
A Simple and High-Throughput Analysis of Amatoxins and Phallotoxins in Human Plasma, Serum and Urine Using UPLC-MS/MS Combined with PRiME HLB μElution Platform

Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of LC-MS/MS
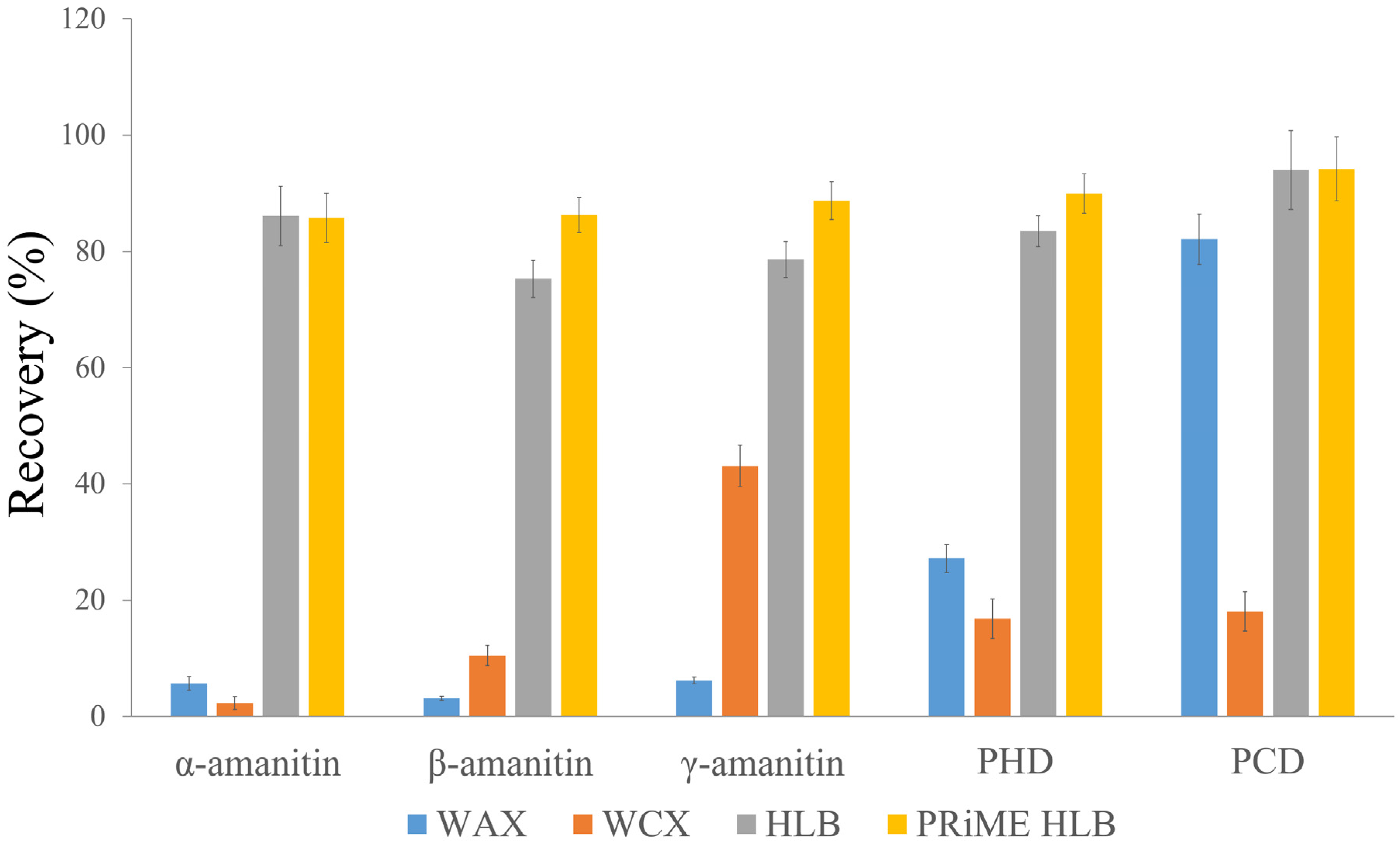
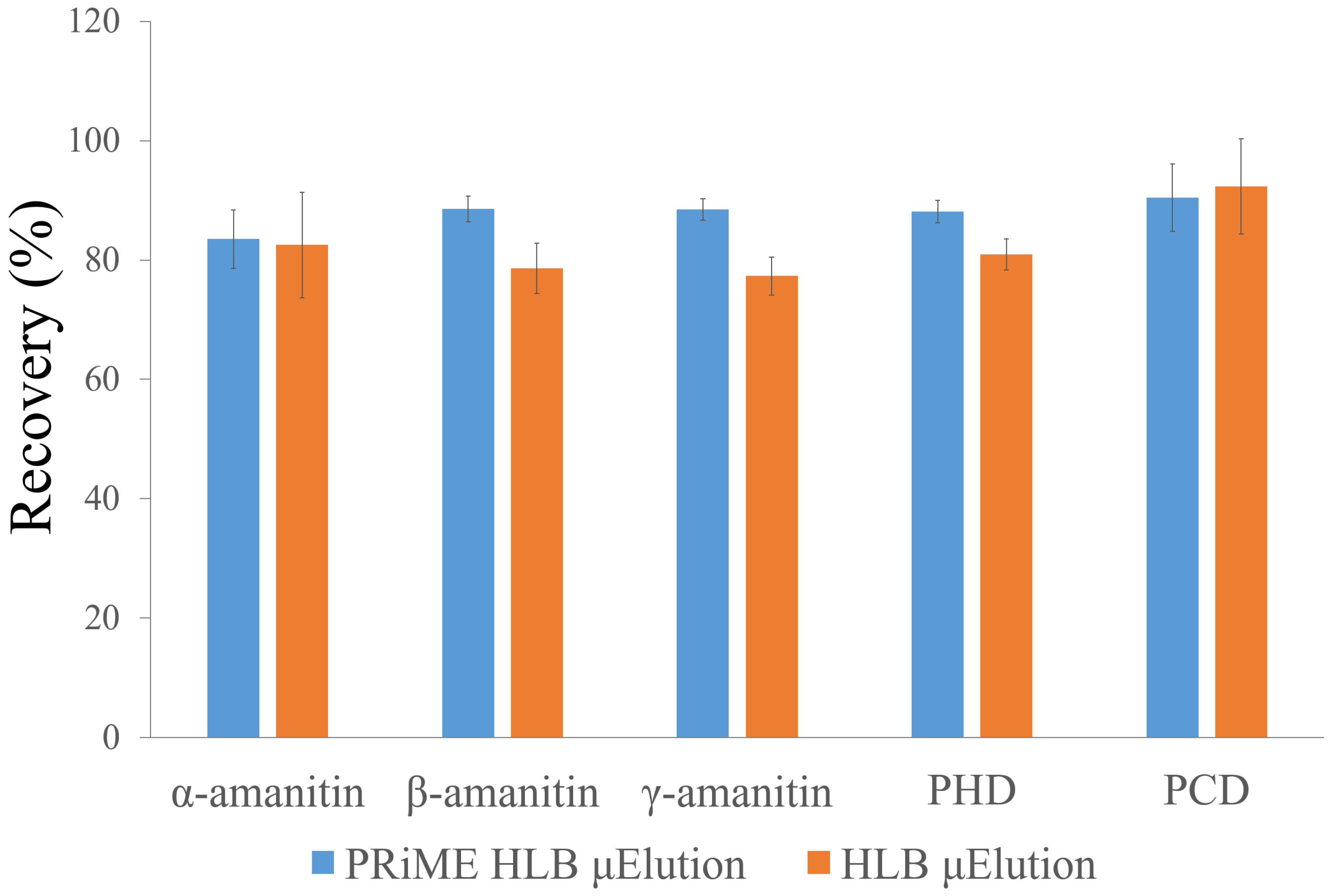
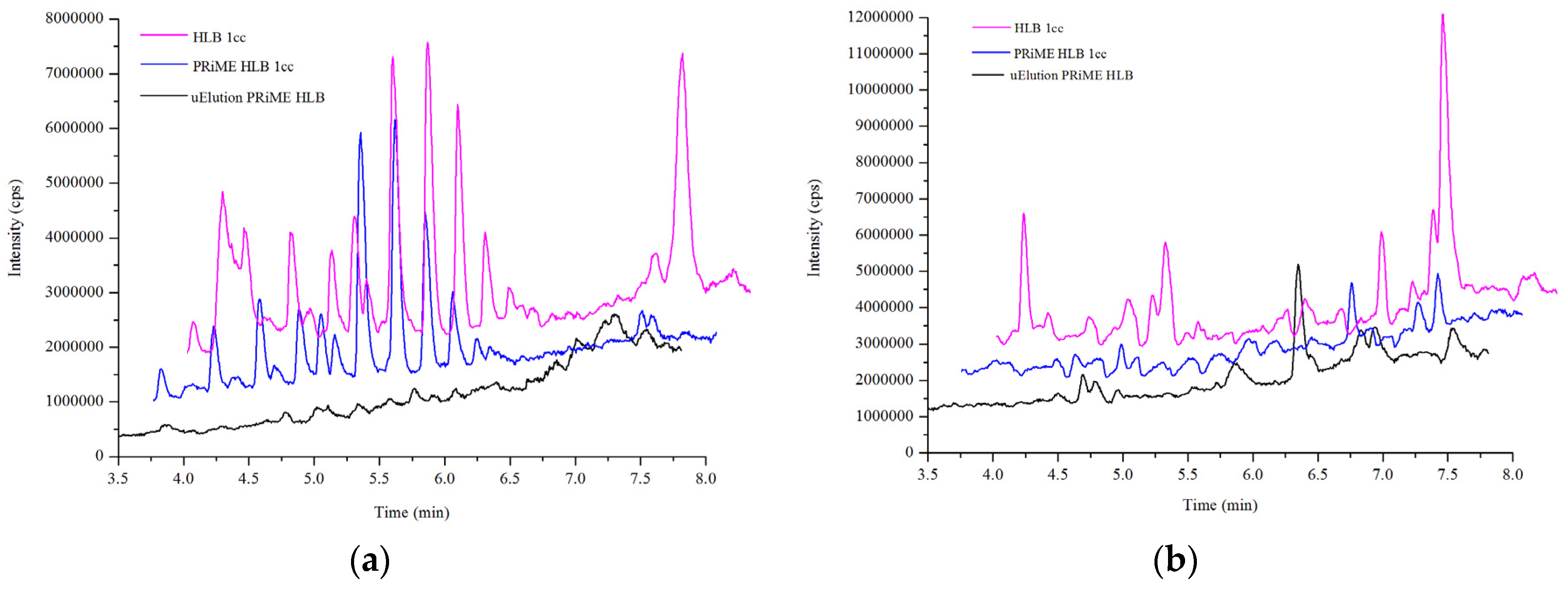
2.2. Sample Preparation
2.3. Method Validation
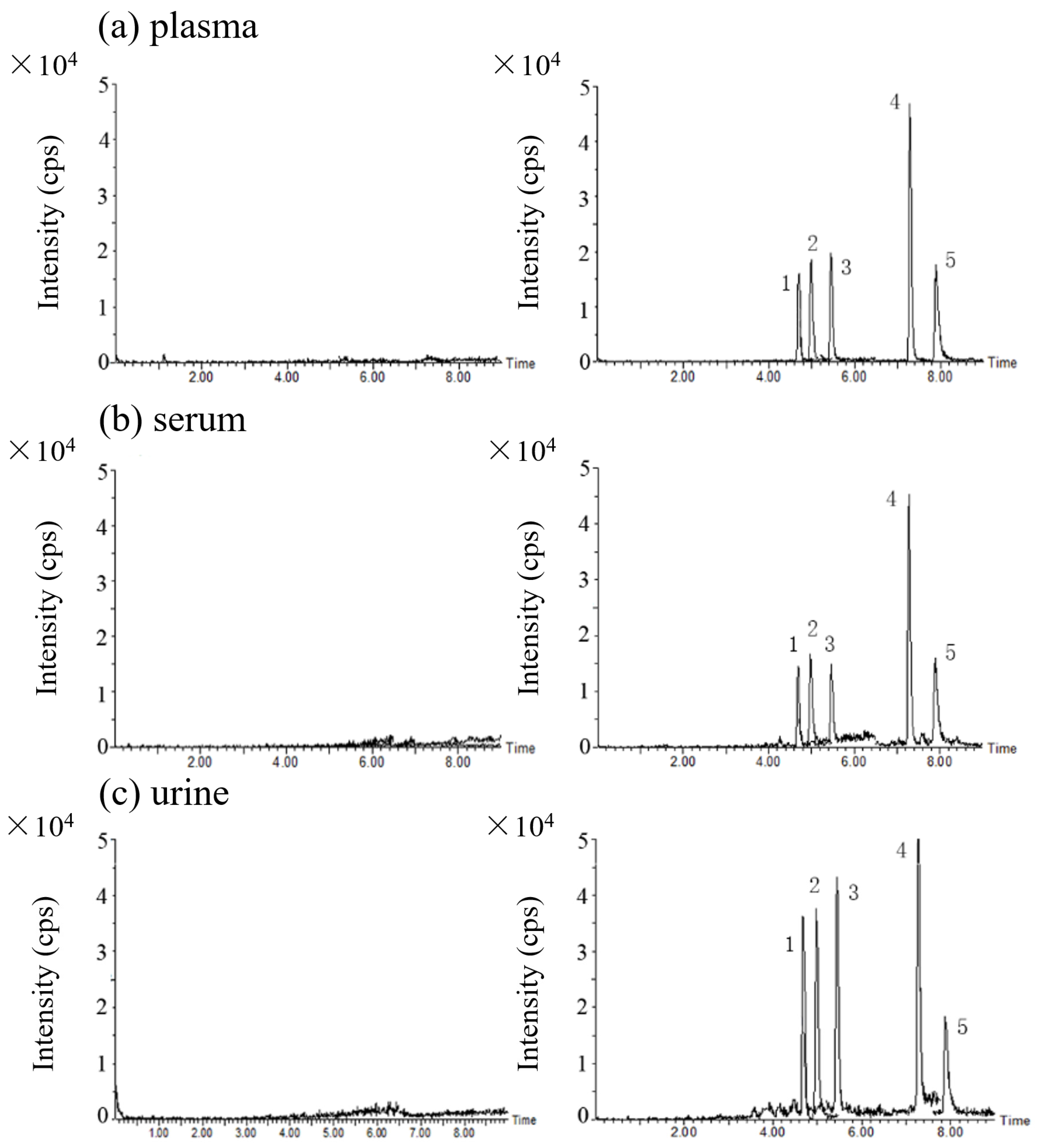
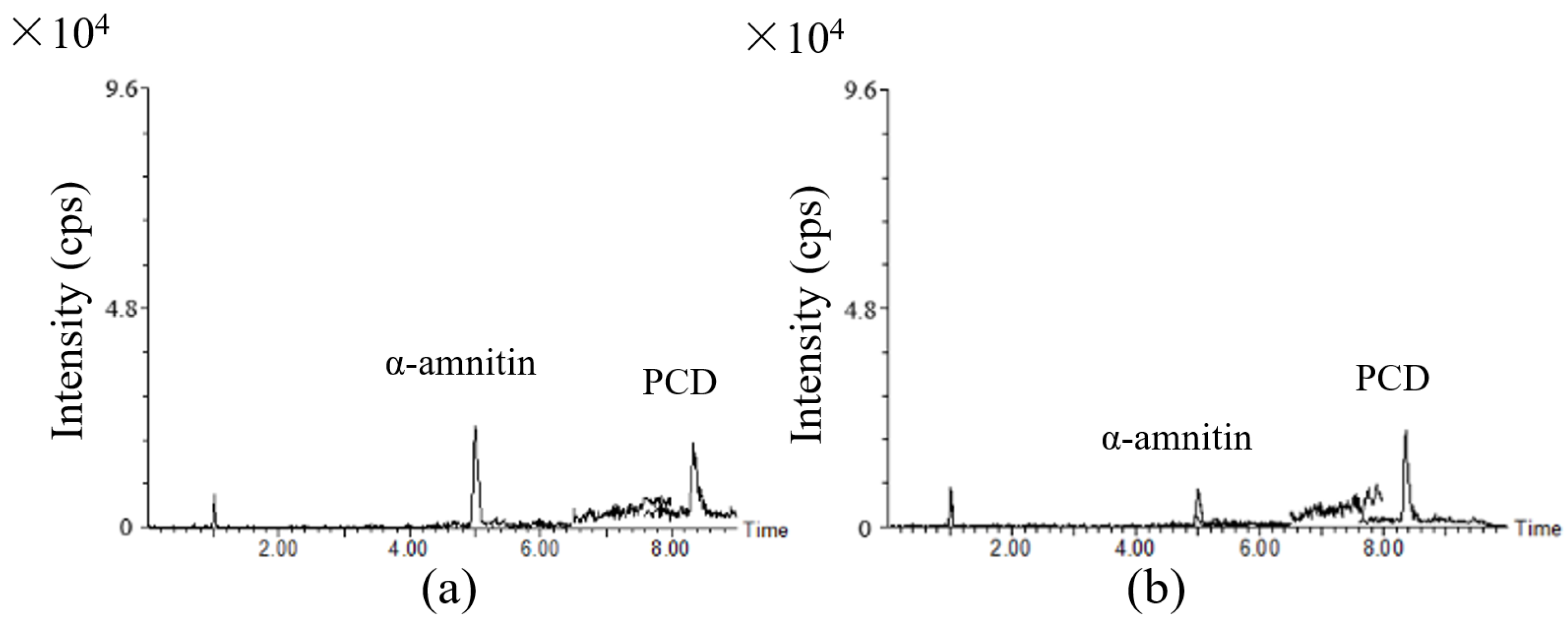
2.3.1. Specificity/Selectivity
2.3.2. Recovery, Matrix Effects, and Process Efficiency
2.3.3. Linearity/Work Range
2.3.4. Limit of Detection and Lower Limit of Quantification
2.3.5. Stability
3. Materials and Methods
3.1. Reagents and Materials
3.2. Preparation of Calibration Standards
3.3. Sample Preparation
3.3.1. Urine Samples
3.3.2. Plasma and Serum Samples
3.4. LC-MS/MS Analysis
4. Method Application
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, Z.H.; Zhang, P.; Zhang, Z.G. Investigation and analysis of 102 mushroom poisoning casesin Southern China from 1994 to 2012. Fungal Divers. 2014, 64, 123–131. [Google Scholar] [CrossRef]
- Wang, R.; Gao, Y.J.; Ding, F.; Xie, R.H.; Wang, X.Y.; Li, Q. Epidemiological analysis on mushroom poisoning in China 2004–2011. Chin. J. Public Health 2014, 30, 158–161. [Google Scholar]
- Wieland, T. The toxic peptides from Amanita mushrooms. Int. J. Peptideprotein Res. 1983, 22, 257–276. [Google Scholar] [CrossRef]
