
Recombinant HSA-CMG2 Is a Promising Anthrax Toxin Inhibitor

 ,
,
Abstract
:1. Introduction
2. Results
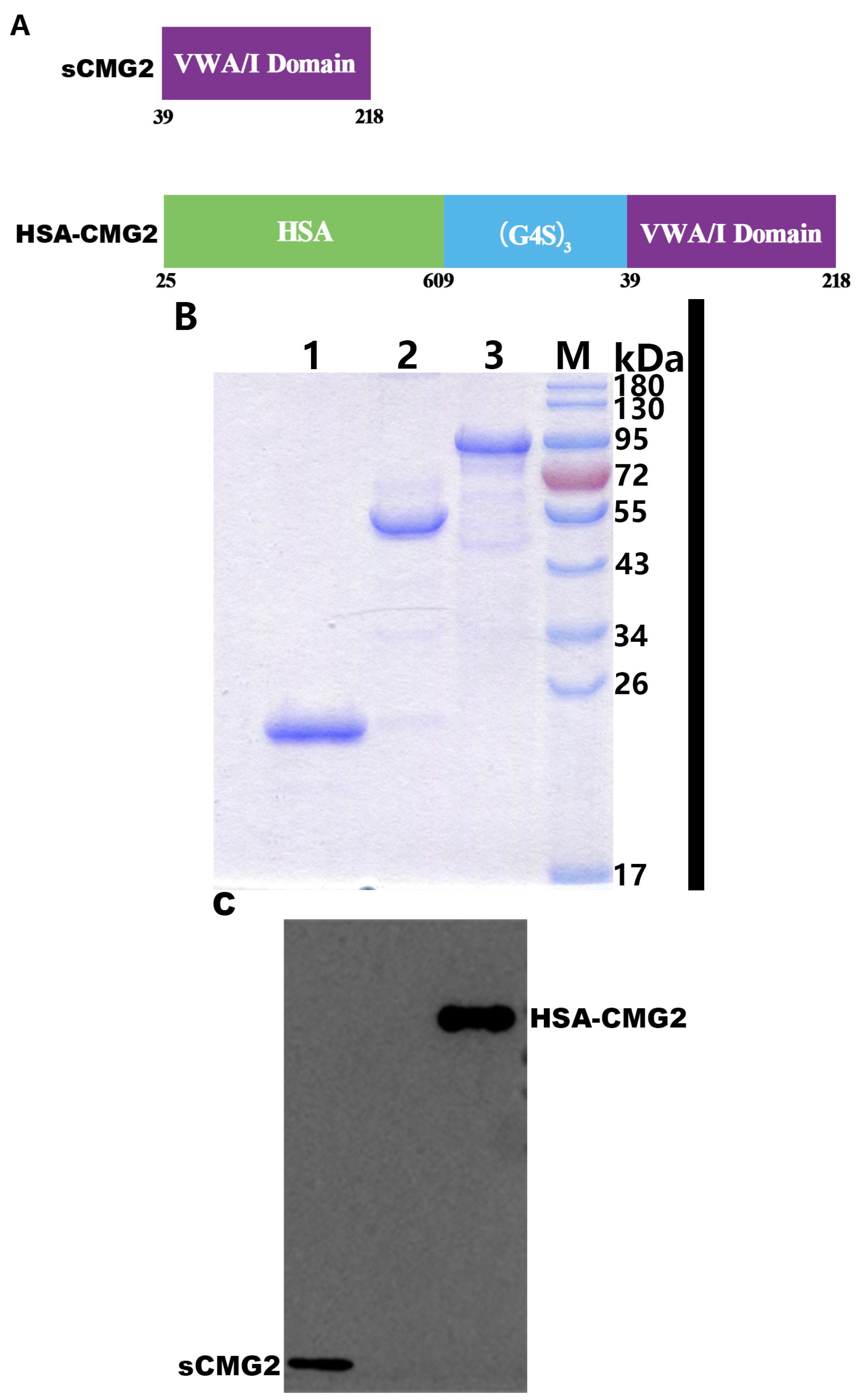
2.1. Expression and Purification of HSA-CMG2

2.2. Affinity of HSA-CMG2 for rPA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor Decoys | ka (1/Ms × 106) | kd (1/s × 102) | KD (nmol/L) |
|---|---|---|---|
| sCMG2 | 7.98 ± 5.80 | 1.38 ± 1.02 | 1.67 ± 0.26 |
| HSA-CMG2 | 0.26 ± 0.21 | 0.12 ± 0.06 | 5.76 ± 2.22 |
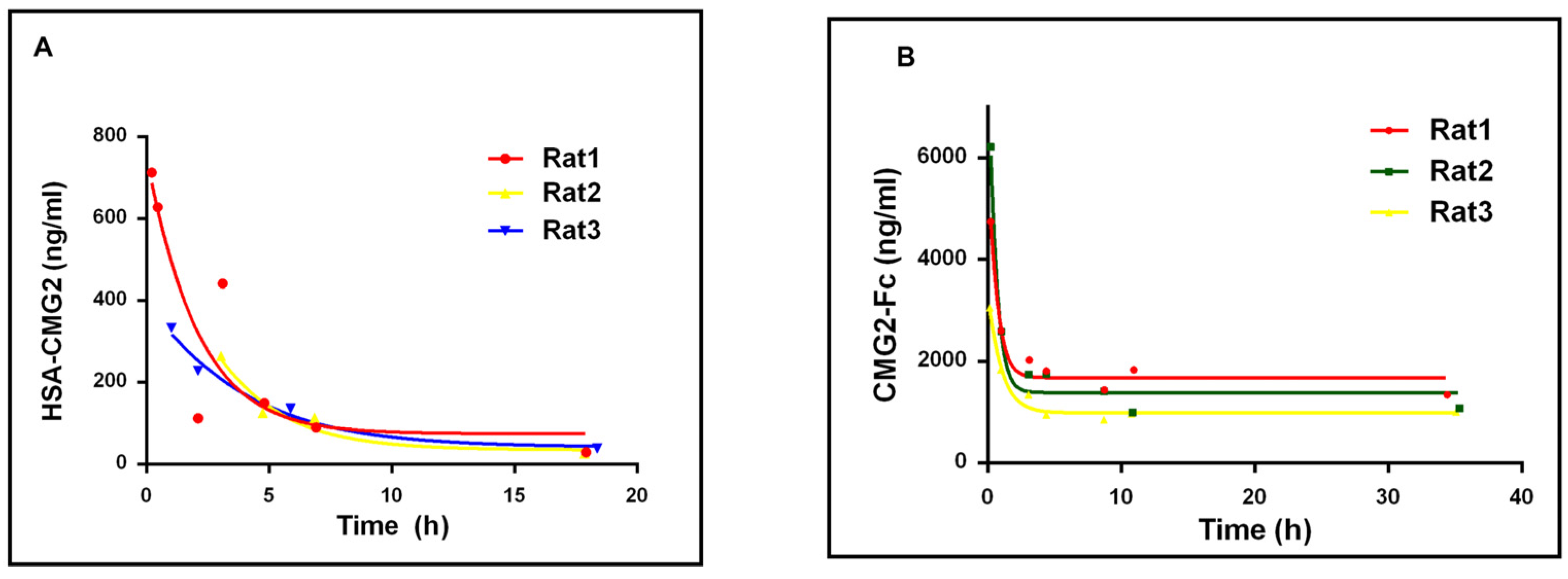
2.3. Half-Life of HSA-CMG2 and CMG2-Fc

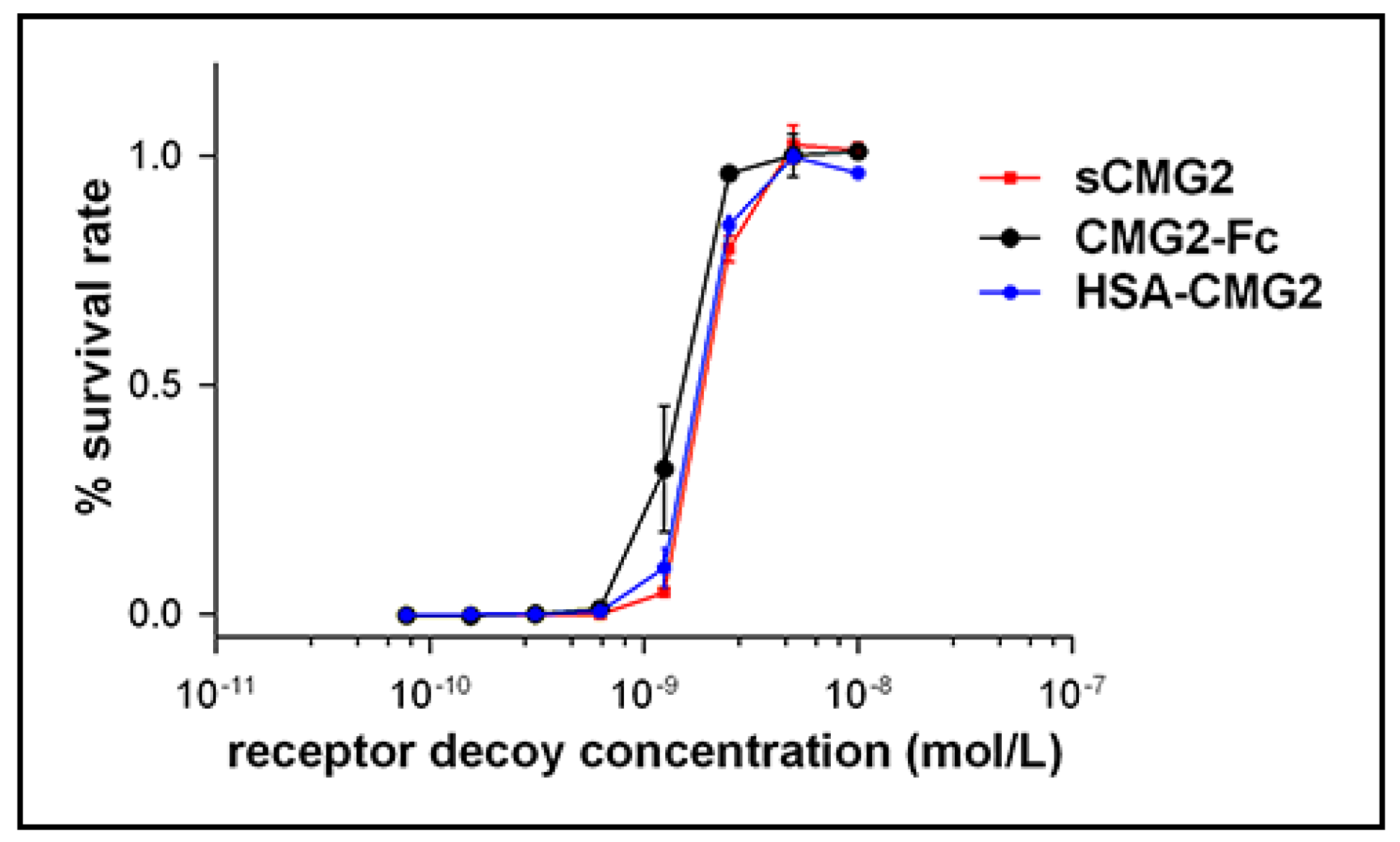
2.4. In Vitro Toxin Inhibition Activity of HSA-CMG2

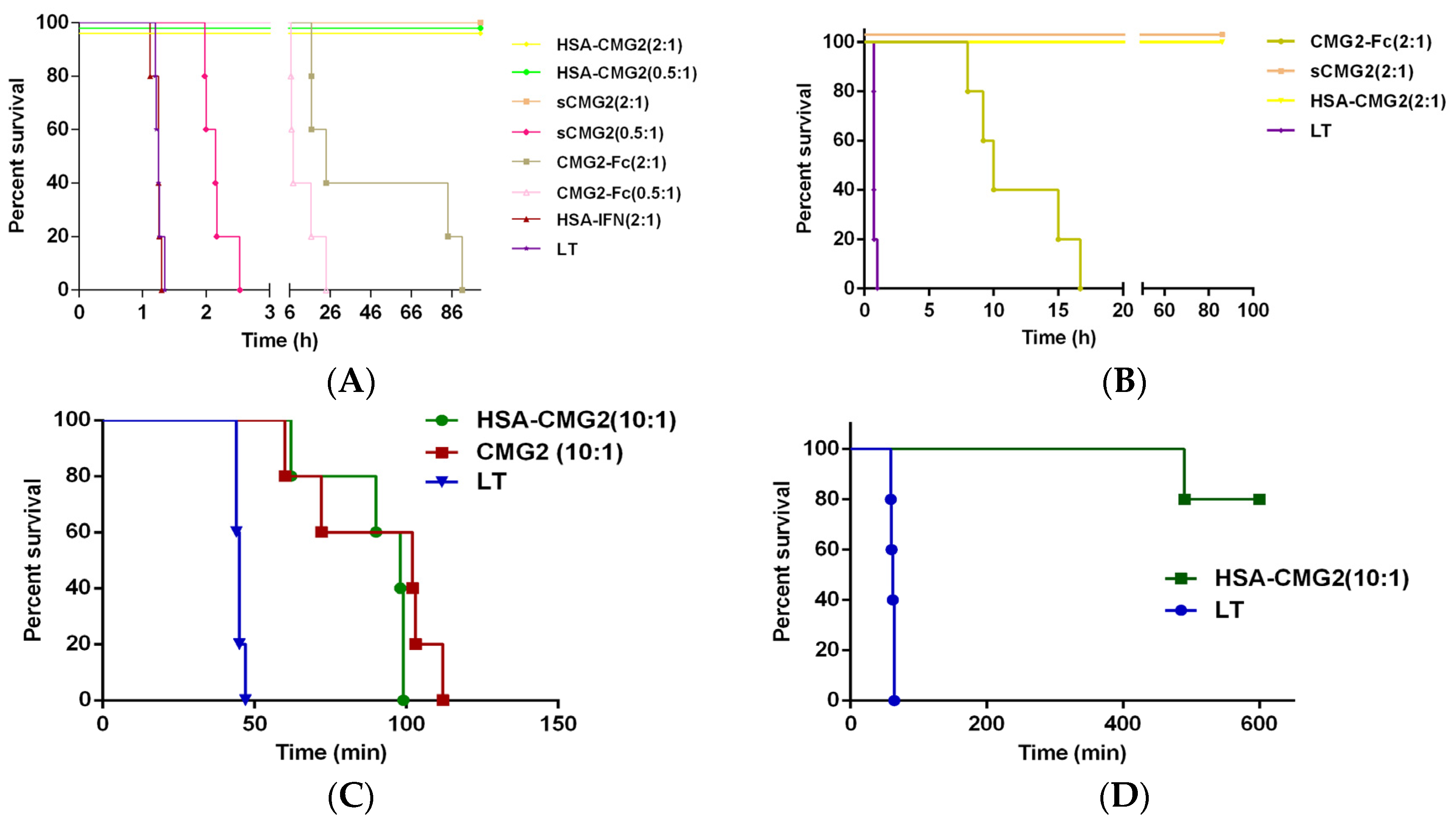
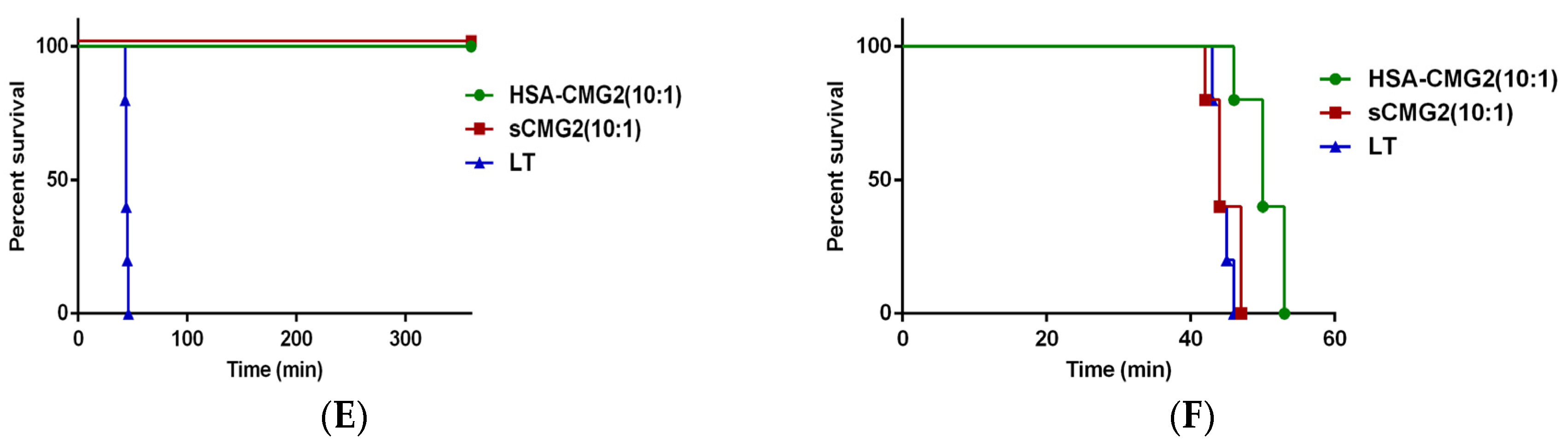
2.5. Toxin Neutralization of HSA-CMG2 in Vivo


| Inhibitor | Receptor Decoy:PA Moral Ratio | Survivors/Total | Average Time to Death (h) | Comparison of TTD (Unpaired t-Test) a | Comparison of Survival Curve (Log-Rank Mantel-Cox) b |
|---|---|---|---|---|---|
| HSA-CMG2 | 0.5:1 | 5/5 | NA | - | p = 0.0018 |
| HSA-CMG2 | 2:1 | 5/5 | NA | - | p = 0.0018 |
| sCMG2 | 0.5:1 | 0/5 | 2.17 | p < 0.0001 | p = 0.0018 |
| sCMG2 | 2:1 | 5/5 | NA | - | p = 0.0018 |
| CMG2-Fc | 0.5:1 | 0/5 | 12.37 | p = 0.0122 | p = 0.0018 |
| CMG2-Fc | 2:1 | 0/5 | 46.50 | p = 0.0276 | p = 0.0018 |
| HSA-IFN | 2:1 | 0/5 | 1.24 | p = 0.5973 | p = 0.7167 |
| LT c | - | 0/5 | 1.34 | - | - |
3. Discussion
4. Experimental Section
4.1. Ethics Statement
4.2. Recombinant Anthrax Toxins
