
Metal Ion Activation of Clostridium sordellii Lethal Toxin and Clostridium difficile Toxin B

Abstract
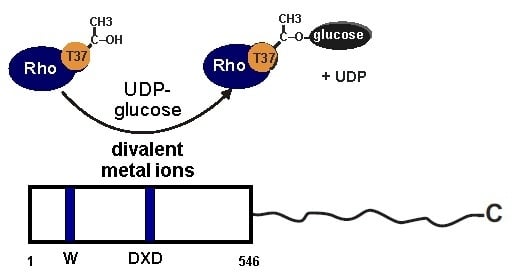
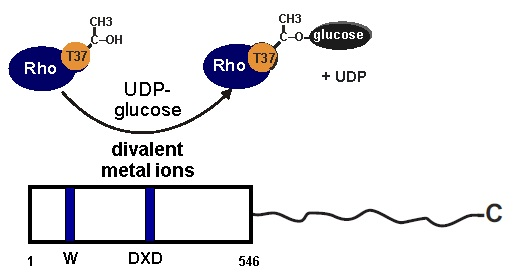
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Divalent Metal Ion Dependency of TcsL
2.2. Essential Role of the DxD Motif for Mn2+-Activated UDP-Glucose Hydrolysis Activity
2.3. Monovalent Metal Ion Dependency of rN-TcsL UDP-Glucose Hydrolysis Activity
2.4. Metal Ion Dependency of rN-TcdB
2.5. Prebound Divalent Metal Ions Are Dispensible for Cytopathic Activity of TcsL and TcdB
3. Conclusions
4. Materials and Methods
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| LCGT | Large clostridial glycosylating toxin |
| PEI | Polyethylenimine-modified |
| TcdA | C. difficile toxin A |
| TcdB | C. difficile toxin B |
| Tcnα | C. novyi alpha-toxin from, and the glycosylating toxin |
| TcpE | Glycosylating toxin from C. perfringens type E strain |
| TcsL | C. sordellii lethal toxin |
| TcsH | C. sordellii hemorrhagic toxin |
References
- Popoff, M.R.; Geny, B. Rho/Ras-GTPase-dependent and -independent activity of clostridial glucosylating toxins. J. Med. Microbiol. 2011, 60, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Jank, T.; Belyi, Y.; Aktories, K. Bacterial glycosyltransferase toxins. Cell. Microbiol. 2015, 17, 1752–1765. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Pauillac, S.; Schelle, I.; Bouvet, P.; Bouchier, C.; Varela-Chavez, C.; Just, I.; Popoff, M.R. Haemorrhagic toxin and lethal toxin from Clostridium sordellii strain vpi9048: Molecular characterization and comparative analysis of substrate specificity of the large clostridial glucosylating toxins. Cell. Microbiol. 2014, 16, 1706–1721. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, S.C.; Klose, I.; Reichenbach, M.; Huelsenbeck, J.; Genth, H. Distinct kinetics of (H/K/N)Ras glucosylation and Rac1 glucosylation catalysed by Clostridium sordellii lethal toxin. FEBS Lett. 2009, 583, 3133–3139. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Just, I. Functional implications of lethal toxin-catalysed glucosylation of (H/K/N)Ras and Rac1 in Clostridium sordellii-associated disease. Eur. J. Cell Biol. 2011, 90, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.R.; Hofmann, F.; Wohlgemuth, S.; Herrmann, C.; Just, I. Structural consequences of mono-glucosylation of Ha-Ras by Clostridium sordellii lethal toxin. J. Mol. Biol. 2000, 301, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.; Wilde, C.; Selzer, J.; Aktories, K.; Kalbitzer, H.R. Glucosylation of Ras by Clostridium sordellii lethal toxin: Consequences for effector loop conformations observed by NMR spectroscopy. Biochemistry 2003, 42, 11951–11959. [Google Scholar] [CrossRef] [PubMed]
- May, M.; Kolbe, T.; Wang, T.; Schmidt, G.; Genth, H. Increased cell-matrix adhesion upon constitutive activation of rho proteins by cytotoxic necrotizing factors from E. coli and Y. pseudotuberculosis. J. Signal Transduct. 2012, 2012, 570183. [Google Scholar] [CrossRef] [PubMed]
- Geny, B.; Grassart, A.; Manich, M.; Chicanne, G.; Payrastre, B.; Sauvonnet, N.; Popoff, M.R. Rac1 inactivation by lethal toxin from Clostridium sordellii modifies focal adhesions upstream of actin depolymerization. Cell. Microbiol. 2010, 12, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Boehm, C.; Gibert, M.; Geny, B.; Popoff, M.R.; Rodriguez, P. Modification of epithelial cell barrier permeability and intercellular junctions by Clostridium sordellii lethal toxins. Cell. Microbiol. 2006, 8, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Petit, P.; Bréard, J.; Montalescot, V.; El Hadj, N.B.; Levade, T.; Popoff, M.; Geny, B. Lethal toxin from Clostridium sordellii induces apoptotic cell death by disruption of mitochondrial homeostasis in HL-60 cells. Cell. Microbiol. 2003, 5, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Dreger, S.C.; Schulz, F.; Huelsenbeck, J.; Gerhard, R.; Hofmann, F.; Just, I.; Genth, H. Killing of rat basophilic leukemia cells by lethal toxin from Clostridium sordellii: Critical role of phosphatidylinositide 3′-OH kinase/Akt signaling. Biochemistry 2009, 48, 1785–1792. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.; May, M.; Schulz, F.; Schelle, I.; Ronkina, N.; Hohenegger, M.; Fritz, G.; Just, I.; Gerhard, R.; Genth, H. Cytoprotective effect of the small GTPase RhoB expressed upon treatment of fibroblasts with the Ras-glucosylating Clostridium sordellii Lethal Toxin. FEBS Lett. 2012, 586, 3665–3673. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.; Dreger, S.C.; Gerhard, R.; Fritz, G.; Just, I.; Genth, H. Upregulation of the immediate early gene product RhoB by exoenzyme C3 from Clostridium limosum and toxin B from Clostridium difficile. Biochemistry 2007, 46, 4923–4931. [Google Scholar] [CrossRef] [PubMed]
