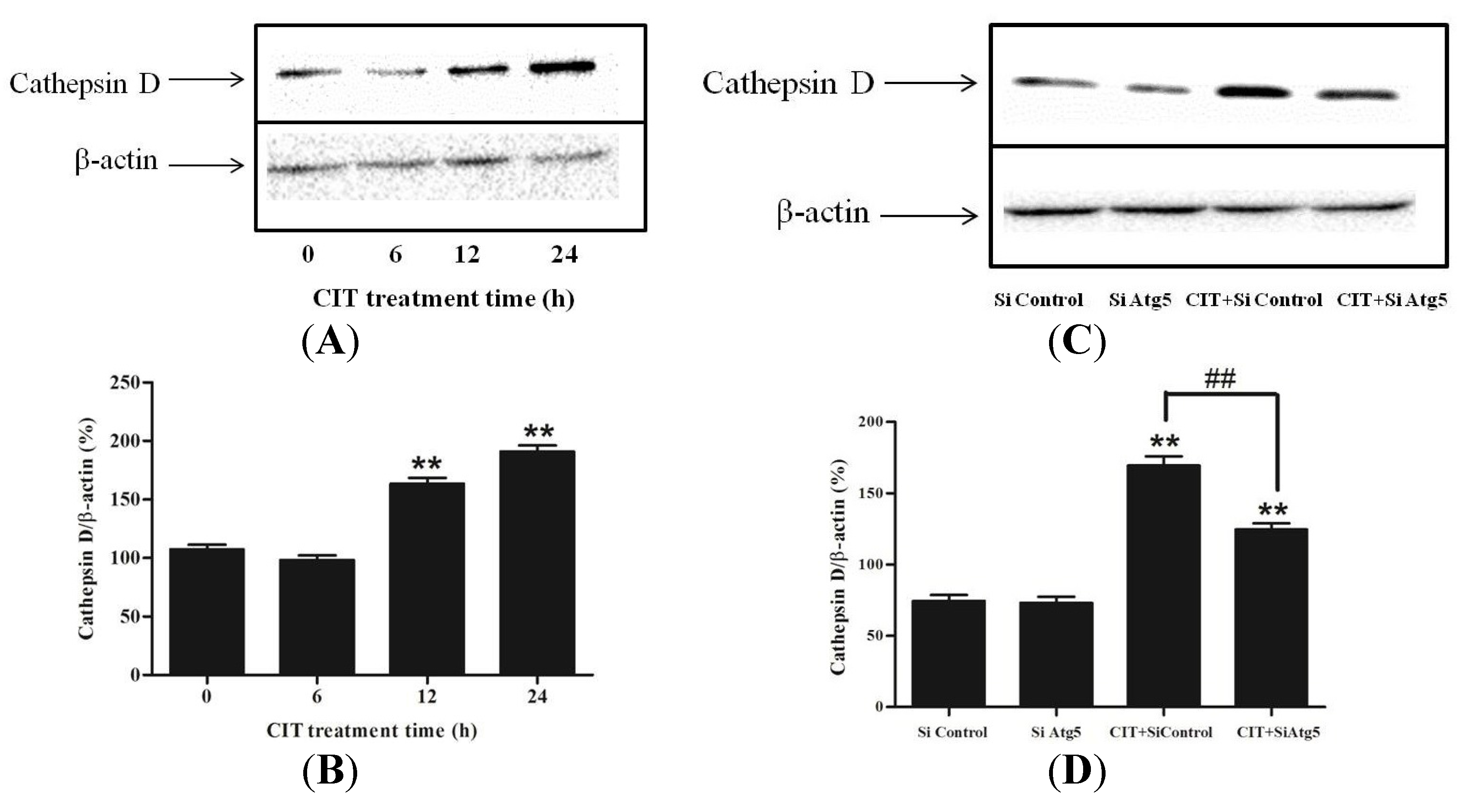
2.1. CIT Increased the Expression of LC3-II from the Early Stage of Treatment

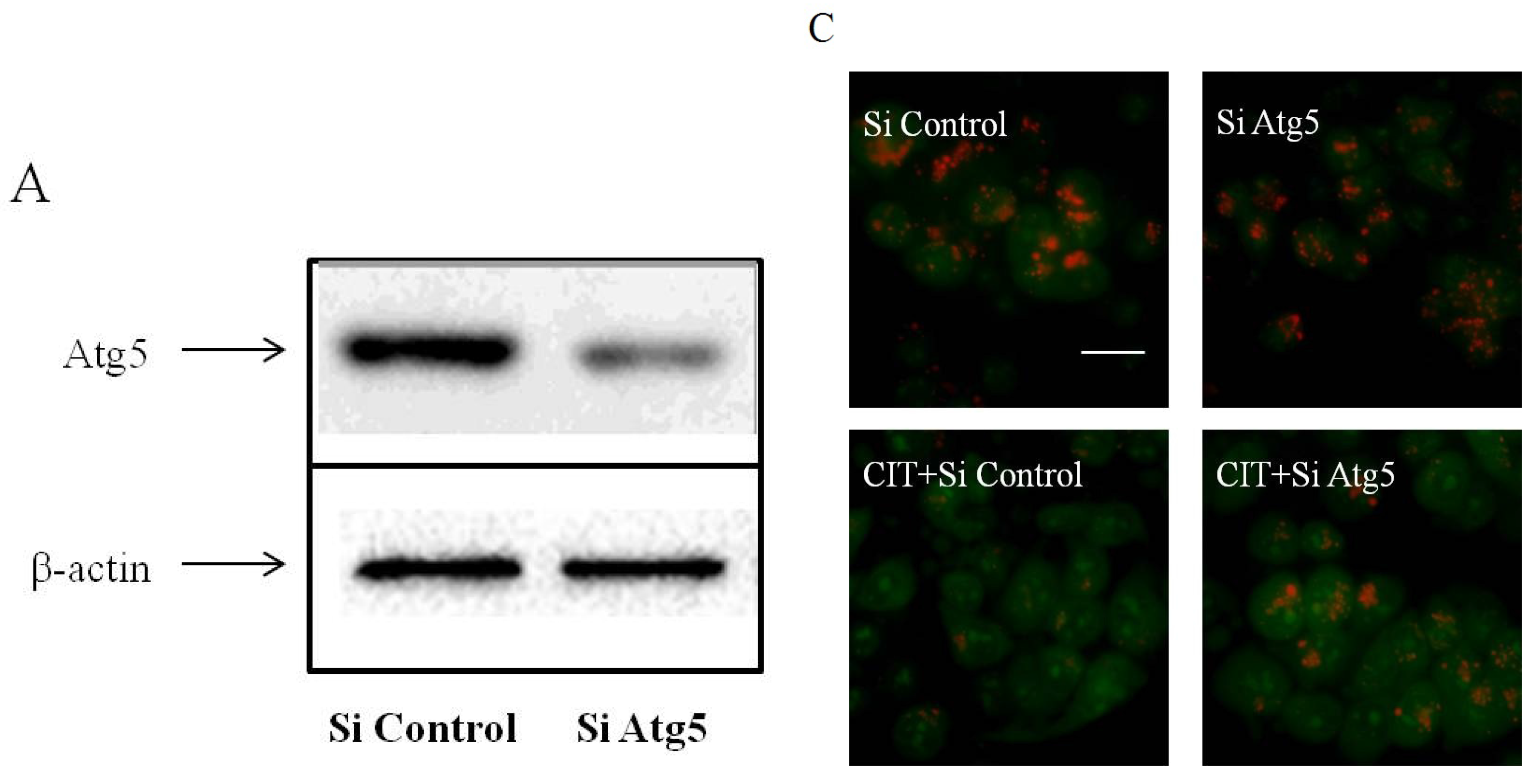
2.2. CIT-Induced LMP Was Autophagy Dependent

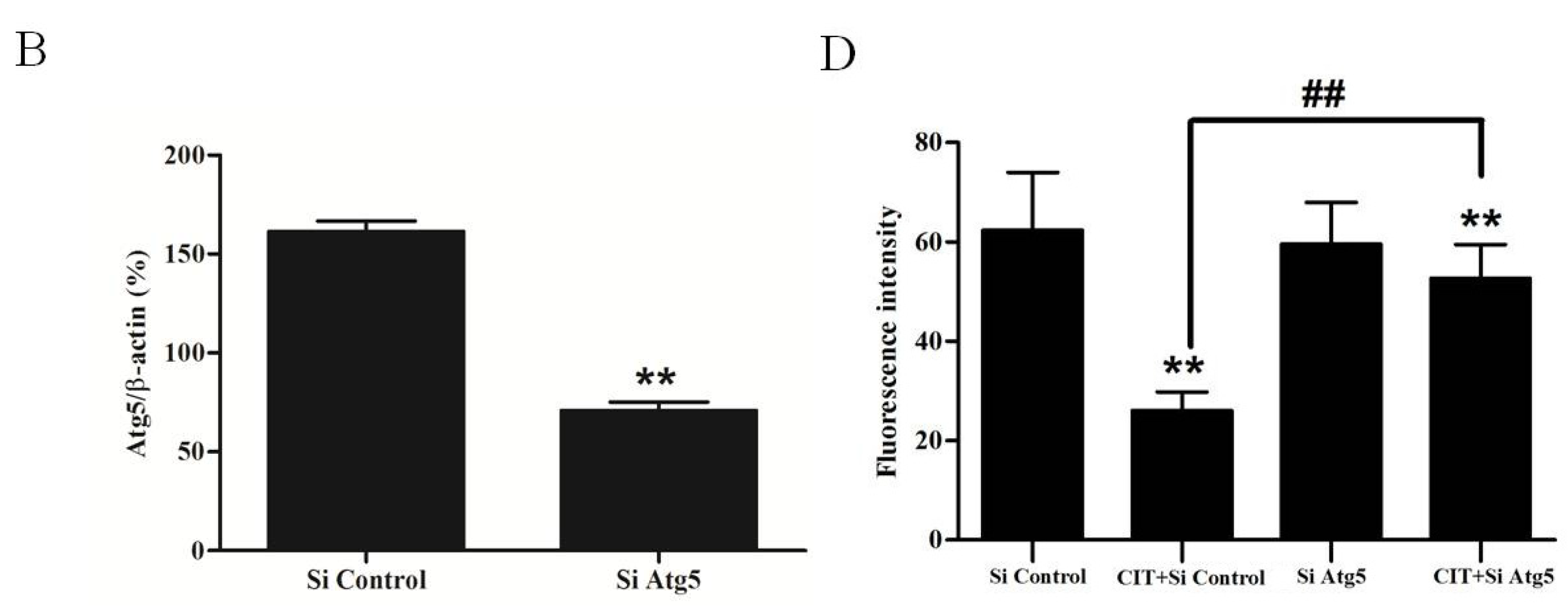
2.3. Inhibition of Autophagosome Formation Alleviated CIT-induced Lysosomal Release of Cathepsin D



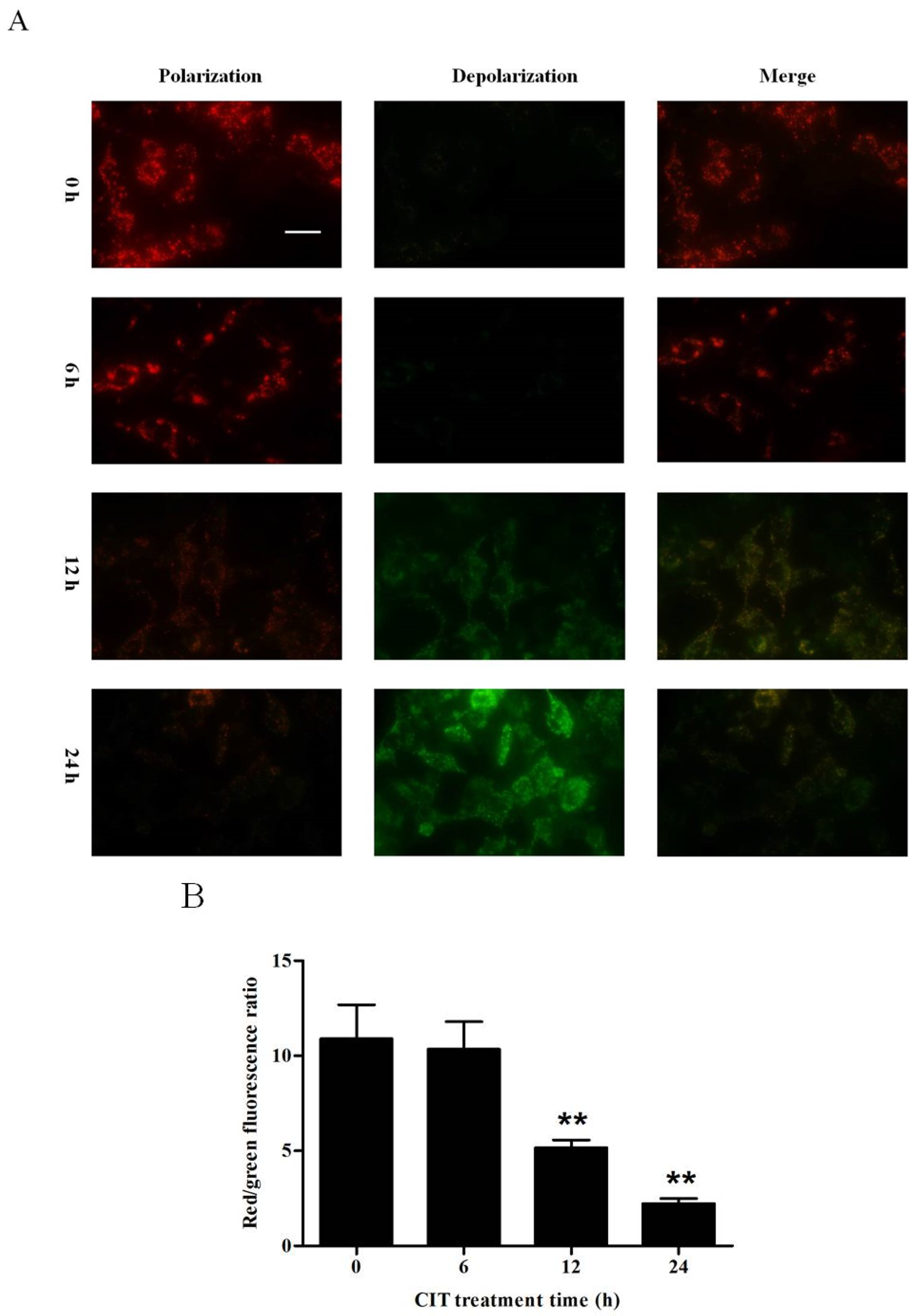
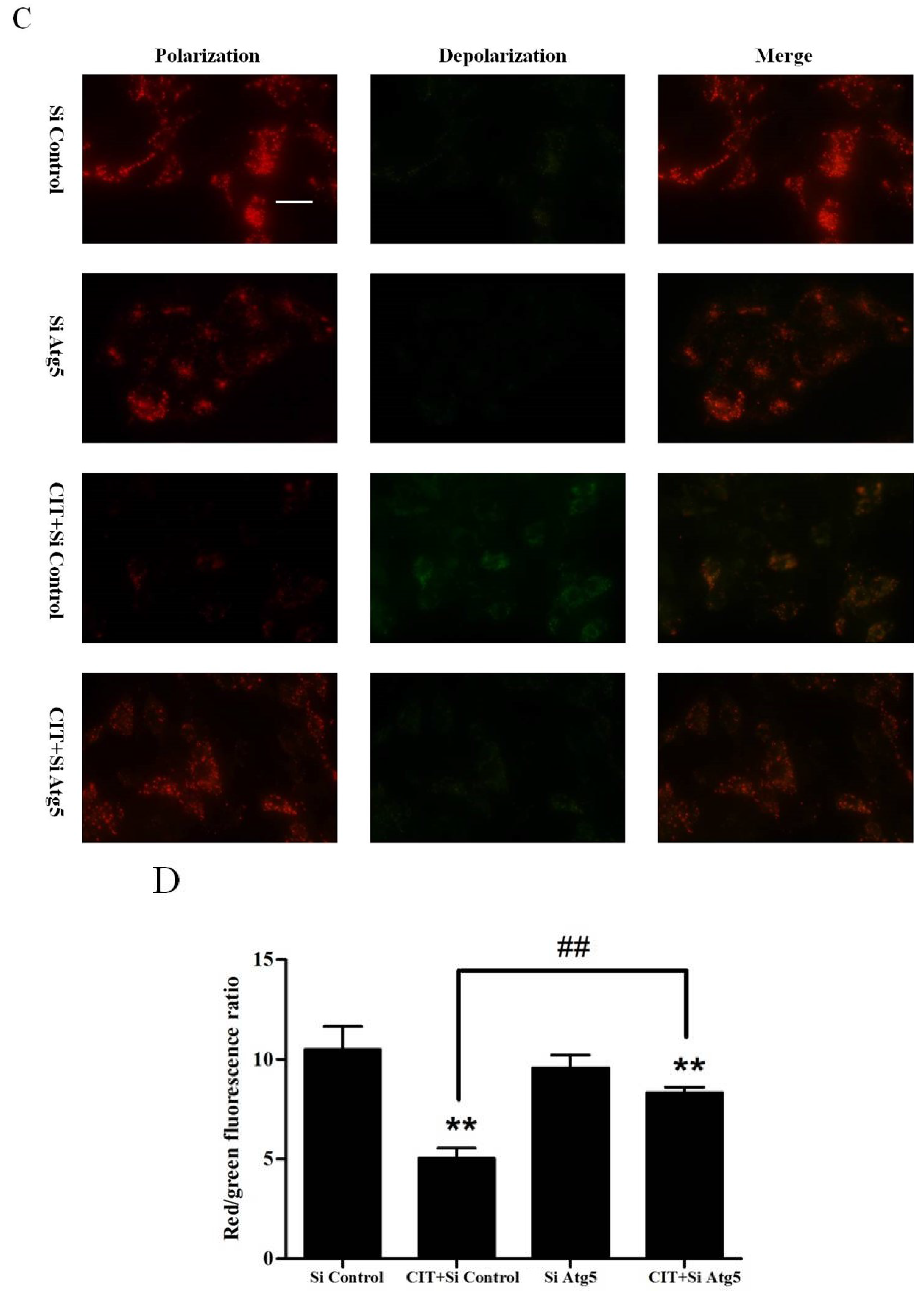
2.4. CIT-induced Collapse of Mitochondrial Transmembrane Potential (ΔΨm) Was Autophagy Dependent


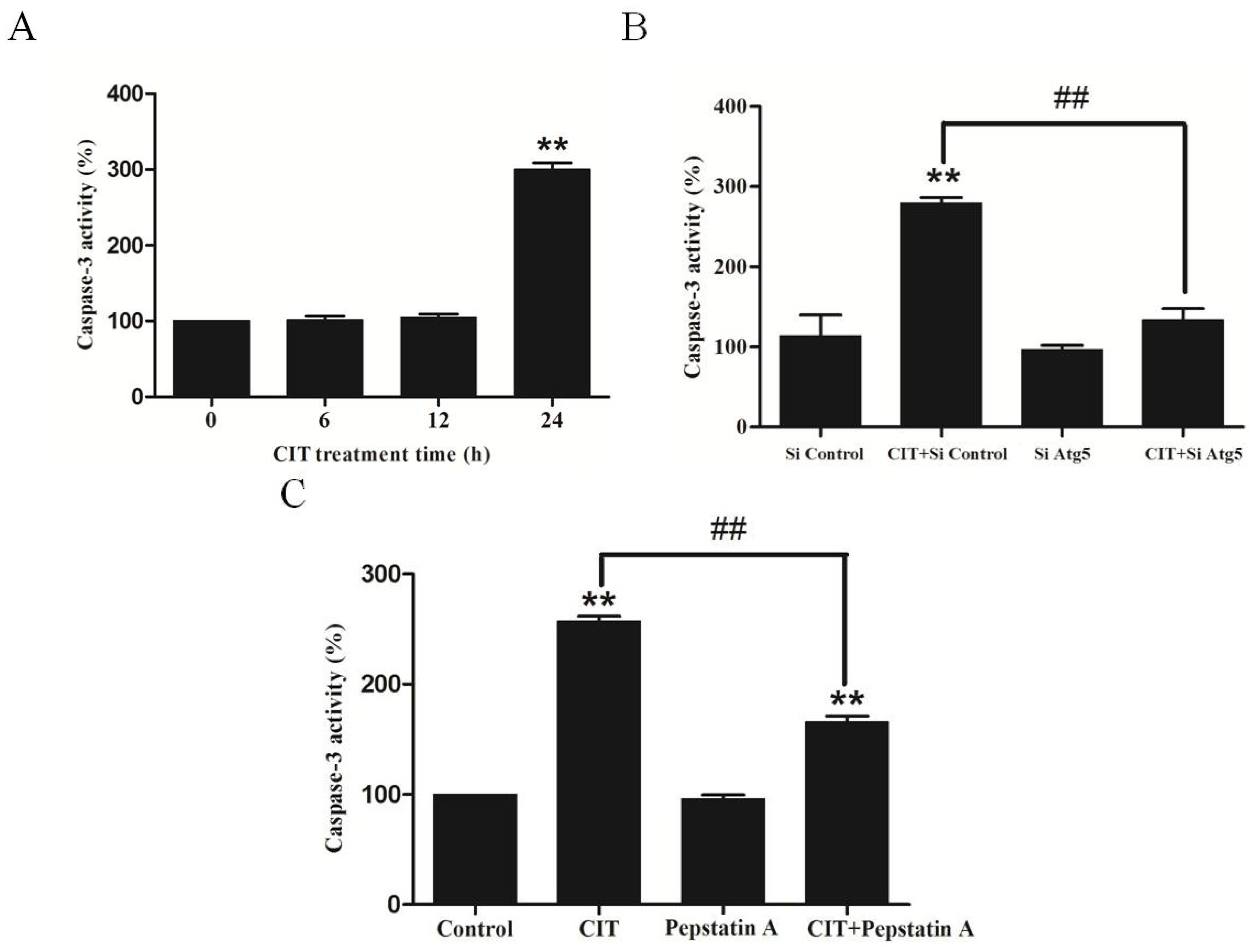
2.5. CIT Induced Autophagy-Dependent Apoptosis in HepG2 Cells

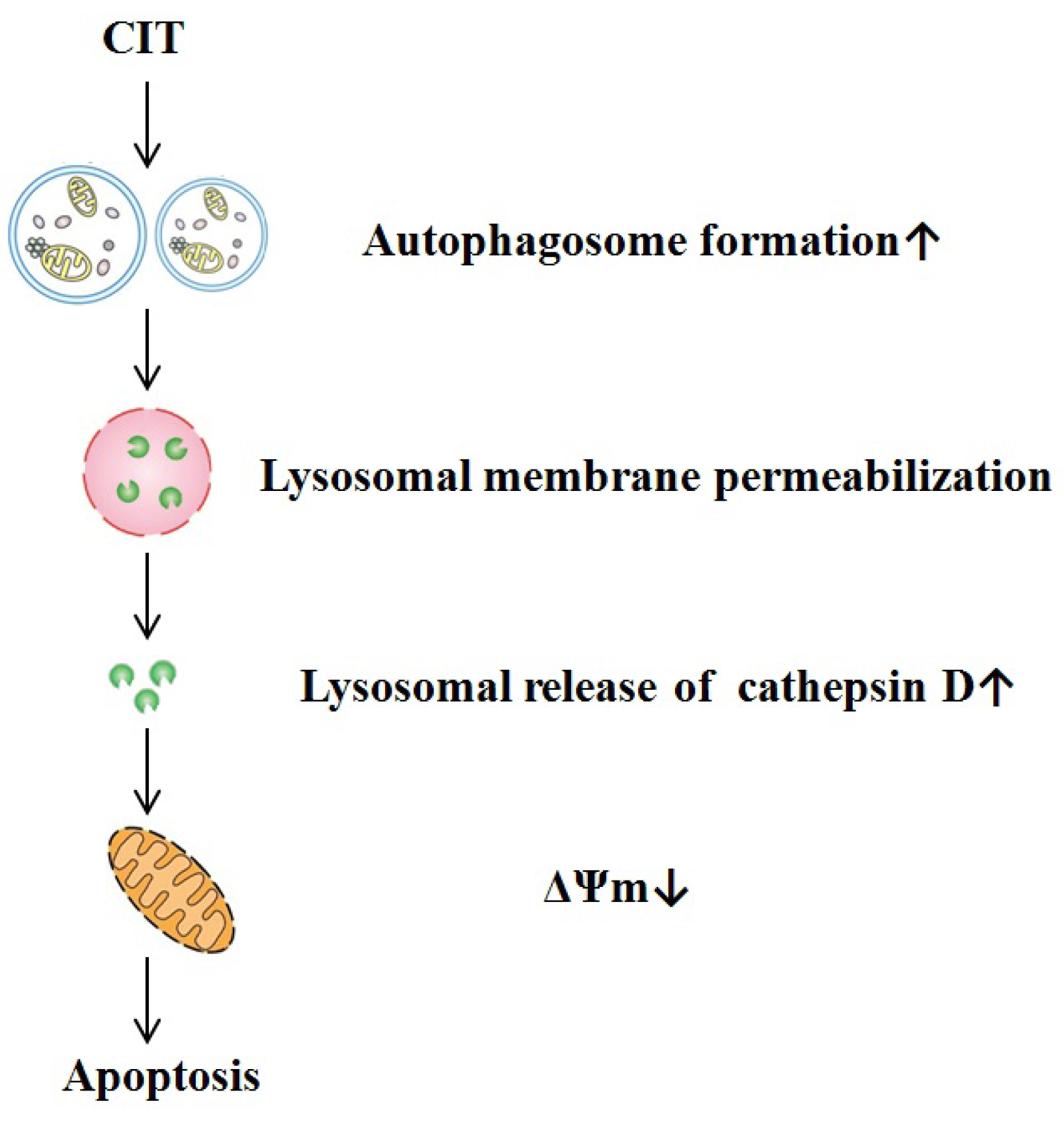
3. Discussion

4. Experimental Section
4.1. Cell Culture and Treatment
4.2. RNA Interference
4.3. Western Blot Analysis
4.4. Measurement of Lysosomal Membrane Stability by AO Staining
4.5. Measurement of ΔΨm
4.6. Caspase-3 Assay
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lima, H.C.; Porto, E.A.; Marins, J.R.; Alves, R.M.; Machado, R.R.; Braga, K.N.; de Paiva, F.B.; Carmo, G.M.; Silva e Santelli, A.C.; Sobel, J. Outbreak of beriberi in the state of Maranhão, Brazil: revisiting