
Yeast Killer Toxin K28: Biology and Unique Strategy of Host Cell Intoxication and Killing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
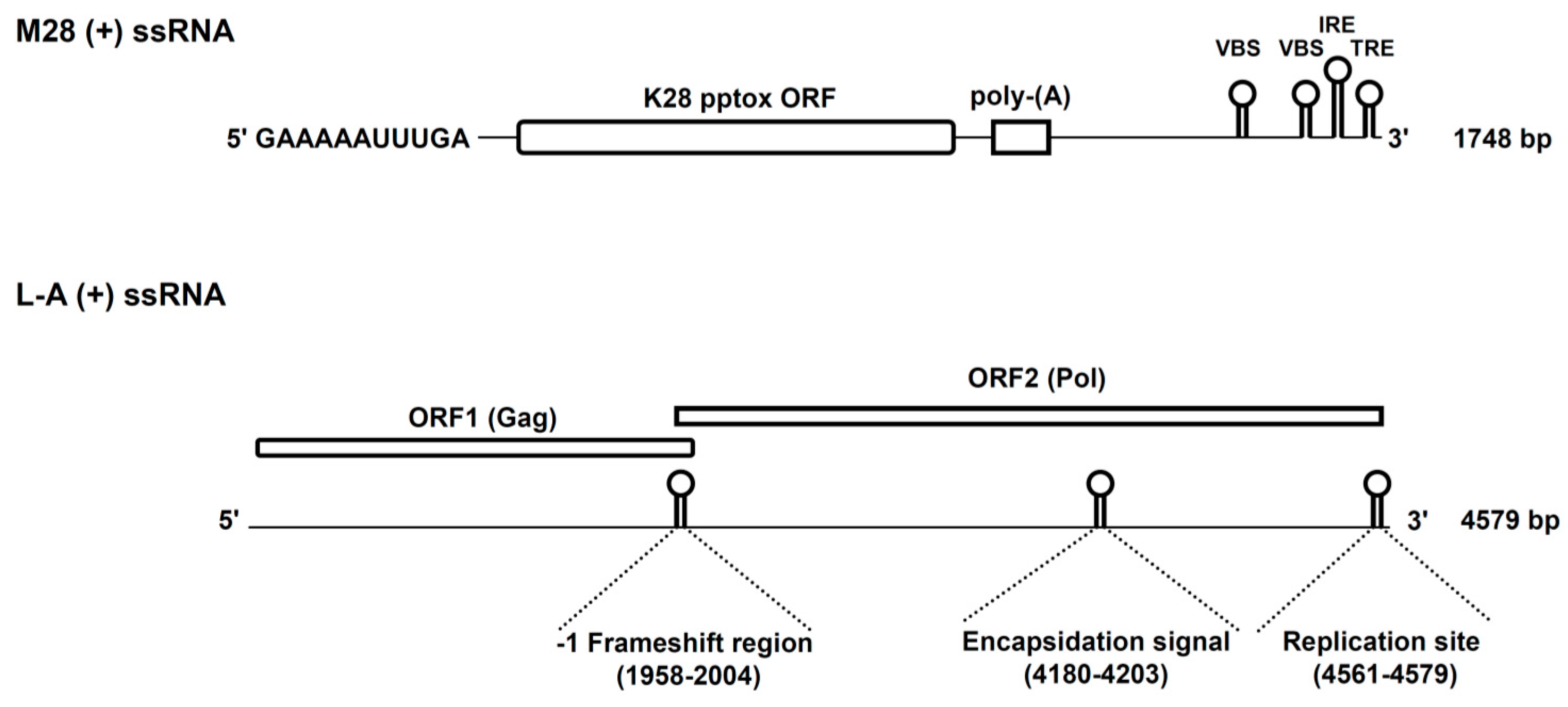
2. K28 Phenotype: Origin, Genomic Organization and Viral Replication
3. Viral Preprotoxin Processing and Toxin Secretion
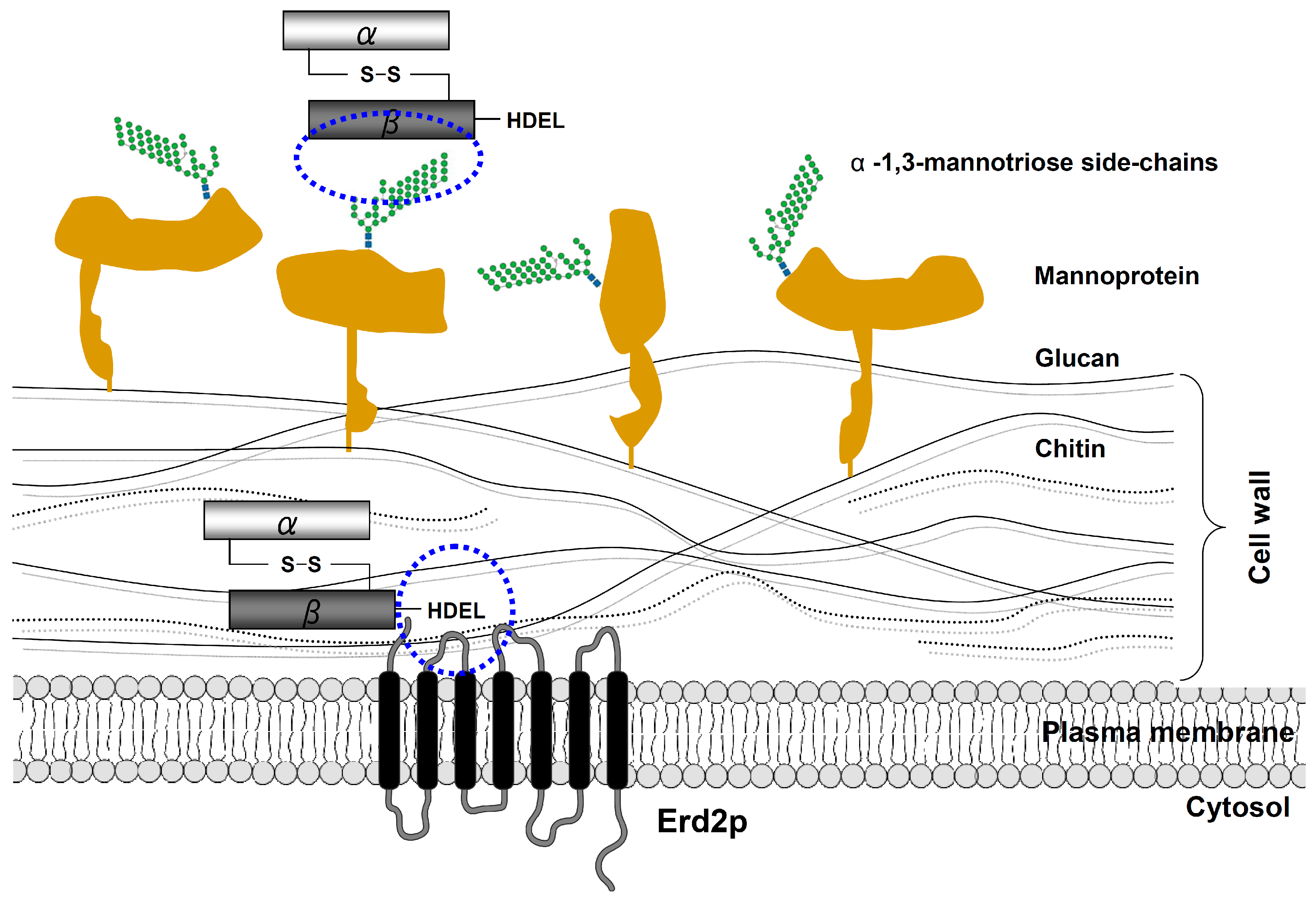
4. Cell Wall Binding and Endocytosis of K28
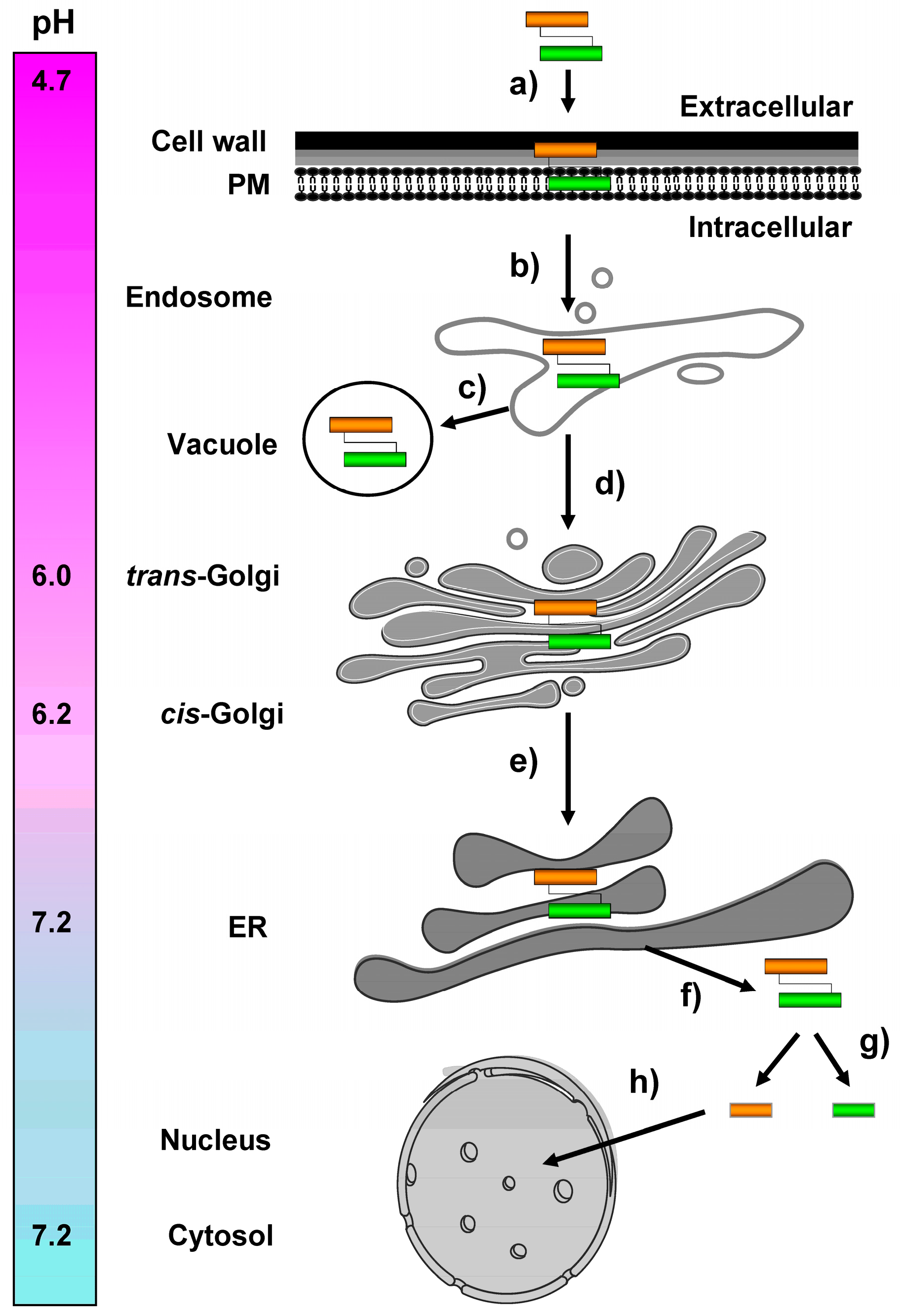
5. Intracellular K28 Trafficking from the Plasma Membrane to the ER
6. ER-To-Cytosol Retrotranslocation
7. Mode of K28 Toxicity
8. Toxin Immunity
9. Open Questions and Future Perspectives
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bevan, E.A.; Makower, M. The physiological basis of the killer character in yeast. In Proceedings of the 11th International Congress Genet, The Hague, The Netherlands, September 1963; Volume 1, pp. 202–203. [Google Scholar]
- Bevan, E.A.; Herring, A.J.; Mitchell, D.J. Preliminary characterization of two species of dsrna in yeast and their relationship to the “Killer” Character. Nature 1973, 245, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Magliani, W.; Conti, S.; Gerloni, M.; Bertolotti, D.; Polonelli, L. Yeast killer systems. Clin. Microbiol. Rev. 1997, 10, 369–400. [Google Scholar] [PubMed]
- Schmitt, M.J.; Breinig, F. The viral killer system in yeast: From molecular biology to application. FEMS Microbiol. Rev. 2002, 26, 257–276. [Google Scholar] [CrossRef] [PubMed]
- Theisen, S.; Molkenau, E.; Schmitt, M.J. Wicaltin, a new protein toxin secreted by the yeast williopsis californica and its broad-spectrum antimycotic potential. J. Microbiol. Biotechnol. 2000, 10, 547–550. [Google Scholar]
- Schaffrath, R.; Meinhardt, F. Kluyveromyces lactis zymocin and related plasmid-encoded yeast killer toxins. In Topics in Current Genetics: Microbial Protein Toxins; Springer: Berlin/Heidelberg, Germany, 2005; pp. 133–155. [Google Scholar]
- Drinnenberg, I.A.; Fink, G.R.; Bartel, D.P. Compatibility with killer explains the rise of rnai-deficient fungi. Science 2011, 333, 1592. [Google Scholar] [CrossRef] [PubMed]
- Ganter, P.F.; Starmer, W.T. Killer factor as a mechanism of interference competition in yeasts associated with cacti. Ecology 1992, 73, 54–67. [Google Scholar] [CrossRef]