- Luo, H.; Hallen-Adams, H.E.; Scott-Craig, J.S.; Walton, J.D. Ribosomal biosynthesis of α-amanitin in Galerina marginata. Fungal Genet. Biol. 2012, 49, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Mas, A. Mushrooms, amatoxins and the liver. J. Hepatol. 2005, 42, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Vargas, N.; Bernal, A.; Sarria, V.; Franco-Molano, A.; Restrepo, S. Amatoxin and phallotoxin composition in species of the genus Amanitain Colombia: A taxonomic perspective. Toxicon 2011, 58, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Sgambelluri, R.M.; Epis, S.; Sassera, D.; Luo, H.; Angelos, E.R.; Walton, J.D. Profiling of amatoxins and phallotoxins in the genus lepiota by liquid chromatography combined with UV absorbance and mass spectrometry. Toxins 2014, 6, 2336–2347. [Google Scholar] [CrossRef] [PubMed]
- Enjalbert, F.; Rapior, S.; Nouguier-Soulé, J.; Guillon, S.; Amouroux, N.; Cabot, C. Treatment of amatoxin poisoning: 20-year retrospective analysis. J. Toxicol. 2002, 40, 715–757. [Google Scholar] [CrossRef]
- Jaeger, A.; Jehl, F.; Flesch, F.; Sauder, P.; Kopferschmitt, J. Kinetics of amatoxins in human poisoning: Therapeutic implications. J. Clin. Toxicol. 1993, 31, 63–80. [Google Scholar] [CrossRef]
- Rentsch, K.M. Laboratory diagnostics in acute poisoning: Critical overview. Clin. Chem. Lab. Med. 2010, 48, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Faulstich, H.; Zobeley, S.; Trischmann, H. A rapid radioimmunoassay, using a nylon support, for amatoxins from Amanita mushrooms. Toxicon 1982, 20, 913–924. [Google Scholar] [CrossRef]
