
Microvesicle Involvement in Shiga Toxin-Associated Infection


Abstract
:1. Introduction
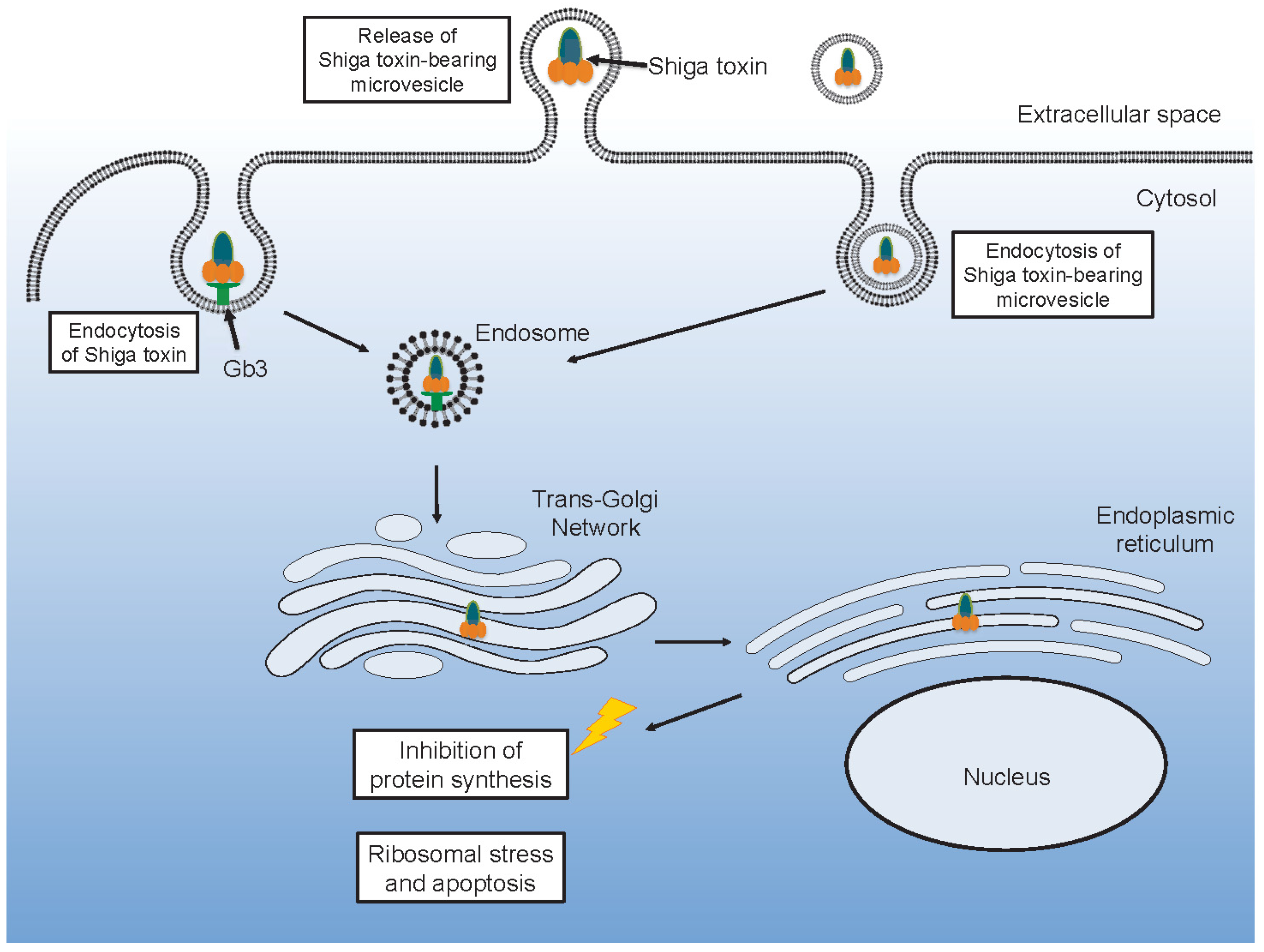
2. Shiga Toxin
2.1. Cytotoxicity of Shiga Toxin
2.2. Inflammatory Effects of Shiga Toxin and Lipopolysaccharide in the Intestine