- Hannig, E.M.; Leibowitz, M.J. Structure and expression of the M 2 genomic segment of a type 2 killer virus of yeast. Nucleic Acids Res. 1985, 13, 4379–4400. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Cousino, N.; Maqueda, M.; Ambrona, J.; Zamora, E.; Esteban, R.; Ramirez, M. A new wine Saccharomyces cerevisiae killer toxin (klus), encoded by a double-stranded RNA virus, with broad antifungal activity is evolutionarily related to a chromosomal host gene. Appl. Environ. Microbiol. 2011, 77, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.J.; Tipper, D.J. K28, a unique double-stranded RNA killer virus of Saccharomyces cerevisiae. Mol. Cell. Biol. 1990, 10, 4807–4815. [Google Scholar] [CrossRef] [PubMed]
- Bostian, K.A.; Jayachandran, S.; Tipper, D.J. A glycosylated protoxin in killer yeast: Models for its structure and maturation. Cell 1983, 32, 169–180. [Google Scholar] [CrossRef]
- Schmitt, M.J.; Schernikau, G. Construction of a cdna-based k1/k2/k28 triple killer strain of Saccharomyces cerevisiae. Food Technol. Biotechnol. 1997, 35, 281–285. [Google Scholar]
- Schmitt, M.J.; Breinig, F. Yeast viral killer toxins: Lethality and self-protection. Nat. Rev. Microbiol. 2006, 4, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.J.; Tipper, D.J. Genetic analysis of maintenance and expression of l and m double-stranded RNAs from yeast killer virus k28. Yeast 1992, 8, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.J.; Tipper, D.J. Sequence of the M28 dsRNA: Preprotoxin is processed to an alpha/beta heterodimeric protein toxin. Virology 1995, 213, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Icho, T.; Wickner, R.B. The double-stranded RNA genome of yeast virus L-A encodes its own putative RNA polymerase by fusing two open reading frames. J. Biol. Chem. 1989, 264, 6716–6723. [Google Scholar] [PubMed]
- Dinman, J.D.; Icho, T.; Wickner, R.B. A −1 ribosomal frameshift in a double-stranded RNA virus of yeast forms a gag-pol fusion protein. Proc. Natl. Acad. Sci. USA 1991, 88, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Ribas, J.C.; Makhov, A.M.; Wickner, R.B. Pol of gag-pol fusion protein required for encapsidation of viral RNA of yeast L-A virus. Nature 1992, 359, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Cousino, N.; Gomez, P.; Esteban, R. L-a-lus, a new variant of the l-a totivirus found in wine yeasts with klus killer toxin-encoding mlus double-stranded rna: Possible role of killer toxin-encoding satellite rnas in the evolution of their helper viruses. Appl. Environ. Microbiol. 2013, 79, 4661–4674. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B. Prions and RNA viruses of Saccharomyces cerevisiae. Annu. Rev. Genet. 1996, 30, 109–139. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B. Double-stranded RNA viruses of Saccharomyces cerevisiae. Microbiol. Rev. 1996, 60, 250–265. [Google Scholar] [PubMed]