- Faulstich, H.; Trischmann, H.; Zobeley, S. A radioimmunoassay for amanitin. FEBS Lett. 1975, 56, 312–315. [Google Scholar] [CrossRef]
- Homann, J.; Rawer, P.; Bleyl, H.; Matthes, K.; Heinrich, D. Early detection of amatoxins in human mushroom poisoning. Arch. Toxicol. 1986, 59, 190–191. [Google Scholar] [CrossRef] [PubMed]
- Abuknesha, R.A.; Maragkou, A. A highly sensitive and specific enzyme immunoassay for detection of beta-amanitin in biological fluids. Anal. Bioanal. Chem. 1993, 379, 853–860. [Google Scholar]
- Butera, R.; Locatelli, C.; Coccini, T.; Manzo, L. Diagnostic accuracy of urinary amanitin in suspected mushroom poisoning: A pilot study. J. Toxicol. Clin. Toxicol. 2004, 42, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Bruggemann, O.; Meder, M.; Freitag, R. Analysis of amatoxins α-amanitin and β-amanitin in toadstool extracts and body fluids by capillary zone electrophoresis with photodiode array detection. J. Chromatogr. A 1996, 744, 167–176. [Google Scholar] [CrossRef]
- Robinson-Fuentes, V.; Jaimes-Sánchez, J.; García-Aguilar, L.; Gómez-Peralta, M.; Vázquez-Garciduenas, M.; Vázquez-Marrfo, G. Determination of α- and β-amanitin in clinical urine samples by Capillary Zone Electrophoresis. J. Pharm. Biomed. Anal. 2008, 47, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Rieck, W.; Platt, D. High-performance liquid chromatographic method for the determination of α-amanitin and phalloidin in human plasma using the column-switching technique and its application in suspected cases of poisoning by the green species of amanita mushroom (Amanita phalloides). J. Chromatogr. 1988, 425, 121–134. [Google Scholar] [PubMed]