4.3. Receptor Decoys Cloning
| Name | Sequences (5′-3′) |
|---|---|
| H-1F | GCCACTCGAGAAAAGAGATGCACACAAGAGTGAGGTTGCTCATC |
| H-1R | CCGCCTGAACCGCCTCCACCTAAGCCTAAGGCAGCTTG |
| CMG2-F | GTGGCTCTGGCGGTGGCGGATCGTGCAGAAGAGCCTTTG |
| CMG2-R | GCTAGCCGAGCGGCCGCTTAACATGACTGAGCTAGTATAG |
| H-2R | CACCGCCAGAGCCACCTCCGCCTGAACCGCC |
| C-2F | TCAGGCGGAGGTGGCTCTGGCGGTG |
| H-3F | CATTCTCGAGAAAAGAGATGCACACAAGAGTG |
| C-3R | GAACGCGGCCGCTTAACATGACTG |
4.4. Receptor Decoys Expression and Purification
4.5. Surface Plasmon Resonance (SPR)
4.6. Half-Life Assay
4.7. In Vitro Neutralization Assay (TNA)
4.8. In Vivo Protection against Intoxication
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Holty, J.E.; Bravata, D.M.; Liu, H.; Olshen, R.A.; McDonald, K.M.; Owens, D.K. Systematic review: A century of inhalational anthrax cases from 1900 to 2005. Ann. Intern. Med. 2006, 144, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Visca, P.; Ippolito, G.; Spallarossa, A.; Bolognesi, M.; Montecucco, C. Anthrax toxin: A tripartite lethal combination. FEBS Lett. 2002, 531, 384–388. [Google Scholar] [CrossRef]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Friedlander, A.; Dreier, T.; Ezzell, J.; Leppla, S. Effects of anthrax toxin components on human neutrophils. Infect. Immun. 1985, 47, 306–310. [Google Scholar] [PubMed]