- Ohtake, Y.; Wickner, R.B. Yeast virus propagation depends critically on free 60s ribosomal subunit concentration. Mol. Cell. Biol. 1995, 15, 2772–2781. [Google Scholar] [CrossRef] [PubMed]
- Ridley, S.P.; Sommer, S.S.; Wickner, R.B. Superkiller mutations in Saccharomyces cerevisiae suppress exclusion of M2 double-stranded RNA by L-A-HN and confer cold sensitivity in the presence of m and L-A-HN. Mol. Cell. Biol. 1984, 4, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Toh, E.A.; Wickner, R.B. “Superkiller” Mutations suppress chromosomal mutations affecting double-stranded RNA killer plasmid replication in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1980, 77, 527–530. [Google Scholar] [CrossRef]
- Toh, E.A.; Wickner, R.B. A mutant killer plasmid whose replication depends on a chromosomal “Superkiller” Mutation. Genetics 1979, 91, 673–682. [Google Scholar]
- Tipper, D.J.; Schmitt, M.J. Yeast dsrna viruses: Replication and killer phenotypes. Mol. Microbiol. 1991, 5, 2331–2338. [Google Scholar] [CrossRef] [PubMed]
- Toh, E.A.; Guerry, P.; Wickner, R.B. Chromosomal superkiller mutants of Saccharomyces cerevisiae. J. Bacteriol. 1978, 136, 1002–1007. [Google Scholar]
- Cheng, R.H.; Caston, J.R.; Wang, G.J.; Gu, F.; Smith, T.J.; Baker, T.S.; Bozarth, R.F.; Trus, B.L.; Cheng, N.; Wickner, R.B.; et al. Fungal virus capsids, cytoplasmic compartments for the replication of double-stranded rna, formed as icosahedral shells of asymmetric gag dimers. J. Mol. Biol. 1994, 244, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Ribas, J.C.; Wickner, R.B. The gag domain of the gag-pol fusion protein directs incorporation into the L-A double-stranded RNA viral particles in Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 9306–9311. [Google Scholar] [CrossRef] [PubMed]
- Naitow, H.; Tang, J.; Canady, M.; Wickner, R.B.; Johnson, J.E. L-A virus at 3.4 a resolution reveals particle architecture and mRNA decapping mechanism. Nat. Struct. Biol. 2002, 9, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Bruenn, J.A. Relationships among the positive strand and double-strand RNA viruses as viewed through their RNA-dependent RNA polymerases. Nucleic Acids Res. 1991, 19, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Esteban, R.; Wickner, R.B. In vitro L-A double-stranded RNA synthesis in virus-like particles from Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1986, 83, 4433–4437. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Wickner, R.B. Replicase of L-A virus-like particles of Saccharomyces cerevisiae. In vitro conversion of exogenous L-A and m1 single-stranded rnas to double-stranded form. J. Biol. Chem. 1988, 263, 454–460. [Google Scholar] [PubMed]
- Esteban, R.; Fujimura, T.; Wickner, R.B. Internal and terminal cis-acting sites are necessary for in vitro replication of the L-A double-stranded RNA virus of yeast. EMBO J. 1989, 8, 947–954. [Google Scholar] [PubMed]
- Fujimura, T.; Wickner, R.B. Gene overlap results in a viral protein having an RNA binding domain and a major coat protein domain. Cell 1988, 55, 663–671. [Google Scholar] [CrossRef]
- Esteban, R.; Wickner, R.B. Three different M1 RNA-containing virus like particle types in Saccharomyces cerevisiae: In vitro M1 double-stranded RNA synthesis. Mol. Cell. Biol. 1986, 6, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Bostian, K.A.; Sturgeon, J.A.; Tipper, D.J. Encapsidation of yeast killer double-stranded ribonucleic acids: Dependence of m on l. J. Bacteriol. 1980, 143, 463–470. [Google Scholar] [PubMed]
- Fujimura, T.; Esteban, R.; Esteban, L.M.; Wickner, R.B. Portable encapsidation signal of the L-A double-stranded RNA virus of S. cerevisiae. Cell 1990, 62, 819–828. [Google Scholar] [CrossRef]
- Breinig, F.; Sendzik, T.; Eisfeld, K.; Schmitt, M.J. Dissecting toxin immunity in virus-infected killer yeast uncovers an intrinsic strategy of self-protection. Proc. Natl. Acad. Sci. USA 2006, 103, 3810–3815. [Google Scholar] [CrossRef] [PubMed]
- Fuller, R.S.; Brake, A.J.; Thorner, J. Intracellular targeting and structural conservation of a prohormone-processing endoprotease. Science 1989, 246, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Schwartz, S.L.; Mueller, N.C.; Schmitt, M.J. Cysteine residues in a yeast viral a/b toxin crucially control host cell killing via ph-triggered disulfide rearrangements. Mol. Biol. Cell 2017, 28, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Fuller, R.S.; Brake, A.; Thorner, J. Yeast prohormone processing enzyme (kex2 gene product) is a Ca2+-dependent serine protease. Proc. Natl. Acad. Sci. USA 1989, 86, 1434–1438. [Google Scholar] [CrossRef] [PubMed]