- Zhou, Z.; Cao, M.; Zhou, L.; Zuo, X.; Tang, Y. Determination of α-amanitin in human serum by solid-phase extraction coupled with HPLC–UV. LC GC N. Am. 2011, 29, 672–677. [Google Scholar]
- Tagliaro, F.; Schiavon, G.; Bontempelli, G.; Carli, G.; Marigo, M. Improved high-performance liquid chromatographic determination with amperometric detection of α-amanitin in human plasma based on its voltammetric study. J. Chromatogr. 1991, 563, 299–311. [Google Scholar] [CrossRef]
- Defendenti, C.; Bonacina, E.; Mauroni, M.; Gelosa, L. Validation of a high performance liquid chromatographic method for alpha amanitin determination in urine. Forensic Sci. Int. 1998, 92, 59–68. [Google Scholar] [CrossRef]
- Garcia, J.; Costa, V.M.; Baptista, P.; Bastos, M.L.; Carvalho, F. Quantification of alpha-amanitin in biological samples by HPLC using simultaneous UV-diode array and electrochemical detection. J. Chromatogr. B 2015, 997, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Leite, M.; Freitas, A.; Azul, A.M.; Barbosa, J.; Costa, S.; Ramos, F. Development, optimization and application of an analytical methodology by ultra performance liquid chromatography—Tandem mass spectrometry for determination of amanitins in urine and liver samples. Anal. Chim. Acta 2013, 799, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Maurer, H.H.; Schmitt, C.J.; Weber, A.A.; Kraemer, T. Validated