the mycotoxin aetiologic hypothesis. Trop. Doct. 2010, 40, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Rosa, C.A.; Keller, K.M.; Oliveira, A.A.; Almeida, T.X.; Keller, L.A.; Marassi, A.C.; Kruger, C.D.; Deveza, M.V.; Monteiro, B.S.; Nunes, L.M.; et al. Production of citreoviridin by Penicillium citreonigrum strains associated with rice consumption and beriberi cases in the Maranhão State, Brazil. Food Addit. Contam. Part A 2010, 27, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.I.; Almeida, N.G.; Carvalho, K.L.; Gonçalves, G.A.; Silva, C.N.; Santos, E.A.; Garcia, J.C.; Vargas, E.A. Co-occurrence of aflatoxins B1, B2, G1 and G2, ochratoxin A, zearalenone, deoxynivalenol, and citreoviridin in rice in Brazil. Food Addit. Contam. Part A 2012, 29, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, R.E.; Vesonder, R.F. Teratogenic potential of the mycotoxin, citreoviridin, in rats. Food Chem. Toxicol. 1986, 24, 1315–1320. [Google Scholar] [CrossRef]
- Liu, Y.N.; Wang, Y.X.; Liu, X.F.; Jiang, L.P.; Yang, G.; Sun, X.C.; Geng, C.Y.; Li, Q.J.; Chen, M.; Yao, X.F. Citreoviridin induces ROS-dependent autophagic cell death in human liver HepG2 