- Riffer, F.; Eisfeld, K.; Breinig, F.; Schmitt, M.J. Mutational analysis of k28 preprotoxin processing in the yeast Saccharomyces cerevisiae. Microbiology 2002, 148, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Bostian, K.A.; Elliott, Q.; Bussey, H.; Burn, V.; Smith, A.; Tipper, D.J. Sequence of the preprotoxin dsrna gene of type i killer yeast: Multiple processing events produce a two-component toxin. Cell 1984, 36, 741–751. [Google Scholar] [CrossRef]
- Pelham, H.R.; Hardwick, K.G.; Lewis, M.J. Sorting of soluble er proteins in yeast. EMBO J. 1988, 7, 1757–1762. [Google Scholar] [PubMed]
- Sandvig, K.; van Deurs, B. Membrane traffic exploited by protein toxins. Annu. Rev. Cell Dev. Biol. 2002, 18, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.J.; Brendel, M.; Schwarz, R.; Radler, F. Inhibition of DNA synthesis in S. cerevisiae by yeast killer toxin kt28. J. Gen. Microbiol. 1989, 135, 1529–1535. [Google Scholar] [CrossRef]
- Schmitt, M.; Radler, F. Mannoprotein of the yeast cell wall as primary receptor for the killer toxin of Saccharomyces cerevisiae strain 28. J. Gen. Microbiol. 1987, 133, 3347–3354. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Radler, F. Molecular structure of the cell wall receptor for killer toxin kt28 in Saccharomyces cerevisiae. J. Bacteriol. 1988, 170, 2192–2196. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.J.; Radler, F. Blockage of cell wall receptors for yeast killer toxin kt28 with antimannoprotein antibodies. Antimicrob. Agents Chemother. 1990, 34, 1615–1618. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.Y.; Stirling, P.C.; Stimpson, H.E.; Giesselmann, E.; Schmitt, M.J.; Drubin, D.G. A yeast killer toxin screen provides insights into a/b toxin entry, trafficking, and killing mechanisms. Dev. Cell 2009, 17, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Vadasz, A.S.; Jagganath, D.B.; Pretorius, I.S.; Gupthar, A.S. Electron microscopy of the k2 killer effect of Saccharomyces cerevisiae t206 on a mesophilic wine yeast. Antonie Van Leeuwenhoek 2000, 78, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Martinac, B.; Zhu, H.; Kubalski, A.; Zhou, X.L.; Culbertson, M.; Bussey, H.; Kung, C. Yeast k1 killer toxin forms ion channels in sensitive yeast spheroplasts and in artificial liposomes. Proc. Natl. Acad. Sci. USA 1990, 87, 6228–6232. [Google Scholar] [CrossRef] [PubMed]