- May, M.; Wang, T.; Muller, M.; Genth, H. Difference in F-actin depolymerization induced by toxin B from the Clostridium difficile strain VPI 10463 and toxin B from the variant Clostridium difficile serotype F strain 1470. Toxins (Basel). 2013, 5, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, M.O.; Jank, T.; Aktories, K.; Schulz, G.E. Conformational changes and reaction of clostridial glycosylating toxins. J. Mol. Biol. 2008, 377, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Reinert, D.J.; Jank, T.; Aktories, K.; Schulz, G.E. Structural basis for the function of Clostridium difficile toxin B. J. Mol. Biol. 2005, 351, 973–981. [Google Scholar] [CrossRef] [PubMed]
- D'Urzo, N.; Malito, E.; Biancucci, M.; Bottomley, M.J.; Maione, D.; Scarselli, M.; Martinelli, M. The structure of Clostridium difficile toxin A glucosyltransferase domain bound to Mn2+ and UDP provides insights into glucosyltransferase activity and product release. FEBS J. 2012, 279, 3085–3097. [Google Scholar] [CrossRef] [PubMed]
- Ciesla, W.P., Jr; Bobak, D.A. Clostridium difficile toxins A and B are cation-dependent UDP-glucose hydrolases with differing catalytic activities. J. Biol. Chem. 1998, 273, 16021–16026. [Google Scholar] [CrossRef] [PubMed]
- Busch, C.; Hofmann, F.; Selzer, J.; Munro, J.; Jeckel, D.; Aktories, K. A common motif of eukaryotic glycosyltransferases is essential for the enzyme activity of large clostridial cytotoxins. J. Biol. Chem. 1998, 273, 19566–19572. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.T.; Brew, K. Affinity labeling of bovine colostrum galactosyltransferase with a uridine 5′-diphosphate derivative. Biochemistry 1976, 15, 3499–3505. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, C.A.R.; Munro, S. Activity of the yeast MNN1 a-1,3-mannosyltransferase requires a motif conserved in many other families of glycosyltransferases. Proc. Natl. Acad. Sci. USA 1998, 95, 7945–7950. [Google Scholar] [CrossRef] [PubMed]
- Di, C.E. A structural perspective on enzymes activated by monovalent cations. J. Biol. Chem. 2006, 281, 1305–1308. [Google Scholar]
- Reineke, J.; Tenzer, S.; Rupnik, M.; Koschinski, A.; Hasselmayer, O.; Schrattenholz, A.; Schild, H.; von Eichel-Streiber, C. Autocatalytic cleavage of Clostridium difficile toxin B. Nature 2007, 446, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Egerer, M.; Giesemann, T.; Jank, T.; Satchell, K.J.; Aktories, K. Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J. Biol. Chem. 2007, 282, 25314–25321. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, P.; Zamboglou, C.; Genisyuerek, S.; Guttenberg, G.; Aktories, K. Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis. PLoS ONE 2010, 5, e10673. [Google Scholar] [CrossRef] [PubMed]
- Zak, J.; Schneider, S.W.; Eue, I.; Ludwig, T.; Oberleithner, H. High-resistance MDCK-C7 monolayers used for measuring invasive potency of tumour cells. Pflug. Arch. 2000, 440, 179–183. [Google Scholar] [CrossRef]
- Feng, Y.; Cohen, S.N. Upregulation of the host SLC11A1 gene by Clostridium difficile toxin B facilitates glucosylation of Rho GTPases and enhances toxin lethality. Infect. Immun. 2013, 81, 2724–2732. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Selzer, J.; Busch, C.; Dumbach, J.; Hofmann, F.; Aktories, K.; Just, I. New method to generate enzymatically deficient Clostridium difficile toxin B as an antigen for immunization. Infect. Immun. 2000, 68, 1094–1101. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genth, H.; Schelle, I.; Just, I. Metal Ion Activation of Clostridium sordellii Lethal Toxin and Clostridium difficile Toxin B. Toxins 2016, 8, 109. https://doi.org/10.3390/toxins8040109
Genth H, Schelle I, Just I. Metal Ion Activation of Clostridium sordellii Lethal Toxin and Clostridium difficile Toxin B. Toxins. 2016; 8(4):109. https://doi.org/10.3390/toxins8040109
Chicago/Turabian StyleGenth, Harald, Ilona Schelle, and Ingo Just. 2016. "Metal Ion Activation of Clostridium sordellii Lethal Toxin and Clostridium difficile Toxin B" Toxins 8, no. 4: 109. https://doi.org/10.3390/toxins8040109
APA StyleGenth, H., Schelle, I., & Just, I. (2016). Metal Ion Activation of Clostridium sordellii Lethal Toxin and Clostridium difficile Toxin B. Toxins, 8(4), 109. https://doi.org/10.3390/toxins8040109