cells. Toxicon 2015, 95, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Young, M.M.; Takahashi, Y.; Khan, O.; Park, S.; Hori, T.; Yun, J.; Sharma, A.K.; Amin, S.; Hu, C.D.; Zhang, J.; et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2012, 287, 12455–12468. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D.H.; Price, M.; Reiners, J.J., Jr. ATG7 deficiency suppresses apoptosis and cell death induced by lysosomal photodamage. Autophagy 2012, 8, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, S.; Ryu, H.Y.; Chung, K.M.; Baek, S.H.; Kim, E.K.; Yu, S.W. Regulation of autophagic cell death by glycogen synthase kinase-3β in adult hippocampal neural stem cells following insulin withdrawal. Mol. Brain 2015, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.C.; Appelqvist, H.; Nilsson, C.; Kågedal, K.; Roberg, K.; Ollinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 2010, 15, 527–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granato, M.; Lacconi, V.; Peddis, M.; Lotti, L.V.; Di Renzo, L.; Gonnella, R.; Santarelli, R.; Trivedi, P.; Frati, L.; D’Orazi, G.; et al. HSP70 inhibition by 2-phenylethynesulfonamide induces lysosomal cathepsin D release and immunogenic cell death in primary effusion lymphoma. Cell Death Dis. 2013, 4, e730. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, X.; Nie, X.; Sun, L.; Tang, T.S.; Chen, D.; Sun, Q. COX5B regulates MAVS-mediated antiviral signaling through interaction with ATG5 and repressing ROS production. PLoS Pathog. 2012, 8, e1003086. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.F.; Wu, C.L.; Huang, S.C.; Wu, C.M.; Hsiao, J.R.; Yo, Y.T.; Chen, Y.H.; Shiau, A.L.; Chou, C.Y. Cathepsin L mediates resveratrol-induced autophagy and apoptotic cell death in cervical cancer cells. Autophagy 2009, 5, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.L.; Simon, A.K.; Prescott, M.; Menendez, J.A.; Liu, F.; Wang, F.; Wang, C.; Wolvetang, E.; Vazquez-Martin, A.; Zhang, J. Autophagy in stem cells. Autophagy 2013, 9, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Pivtoraiko, V.N.; Stone, S.L.; Roth, K.A.; Shacka, J.J. Oxidative stress and autophagy in the regulation of lysosome-dependent neuron death. Antioxid. Redox. Signal 2009, 11, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Laforge, M.; Limou, S.; Harper, F.; Casartelli, N.; Rodrigues, V.; Silvestre, R.; Haloui, H.; Zagury, J.F.; Senik, A.; Estaquier, J. DRAM