- Orentaite, I.; Poranen, M.M.; Oksanen, H.M.; Daugelavicius, R.; Bamford, D.H. K2 killer toxin-induced physiological changes in the yeast Saccharomyces cerevisiae. FEMS Yeast Res. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Eisfeld, K.; Riffer, F.; Mentges, J.; Schmitt, M.J. Endocytotic uptake and retrograde transport of a virally encoded killer toxin in yeast. Mol. Microbiol. 2000, 37, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.; Blum, A.; Giesselmann, E.; Dausend, J.; Rammo, D.; Muller, N.C.; Tschacksch, E.; Steimer, M.; Spindler, J.; Becherer, U.; et al. H/kdel receptors mediate host cell intoxication by a viral a/b toxin in yeast. Sci. Rep. 2016, 6, 31105. [Google Scholar] [CrossRef] [PubMed]
- Heiligenstein, S.; Eisfeld, K.; Sendzik, T.; Jimenez-Becker, N.; Breinig, F.; Schmitt, M.J. Retrotranslocation of a viral a/b toxin from the yeast endoplasmic reticulum is independent of ubiquitination and erad. EMBO J. 2006, 25, 4717–4727. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.W.; Lewis, M.J.; Pelham, H.R. Ph-dependent binding of kdel to its receptor in vitro. J. Biol. Chem. 1993, 268, 7465–7468. [Google Scholar] [PubMed]
- Sandvig, K.; van Deurs, B. Endocytosis, intracellular transport, and cytotoxic action of shiga toxin and ricin. Physiol. Rev. 1996, 76, 949–966. [Google Scholar] [PubMed]
- Majoul, I.V.; Bastiaens, P.I.; Soling, H.D. Transport of an external lys-asp-glu-leu (kdel) protein from the plasma membrane to the endoplasmic reticulum: Studies with cholera toxin in vero cells. J. Cell. Biol. 1996, 133, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.A.; Watson, P.D.; Marsden, C.J.; Smith, D.C.; Moore, K.A.; Cook, J.P.; Lord, J.M.; Roberts, L.M. Protein disulphide-isomerase reduces ricin to its a and b chains in the endoplasmic reticulum. Biochem. J. 2004, 383, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, P.A. Protein-disulfide isomerase-mediated reduction of the a subunit of cholera toxin in a human intestinal cell line. J. Biol. Chem. 1997, 272, 4591–4599. [Google Scholar] [PubMed]
- Suzuki, Y.; Schmitt, M.J. Redox diversity in erad-mediated protein retrotranslocation from the endoplasmic reticulum: A complex puzzle. Biol. Chem. 2015, 396, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.J.; Reiter, J. Viral induced yeast apoptosis. Biochim. Biophys. Acta 2008, 1783, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.; Herker, E.; Madeo, F.; Schmitt, M.J. Viral killer toxins induce caspase-mediated apoptosis in yeast. J. Cell. Biol. 2005, 168, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.J.; Klavehn, P.; Wang, J.; Schonig, I.; Tipper, D.J. Cell cycle studies on the mode of action of yeast k28 killer toxin. Microbiology 1996, 142 Pt 9, 2655–2662. [Google Scholar] [CrossRef] [PubMed]