electrospray liquid chromatographic–mass spectrometric assay for the determination of the mushroom toxins α- and β-amanitin in urine after immunoaffinity extraction. J. Chromatogr. B 2000, 748, 125–135. [Google Scholar] [CrossRef]
- Chung, W.; Tso, S.; Sze, S. Separation of polar mushroom toxins by mixed-mode hydrophilic and ionicInteraction liquid chromatography—Electrospray ionization-mass spectrometry. J. Chromatogr. Sci. 2007, 45, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, M.; Kaneko, R.; Hirata, Y.; Hamajima, M.; Arinobu, T.; Ogawa, T.; Ishii, A. Simple analysis of α-amanitin and β-amanitin in human plasma by liquid chromatography-mass spectrometry. Forensic Toxicol. 2010, 28, 110–114. [Google Scholar] [CrossRef]
- Nomura, M.; Suzuki, Y.; Kaneko, R.; Ogawa, T.; Hattori, H.; Seno, H.; Ishii, A. Simple and rapid analysis of amatoxins using UPLC–MS–MS. Forensic Toxicol. 2012, 30, 185–192. [Google Scholar] [CrossRef]
- Helfer, A.G.; Meyer, M.R.; Michely, J.A.; Maurer, H.H. Direct analysis of the mushroom poisons α- and β-amanitin in human urine using a novel on-line turbulent flow chromatography modecoupled to liquid chromatography–high resolution-massspectrometry/mass spectrometry. J. Chromatogr. A 2014, 1325, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Tomkova, J.; Ondra, P.; Válka, I. Simultaneous determination of mushroom toxins α-amanitin, β-amanitin and muscarine in human urine by solid-phase extraction and ultra-high-performance liquid chromatography coupled with