triggers lysosomal membrane permeabilization and cell death in CD4(+) T cells infected with HIV. PLoS. Pathog. 2013, 9, e1003328. [Google Scholar] [CrossRef] [PubMed]
- Owa, C.; Messina, M.E., Jr.; Halaby, R. Triptolide induces lysosomal-mediated programmed cell death in MCF-7 breast cancer cells. Int. J. Womens Health 2013, 5, 557–569. [Google Scholar] [PubMed]
- Lorin, S.; Borges, A.; Ribeiro, Dos.; Santos, L.; Souquère, S.; Pierron, G.; Ryan, K.M.; Codogno, P.; Djavaheri-Mergny, M. c-Jun NH2-terminal kinase activation is essential for DRAM-dependent induction of autophagy and apoptosis in 2-methoxyestradiol-treated Ewing sarcoma cells. Cancer Res. 2009, 69, 6924–6931. [Google Scholar] [PubMed]
- Wang, Q.; Chen, Z.; Diao, X.; Huang, S. Induction of autophagy-dependent apoptosis by the survivin suppressant YM155 in prostate cancer cells. Cancer Lett. 2011, 302, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Hanzel, C.E.; Almeira Gubiani, M.F.; Verstraeten, S.V. Endosomes and lysosomes are involved in early steps of Tl(III)-mediated apoptosis in rat pheochromocytoma (PC12) cells. Arch. Toxicol. 