- Ezzell, J.W., Jr.; Abshire, T.G. Serum protease cleavage of bacillus anthracis protective antigen. J. Gen. Microbiol. 1992, 138, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A.T. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Scobie, H.M.; Rainey, G.J.A.; Bradley, K.A.; Young, J.A.T. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar] [CrossRef] [PubMed]
- Scobie, H.M.; Thomas, D.; Marlett, J.M.; Destito, G.; Wigelsworth, D.J.; Collier, R.J.; Young, J.A.T.; Manchester, M. A soluble receptor decoy protects rats against anthrax lethal toxin challenge. J. Infect. Dis. 2005, 192, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Leppla, S.H.; van der Goot, F.G. Receptor palmitoylation and ubiquitination regulate anthrax toxin endocytosis. J. Cell Biol. 2006, 172, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.E.; Mavila, A.; Salazar, R.; Bayless, K.J.; Kanagala, S.; Maxwell, S.A.; Davis, G.E. Differential gene expression during capillary morphogenesis in 3D collagen matrices regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J. Cell Sci. 2001, 114, 2755–2773. [Google Scholar] [PubMed]
- Liu, S.; Zhang, Y.; Hoover, B.; Leppla, S.H. The receptors that mediate the direct lethality of anthrax toxin. Toxins 2013, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.G.; Inglesby, T.V., Jr.; Borio, L. Management of anthrax. Clin. Infect. Dis. 2002, 35, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Mock, M.; Fouet, A. Anthrax. Annu. Rev. Microbiol. 2001, 55, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, A.M. Tackling anthrax. Nature 2001, 414, 160–161. [Google Scholar] [CrossRef] [PubMed]