ultra-high-resolution TOF mass spectrometry. Forensic Sci. Int. 2015, 251, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Gonmori, K.; Minakata, K.; Suzuki, M.; Yamagishi, I.; Nozawa, H.; Hasegawa, K.; Wurita, A.; Watanabe, K.; Suzuki, O. MALDI-TOF mass spectrometric analysis of α-amanitin, β-amanitin, and phalloidin in urine. Forensic Toxicol. 2012, 30, 179–184. [Google Scholar] [CrossRef]
- Vetter, J. Toxins of Amanita phalloides. Toxicon 1997, 36, 13–24. [Google Scholar] [CrossRef]
- Chambers, E.; Wagrowski-Diehl, D.M.; Lu, Z.; Mazzeo, J.R. Systematic and comprehensive strategy for reducing matrix effects in LC/MS/MS analyses. J. Chromatogr. B 2007, 852, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Carmical, J.; Brown, S. The impact of phospholipids and phospholipid removal on bioanalytical method performance. Biomed. Chromatogr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Little, J.L.; Wempe, M.F.; Buchanan, C.M. Liquid chromatography–mass spectrometry/mass spectrometry method development for drug metabolism studies: Examining lipid matrix ionization effects in plasma. J. Chromatogr. B 2006, 833, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, B.K. Standard line slopes as a measure of a relative matrix effect in quantitative HPLC–MS bioanalysis. J. Chromatogr. B 2006, 830, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Guideline on bioanalytical method validation. European Medicines Agency. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf (accessed on 12 February 2016).