2012, 86, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Li, Q.; Zhao, X.H.; Wang, H.G.; Li, N.; Fang, Y.; Wang, K.; Jia, Y.P.; Zhu, P.; Gu, J.; et al. Shiga toxins induce autophagic cell death in intestinal epithelial cells via the endoplasmic reticulum stress pathway. Autophagy 2015, 11, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Guo, X.; Li, J.; Fan, L.; Hu, H. Fumonisin B1 induces autophagic cell death via activation of ERN1-MAPK8/9/10 pathway in monkey kidney MARC-145 cells. Arch. Toxicol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Pan, W.; Zhu, M.; Zhang, M.; Hao, X.; Liang, G.; Feng, Y. Fangchinoline induces autophagic cell death via p53/sestrin2/AMPK signalling in human hepatocellular carcinoma cells. Br. J. Pharmacol. 2011, 164, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Emert-Sedlak, L.; Shangary, S.; Rabinovitz, A.; Miranda, M.B.; Delach, S.M.; Johnson, D.E. Involvement of cathepsin D in chemotherapy-induced cytochrome c release, caspase activation, and cell death. Mol. Cancer Ther. 2005, 4, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cheng, B.; Miao, L.; Mei, Y.; Wu, M. Mutant p53-R273H gains new function in sustained activation of EGFR signaling via suppressing miR-27a expression. Cell Death Dis. 2013, 4, e574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, N.D.; Zhou, F.; Shen, T.; Duan, T.; Zhou, J.; Shi, Y.; Zhu, X.Q.; Shen, H.M. (-)-Epigallocatechin-3-gallate induces non-apoptotic cell death in human cancer cells via ROS-mediated lysosomal membrane permeabilization. PLoS ONE 2012, 7, e46749. [Google Scholar] [CrossRef] [PubMed]
- Stroikin, Y.; Mild, H.; Johansson, U.; Roberg, K.; Ollinger, K. Lysosome-targeted stress reveals increased stability of lipofuscin-containing lysosomes. Age 2008, 30, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, X.; Li, Y.; Leu, C.; Guo, L.; Zheng, X.; Zhu, D. 20-Hydroxyeicosatetraenoic acid inhibits the apoptotic responses in pulmonary artery smooth muscle cells. Eur. J. Pharmacol. 2008, 588, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, R.A.; Geenen, D.L. Mesenchymal Stem Cells Secrete Multiple Cytokines That Promote Angiogenesis and Have Contrasting Effects on Chemotaxis and Apoptosis. PLoS ONE 2012, 7, e35685. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Liu, Y.; Liu, X.; Jiang, L.; Yang, G.; Sun, X.; Geng, C.; Li, Q.; Yao, X.; Chen, M. Citreoviridin Induces Autophagy-Dependent Apoptosis through Lysosomal-Mitochondrial Axis in Human Liver HepG2 Cells. Toxins 2015, 7, 3030-3044. https://doi.org/10.3390/toxins7083030
Wang Y, Liu Y, Liu X, Jiang L, Yang G, Sun X, Geng C, Li Q, Yao X, Chen M. Citreoviridin Induces Autophagy-Dependent Apoptosis through Lysosomal-Mitochondrial Axis in Human Liver HepG2 Cells. Toxins. 2015; 7(8):3030-3044. https://doi.org/10.3390/toxins7083030
Chicago/Turabian StyleWang, Yuexia, Yanan Liu, Xiaofang Liu, Liping Jiang, Guang Yang, Xiance Sun, Chengyan Geng, Qiujuan Li, Xiaofeng Yao, and Min Chen. 2015. "Citreoviridin Induces Autophagy-Dependent Apoptosis through Lysosomal-Mitochondrial Axis in Human Liver HepG2 Cells" Toxins 7, no. 8: 3030-3044. https://doi.org/10.3390/toxins7083030
APA StyleWang, Y., Liu, Y., Liu, X., Jiang, L., Yang, G., Sun, X., Geng, C., Li, Q., Yao, X., & Chen, M. (2015). Citreoviridin Induces Autophagy-Dependent Apoptosis through Lysosomal-Mitochondrial Axis in Human Liver HepG2 Cells. Toxins, 7(8), 3030-3044. https://doi.org/10.3390/toxins7083030