- Nestorovich, E.M.; Bezrukov, S.M. Designing inhibitors of anthrax toxin. Expert Opin. Drug Discov. 2014, 9, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Thomas, D.; Marlett, J.; Manchester, M.; Young, J.A. Efficient neutralization of antibody-resistant forms of anthrax toxin by a soluble receptor decoy inhibitor. Antimicrob. Agents Chemother. 2009, 53, 1210–1212. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Naughton, J.; Cote, C.; Welkos, S.; Manchester, M.; Young, J.A.T. Delayed toxicity associated with soluble anthrax toxin receptor decoy-Ig fusion protein treatment. PLoS ONE 2012, 7, e34611. [Google Scholar] [CrossRef] [PubMed]
- Wycoff, K.L.; Belle, A.; Deppe, D.; Schaefer, L.; Maclean, J.M.; Haase, S.; Trilling, A.K.; Liu, S.; Leppla, S.H.; Geren, I.N.; et al. Recombinant anthrax toxin receptor-Fc fusion proteins produced in plants protect rabbits against inhalational anthrax. Antimicrob. Agents Chemother. 2011, 55, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Soroka, S.D.; Taylor, T.H., Jr.; Stamey, K.L.; Stinson, K.W.; Freeman, A.E.; Abramson, D.R.; Desai, R.; Cronin, L.X.; Oxford, J.W.; et al. Standardized, mathematical model-based and validated in vitro analysis of anthrax lethal toxin neutralization. J. Immunol. Methods 2008, 333, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, K.; Lacy, D.B.; Mogridge, J.; Collier, R.J. Mapping the lethal factor and edema factor binding sites on oligomeric anthrax protective antigen. Proc. Natl. Acad. Sci. USA 2002, 99, 7049–7053. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Bischofberger, M.; Kunz, B.; Groux, R.; van der Goot, F.G. Endocytosis of the anthrax toxin is mediated by clathrin, actin and unconventional adaptors. PLoS Pathog. 2010, 6, e1000792. [Google Scholar] [CrossRef] [PubMed]
- Froude, J.W.; Stiles, B.; Pelat, T.; Thullier, P. Antibodies for biodefense. MAbs 2011, 3, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Vuyisich, M.; Gnanakaran, S.; Lovchik, J.A.; Rick Lyons, C.; Gupta, G. A dual-purpose protein ligand for effective therapy and sensitive diagnosis of anthrax. Protein J. 2008, 27, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Wu, X.; Gao, L.; Shao, Y.; Peng, H.; Chen, H.; Chen, H.; Hu, X.; Yue, J. Improving the anti-toxin abilities of the CMG2-Fc fusion protein with the aid of computational design. PLoS ONE 2014, 9, e104674. [Google Scholar] [CrossRef] [PubMed]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Aspects Med. 2012, 33, 209–290. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.; Karle, A.; Meissburger, B.; Hofig, I.; Stork, R.; Kontermann, R.E. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J. Biol. Chem. 2007, 282, 12650–12660. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.L.; Xue, C.; Wang, Y.; Li, X.Y.; Xiong, X.H.; Yao, X.Q.; Liu, Z.M. Circumventing the heterogeneity and instability of human serum albumin-interferon-alpha2b fusion protein by altering its orientation. J. Biotechnol. 