- Deng, Q.W.; Li, T.H.; Xi, P.G.; Gan, L.X.; Xiao, Z.D.; Jiang, Z.D. Peptide toxin components of Amanita exitialis basidiocarps. Mycologia 2011, 103, 946–949. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MRM Transition | Cone Voltage (V) | Collision Energy (eV) | Retention Times (min) | Ion Abundant Ratio (% of Base Peak) |
|---|---|---|---|---|---|
| α-amanitin | 919.5 > 86.0 1,2 | 20 | 68 | 4.72 ± 0.2 | 38 ± 10 |
| 919.5 > 259.1 1 | 42 | ||||
| β-amanitin | 920.5 > 86.0 1,2 | 20 | 71 | 4.96 ± 0.2 | 47 ± 9 |
| 920.5 > 259.1 1 | 42 | ||||
| γ-amanitin | 903.0 > 86.0 1,2 | 20 | 70 | 5.45 ± 0.2 | 43 ± 8 |
| 903.0 > 243.1 1 | 41 | ||||
| PHD | 789.4 > 157.0 1,2 | 20 | 61 | 7.31 ± 0.2 | 91 ± 4 |
| 789.4 > 86.0 1 | 70 | ||||
| PCD | 847.0 > 157.0 1,2 | 20 | 64 | 7.87 ± 0.2 | 83 ± 5 |
| 847.0 > 86.0 1 | 70 |
| Compound | Matrix | Relative Matrix Effect % (CV%) n = 6 | Process Efficiency % | Recovery % n = 6 | Precision % n = 6 | LOD ng/g n = 20 | LLOQ ng/g n = 20 | |
|---|---|---|---|---|---|---|---|---|
| Intra-day | Inter-day | |||||||
| α-amanitin | plasma | 105.18 (3.05) | 87.84 | 83.51 | 5.86 | 6.68 | 0.5 | 1 |
| serum | 98.25 (6.52) | 80.93 | 82.37 | 4.76 | 5.03 | 0.5 | 1 | |
| urine | 91.92 (6.80) | 77.43 | 84.24 | 3.94 | 4.02 | 1 | 2.5 | |
| β-amanitin | plasma | 103.37 (4.44) | 91.56 | 88.58 | 5.43 | 2.42 | 0.5 | 1 |
| serum | 100.48 (3.08) | 85.50 | 85.10 | 7.01 | 5.46 | 0.5 | 1 | |
| urine | 96.80 (3.85) | 78.11 | 80.69 | 3.68 | 3.22 | 0.5 | 1 | |
| γ-amanitin | plasma | 98.51 (4.82) | 87.17 | 88.49 | 3.01 | 2.57 | 0.5 | 1 |
| serum | 105.47 (3.97) | 95.11 | 90.18 | 2.14 | 4.04 | 1 | 2.5 | |
| urine | 105.99 (4.11) | 87.62 | 82.67 | 3.29 | 5.67 | 1 | 2.5 | |
| PHD | plasma | 103.83 (3.44) | 91.49 | 88.12 | 2.16 | 5.53 | 0.5 | 1 |
| serum | 103.57 (3.39) | 96.21 | 92.90 | 3.92 | 5.71 | 0.5 | 1 | |
| urine | 103.84 (3.36) | 91.01 | 87.65 | 3.44 | 4.03 | 0.5 | 1 | |
| PCD | plasma | 99.03 (3.17) | 99.46 | 90.44 | 6.21 | 8.54 | 0.5 | 1 |
| serum | 105.77 (2.54) | 91.79 | 86.78 | 2.72 | 3.04 | 0.5 | 1 | |
| urine | 94.58 (3.62) | 81.65 | 86.33 | 4.09 | 7.22 | 0.5 | 1 | |
| Compound | Slopes of Set I (CV%), Correlation Coefficient (r2) (n = 6) | Matrix | Slopes of Set II (CV%), Correlation Coefficient (r2) (n = 6) | Slopes of Set III (CV%), Correlation Coefficient (r2) (n = 6) |
|---|---|---|---|---|
| α-amanitin | 1306.37 (0.84), 0.9993 | plasma | 1374.01 (3.05), 0.9996 | 1243.57 (5.86),0.9998 |
| serum | 1294.35 (6.52), 0.9993 | 1011.68 (4.76), 0.9989 | ||
| urine | 1200.82 (6.80), 0.9998 | 1061.60 (3.94), 0.9998 | ||
| β-amanitin | 1534.87 (1.03), 0.9998 | plasma | 1510.67 (4.44), 9.9998 | 1338.17 (2.43), 0.9994 |
| serum | 1542.29 (3.08), 0.9983 | 1306.25 (6.01), 0.9978 | ||
| urine | 1485.85 (3.85), 0.9987 | 1235.27 (2.68), 0.9995 | ||
| γ-amanitin | 952.75 (0.96), 0.9996 | plasma | 938.58 (4.82), 0.9998 | 933.87 (2.01), 0.9992 |
| serum | 1004.89 (3.97), 0.9986 | 875.13 (2.14), 0.9967 | ||
| urine | 1009.82 (4.11), 0.9999 | 906.19 (3.29), 0.9998 | ||