- Belda, I.; Ruiz, J.; Alonso, A.; Marquina, D.; Santos, A. The biology of Pichia membranifaciens killer toxins. Toxins 2017, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- Ivanovska, I.; Hardwick, J.M. Viruses activate a genetically conserved cell death pathway in a unicellular organism. J. Cell. Biol. 2005, 170, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Eiden-Plach, A.; Zagorc, T.; Heintel, T.; Carius, Y.; Breinig, F.; Schmitt, M.J. Viral preprotoxin signal sequence allows efficient secretion of green fluorescent protein by Candida glabrata, Pichia pastoris, Saccharomyces cerevisiae, and Schizosaccharomyces pombe. Appl. Environ. Microbiol. 2004, 70, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Boone, C.; Scidu, A.M.; Wagner, J.; Degre, R.; Sanchez, C.; Bussey, H. Integration of the yeast k1 killer toxin gene into the genome of marked wine yeasts and its effect on vinication. Am. J. Enol. Vitic. 1990, 41, 37–42. [Google Scholar]


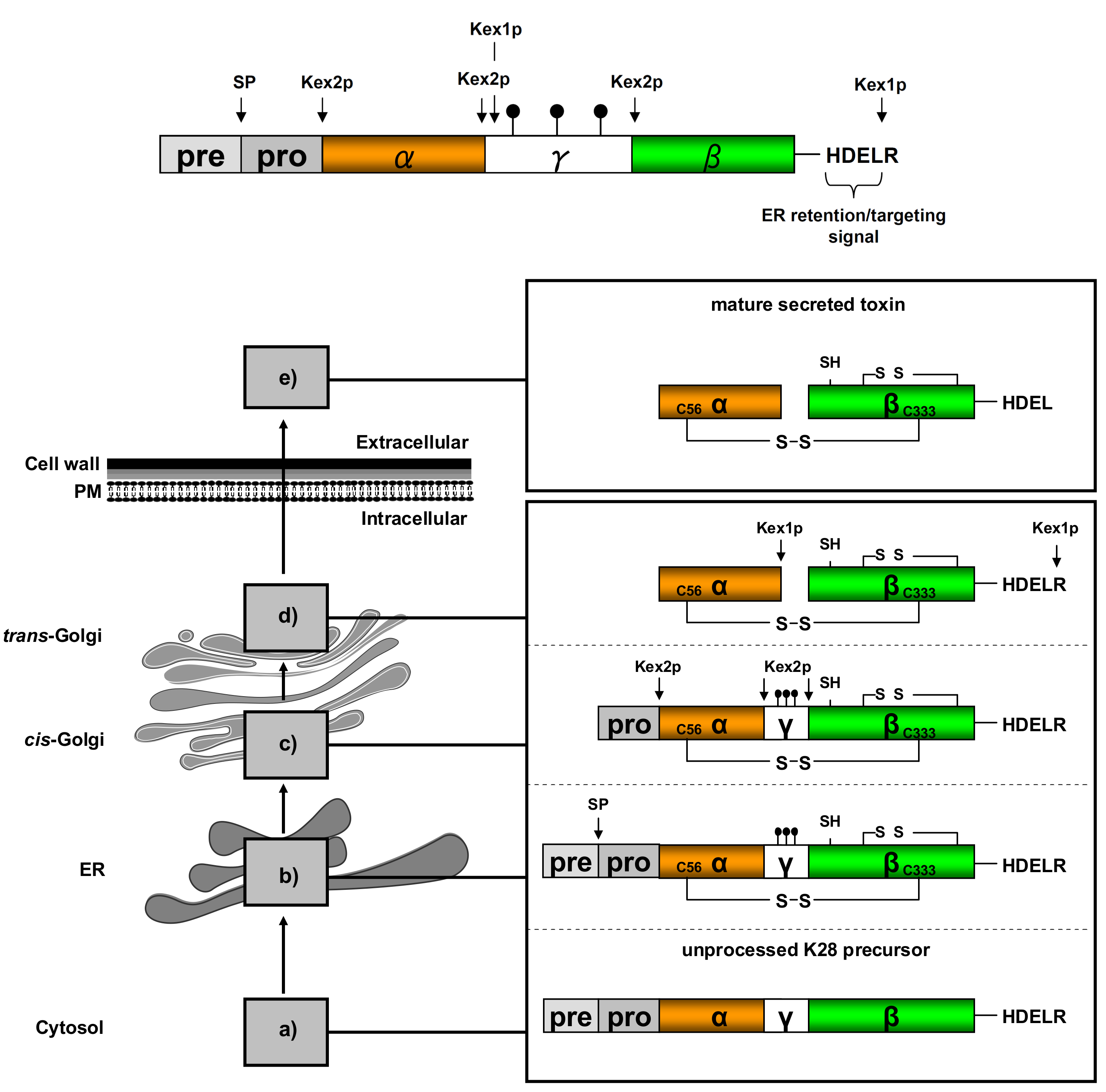
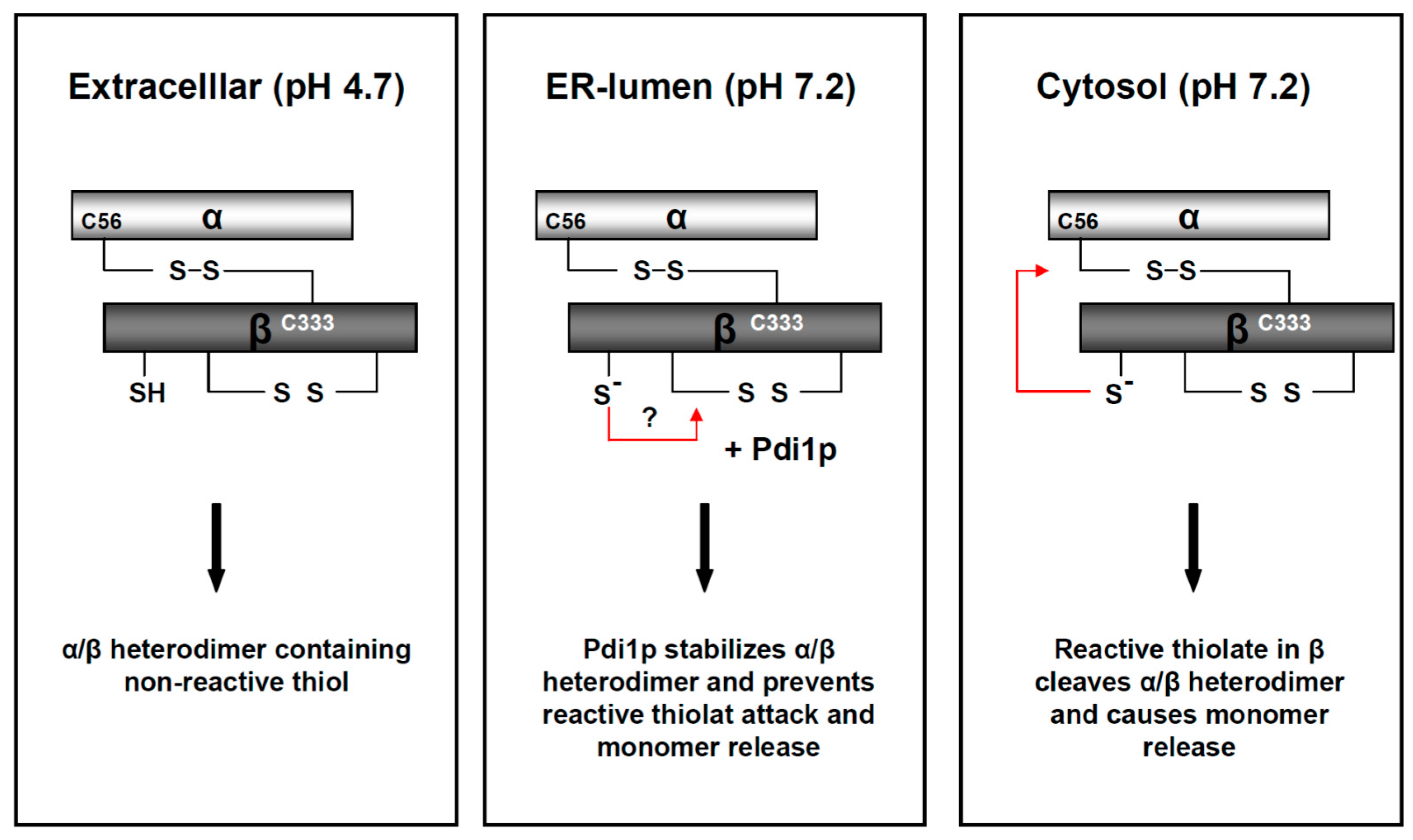
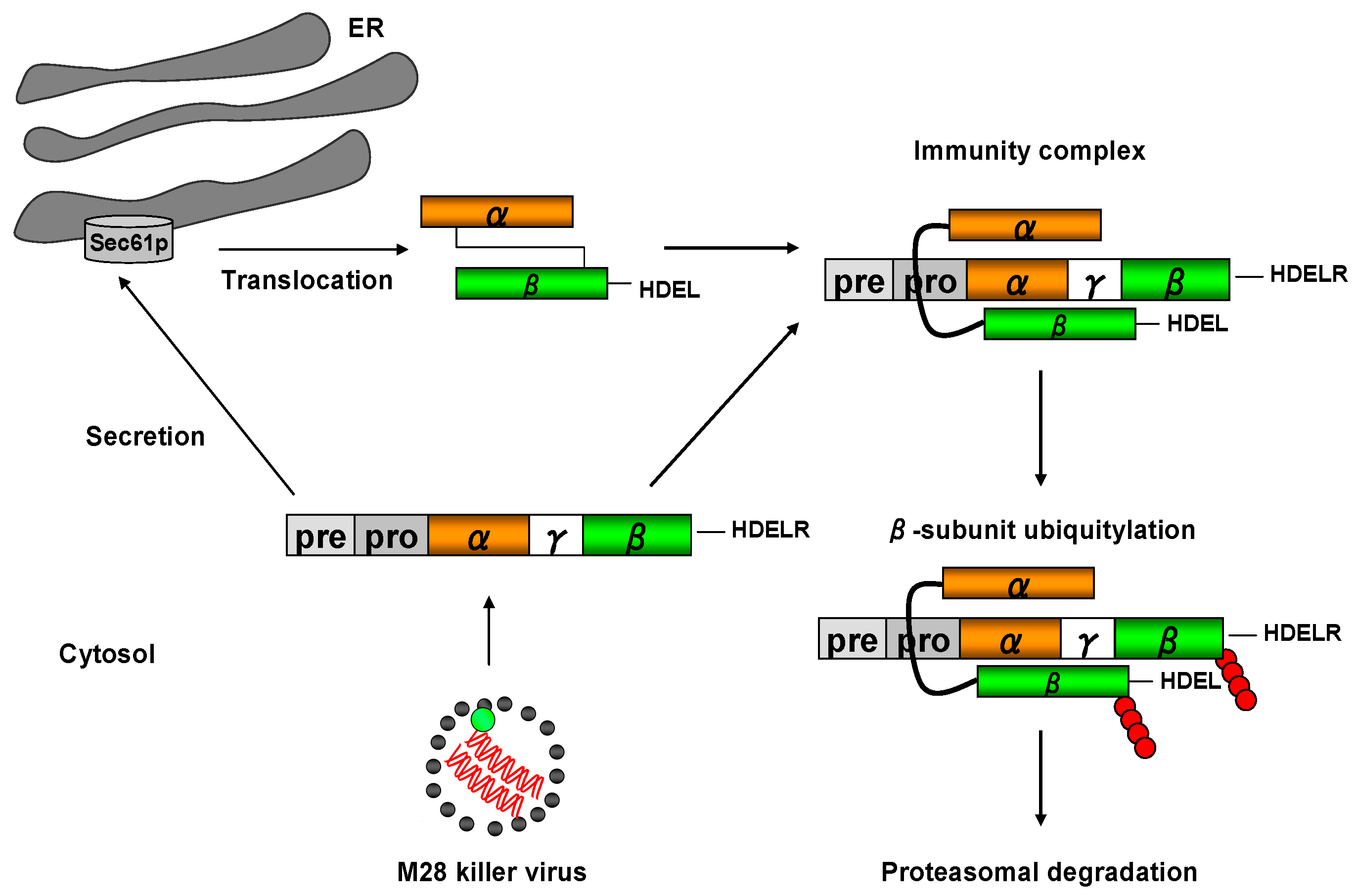
 ) and the connecting disulfide (s-s) between α and β is formed by protein disulfide isomerase (Pdi1p); (c) In the cis-Golgi, the furin-like endopeptidase, Kex2p, removes the pro-sequence and γ-subunit which leads to a disulfide-bonded α/β heterodimer. Although the precise function of the pro-region is still not fully understood, it has been proposed to be important for proper post-translational pptox import into the ER lumen [40]. While the inter-chain disulfide in the heterodimer has clearly been demonstrated to be positioned between the single cysteine in α (Cys56) and Cys333 in β, the exact position of the cysteine residues in β that form the intra-chain disulfide and the single free thiol is still hypothetical [41,42]; (d) In the trans-Golgi, the C-terminus of the α-subunit as well as the C-terminal arginine of the β-subunit are removed by carboxypeptidase Kex1p cleavage, which unmasks the ER retention/targeting motif, HDEL, thereby converting the precursor in its biologically active conformation; (e) K28 is finally secreted as a 21 kDa heterodimer, whose β-C terminus carries a potential ER retention/targeting signal required for toxin uptake in a sensitive target cell [14].