2007, 131, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Chen, Y.; Yuan, H.Y.; Li, H.; Lu, H. Fusion of HSA influences tnf-aneutralizing activity of shTNFRs. Biotechnol. Lett. 2012, 34, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Berger, V.; Richter, F.; Zettlitz, K.; Unverdorben, F.; Scheurich, P.; Herrmann, A.; Pfizenmaier, K.; Kontermann, R.E. An anti-TNFR1 scFv-HSA fusion protein as selective antagonist of TNF action. Protein Eng. Des. Sel. 2013, 26, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Guan, B.; Li, B.; Duan, Z.; Chen, Y.; Li, H.; Jin, J. Expression, purification and characterization of recombinant human interleukin-2-serum albumin (rhiL-2-HSA) fusion protein in pichia pastoris. Protein Expr. Purif. 2012, 84, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Junghans, R.; Anderson, C. The protection receptor for igg catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc. Natl. Acad. Sci. USA 1996, 93, 5512–5516. [Google Scholar] [CrossRef] [PubMed]
- Czajkowsky, D.M.; Hu, J.; Shao, Z.; Pleass, R.J. Fc-fusion proteins: New developments and future perspectives. EMBO Mol. Med. 2012, 4, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.J.; Dong, D.Y.; Song, X.H.; Ge, M.; Li, G.L.; Fu, L.; Zhuang, H.L.; Chen, W. Expression, purification and characterization of the recombinant anthrax protective antigen. Sheng Wu Gong Cheng Xue Bao 2004, 20, 652–655. [Google Scholar] [PubMed]