| PHD | 3434.45(0.95), 0.9998 | plasma | 3595.76 (3.44), 0.9998 | 3600.11 (2.16), 0.9976 |
| serum | 3557.01 (3.39), 0.9997 | 3125.76 (3.92), 0.9994 | ||
| urine | 3566.29 (3.36), 0.9998 | 3304.45 (3.44), 0.9993 | ||
| PCD | 1442.27 (0.99), 0.9998 | plasma | 1428.30 (3.17), 0.9999 | 1434.52 (6.21), 0.9992 |
| serum | 1525.57 (2.54), 0.9997 | 1177.65 (2.72), 0.9987 | ||
| urine | 1364.07 (3.62), 0.9998 | 1339.23 (4.09), 0.9983 |
| Compound | Matrix | One Week | Two Weeks | Four Weeks | Eight Weeks |
|---|---|---|---|---|---|
| Recovery% (Precision%) | |||||
| α-amanitin | plasma | 87.12 (5.52) | 90.95 (3.52) | 94.56 (7.23) | 93.73 (7.31) |
| serum | 83.31 (4.57) | 87.25 (8.74) | 85.15 (6.79) | 84.42 (10.04) | |
| urine | 81.03 (3.25) | 86.32 (5.54) | 83.64 (4.54) | 80.43 (3.40) | |
| β-amanitin | plasma | 82.56 (5.72) | 80.77 (4.72) | 84.78 (8.79) | 83.38 (9.09) |
| serum | 75.15 (2.98) | 83.26 (5.08) | 80.63 (5.08) | 87.55 (4.99) | |
| urine | 87.54 (3.76) | 83.94 (3.60) | 85.42 (2.65) | 89.12 (2.95) | |
| γ-amanitin | plasma | 91.33 (1.98) | 96.68 (11.99) | 94.44 (3.90) | 96.61 (8.30) |
| serum | 85.27 (2.88) | 88.71 (4.58) | 86.04 (6.82) | 83.14 (3.82) | |
| urine | 78.34 (3.25 | 83.42 (6.94) | 85.23 (5.43) | 76.23 (5.43) | |
| PHD | plasma | 95.87 (3.12) | 98.66 (3.22) | 97.42 (6.26) | 93.40 (6.27) |
| serum | 90.03 (7.38) | 97.04 (3.28) | 95.11 (5.24) | 100.86 (6.72) | |
| urine | 80.62 (6.10) | 80.62 (5.05) | 85.24 (9.05) | 90.50 (7.55) | |
| PCD | plasma | 96.41 (4.34) | 90.65 (3.36) | 92.18 (2.35) | 88.35 (4.56) |
| serum | 85.24 (3.66) | 83.49 (5.16) | 82.55 (4.36) | 87.52 (6.01) | |
| urine | 87.55 (5.45) | 84.56 (7.91) | 86.49 (6.88) | 83.92 (3.85) | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Zhao, Y.; Li, H.; Zhou, S.; Chen, D.; Zhang, Y.; Yao, Q.; Sun, C. A Simple and High-Throughput Analysis of Amatoxins and Phallotoxins in Human Plasma, Serum and Urine Using UPLC-MS/MS Combined with PRiME HLB μElution Platform. Toxins 2016, 8, 128. https://doi.org/10.3390/toxins8050128
Zhang S, Zhao Y, Li H, Zhou S, Chen D, Zhang Y, Yao Q, Sun C. A Simple and High-Throughput Analysis of Amatoxins and Phallotoxins in Human Plasma, Serum and Urine Using UPLC-MS/MS Combined with PRiME HLB μElution Platform. Toxins. 2016; 8(5):128. https://doi.org/10.3390/toxins8050128
Chicago/Turabian StyleZhang, Shuo, Yunfeng Zhao, Haijiao Li, Shuang Zhou, Dawei Chen, Yizhe Zhang, Qunmei Yao, and Chengye Sun. 2016. "A Simple and High-Throughput Analysis of Amatoxins and Phallotoxins in Human Plasma, Serum and Urine Using UPLC-MS/MS Combined with PRiME HLB μElution Platform" Toxins 8, no. 5: 128. https://doi.org/10.3390/toxins8050128
APA StyleZhang, S., Zhao, Y., Li, H., Zhou, S., Chen, D., Zhang, Y., Yao, Q., & Sun, C. (2016). A Simple and High-Throughput Analysis of Amatoxins and Phallotoxins in Human Plasma, Serum and Urine Using UPLC-MS/MS Combined with PRiME HLB μElution Platform. Toxins, 8(5), 128. https://doi.org/10.3390/toxins8050128