) and the connecting disulfide (s-s) between α and β is formed by protein disulfide isomerase (Pdi1p); (c) In the cis-Golgi, the furin-like endopeptidase, Kex2p, removes the pro-sequence and γ-subunit which leads to a disulfide-bonded α/β heterodimer. Although the precise function of the pro-region is still not fully understood, it has been proposed to be important for proper post-translational pptox import into the ER lumen [40]. While the inter-chain disulfide in the heterodimer has clearly been demonstrated to be positioned between the single cysteine in α (Cys56) and Cys333 in β, the exact position of the cysteine residues in β that form the intra-chain disulfide and the single free thiol is still hypothetical [41,42]; (d) In the trans-Golgi, the C-terminus of the α-subunit as well as the C-terminal arginine of the β-subunit are removed by carboxypeptidase Kex1p cleavage, which unmasks the ER retention/targeting motif, HDEL, thereby converting the precursor in its biologically active conformation; (e) K28 is finally secreted as a 21 kDa heterodimer, whose β-C terminus carries a potential ER retention/targeting signal required for toxin uptake in a sensitive target cell [14].
) and the connecting disulfide (s-s) between α and β is formed by protein disulfide isomerase (Pdi1p); (c) In the cis-Golgi, the furin-like endopeptidase, Kex2p, removes the pro-sequence and γ-subunit which leads to a disulfide-bonded α/β heterodimer. Although the precise function of the pro-region is still not fully understood, it has been proposed to be important for proper post-translational pptox import into the ER lumen [40]. While the inter-chain disulfide in the heterodimer has clearly been demonstrated to be positioned between the single cysteine in α (Cys56) and Cys333 in β, the exact position of the cysteine residues in β that form the intra-chain disulfide and the single free thiol is still hypothetical [41,42]; (d) In the trans-Golgi, the C-terminus of the α-subunit as well as the C-terminal arginine of the β-subunit are removed by carboxypeptidase Kex1p cleavage, which unmasks the ER retention/targeting motif, HDEL, thereby converting the precursor in its biologically active conformation; (e) K28 is finally secreted as a 21 kDa heterodimer, whose β-C terminus carries a potential ER retention/targeting signal required for toxin uptake in a sensitive target cell [14].
) and the connecting disulfide (s-s) between α and β is formed by protein disulfide isomerase (Pdi1p); (c) In the cis-Golgi, the furin-like endopeptidase, Kex2p, removes the pro-sequence and γ-subunit which leads to a disulfide-bonded α/β heterodimer. Although the precise function of the pro-region is still not fully understood, it has been proposed to be important for proper post-translational pptox import into the ER lumen [40]. While the inter-chain disulfide in the heterodimer has clearly been demonstrated to be positioned between the single cysteine in α (Cys56) and Cys333 in β, the exact position of the cysteine residues in β that form the intra-chain disulfide and the single free thiol is still hypothetical [41,42]; (d) In the trans-Golgi, the C-terminus of the α-subunit as well as the C-terminal arginine of the β-subunit are removed by carboxypeptidase Kex1p cleavage, which unmasks the ER retention/targeting motif, HDEL, thereby converting the precursor in its biologically active conformation; (e) K28 is finally secreted as a 21 kDa heterodimer, whose β-C terminus carries a potential ER retention/targeting signal required for toxin uptake in a sensitive target cell [14].




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becker, B.; Schmitt, M.J. Yeast Killer Toxin K28: Biology and Unique Strategy of Host Cell Intoxication and Killing. Toxins 2017, 9, 333. https://doi.org/10.3390/toxins9100333
Becker B, Schmitt MJ. Yeast Killer Toxin K28: Biology and Unique Strategy of Host Cell Intoxication and Killing. Toxins. 2017; 9(10):333. https://doi.org/10.3390/toxins9100333
Chicago/Turabian StyleBecker, Björn, and Manfred J. Schmitt. 2017. "Yeast Killer Toxin K28: Biology and Unique Strategy of Host Cell Intoxication and Killing" Toxins 9, no. 10: 333. https://doi.org/10.3390/toxins9100333
APA StyleBecker, B., & Schmitt, M. J. (2017). Yeast Killer Toxin K28: Biology and Unique Strategy of Host Cell Intoxication and Killing. Toxins, 9(10), 333. https://doi.org/10.3390/toxins9100333