- Liu, J.; Cai, C.; Guo, Q.; Zhang, J.; Dong, D.; Li, G.; Fu, L.; Xu, J.; Chen, W. Secretory expression and efficient purification of recombinant anthrax toxin lethal factor with full biological activity in E. coli. Protein Expr. Purif. 2013, 89, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Che, J.; Xu, L.; Guo, Q.; Kong, Y.; Fu, L.; Xu, J.; Cheng, Y.; Chen, W. Tumor endothelium marker-8 based decoys exhibit superiority over capillary morphogenesis protein-2 based decoys as anthrax toxin inhibitors. PLoS ONE 2011, 6, e20646. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Guo, Q.; Liu, J.; Zhang, J.; Yin, Y.; Dong, D.; Fu, L.; Xu, J.; Chen, W. Recombinant HSA-CMG2 Is a Promising Anthrax Toxin Inhibitor. Toxins 2016, 8, 28. https://doi.org/10.3390/toxins8010028
Li L, Guo Q, Liu J, Zhang J, Yin Y, Dong D, Fu L, Xu J, Chen W. Recombinant HSA-CMG2 Is a Promising Anthrax Toxin Inhibitor. Toxins. 2016; 8(1):28. https://doi.org/10.3390/toxins8010028
Chicago/Turabian StyleLi, Liangliang, Qiang Guo, Ju Liu, Jun Zhang, Ying Yin, Dayong Dong, Ling Fu, Junjie Xu, and Wei Chen. 2016. "Recombinant HSA-CMG2 Is a Promising Anthrax Toxin Inhibitor" Toxins 8, no. 1: 28. https://doi.org/10.3390/toxins8010028
APA StyleLi, L., Guo, Q., Liu, J., Zhang, J., Yin, Y., Dong, D., Fu, L., Xu, J., & Chen, W. (2016). Recombinant HSA-CMG2 Is a Promising Anthrax Toxin Inhibitor. Toxins, 8(1), 28. https://doi.org/10.3390/toxins8010028