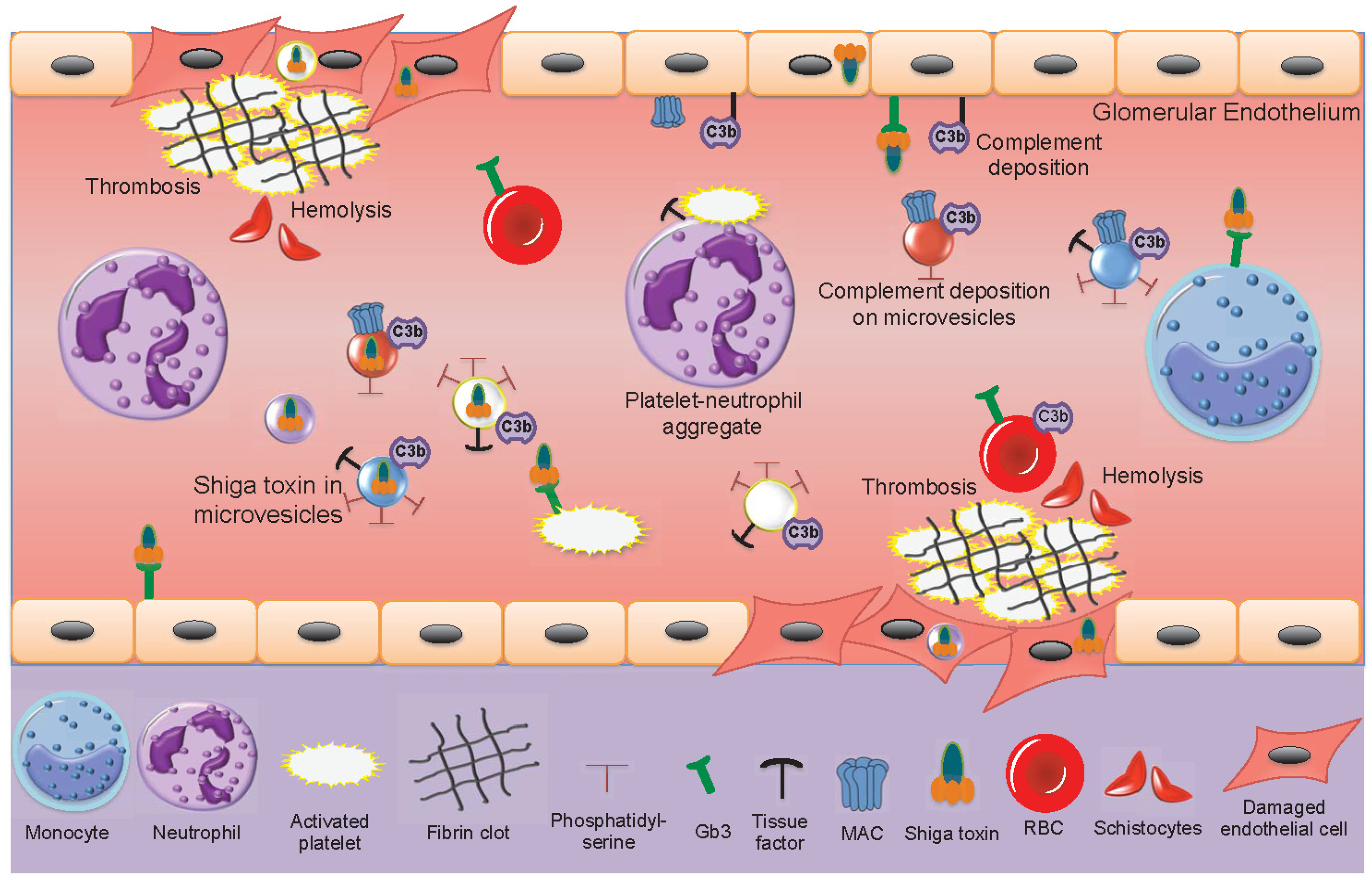
2.3. Interactions between Shiga Toxin and Blood Cells
2.4. Thrombus Formation During HUS
2.5. Shiga Toxin Induces the Release of Blood Cell-Derived Microvesicles
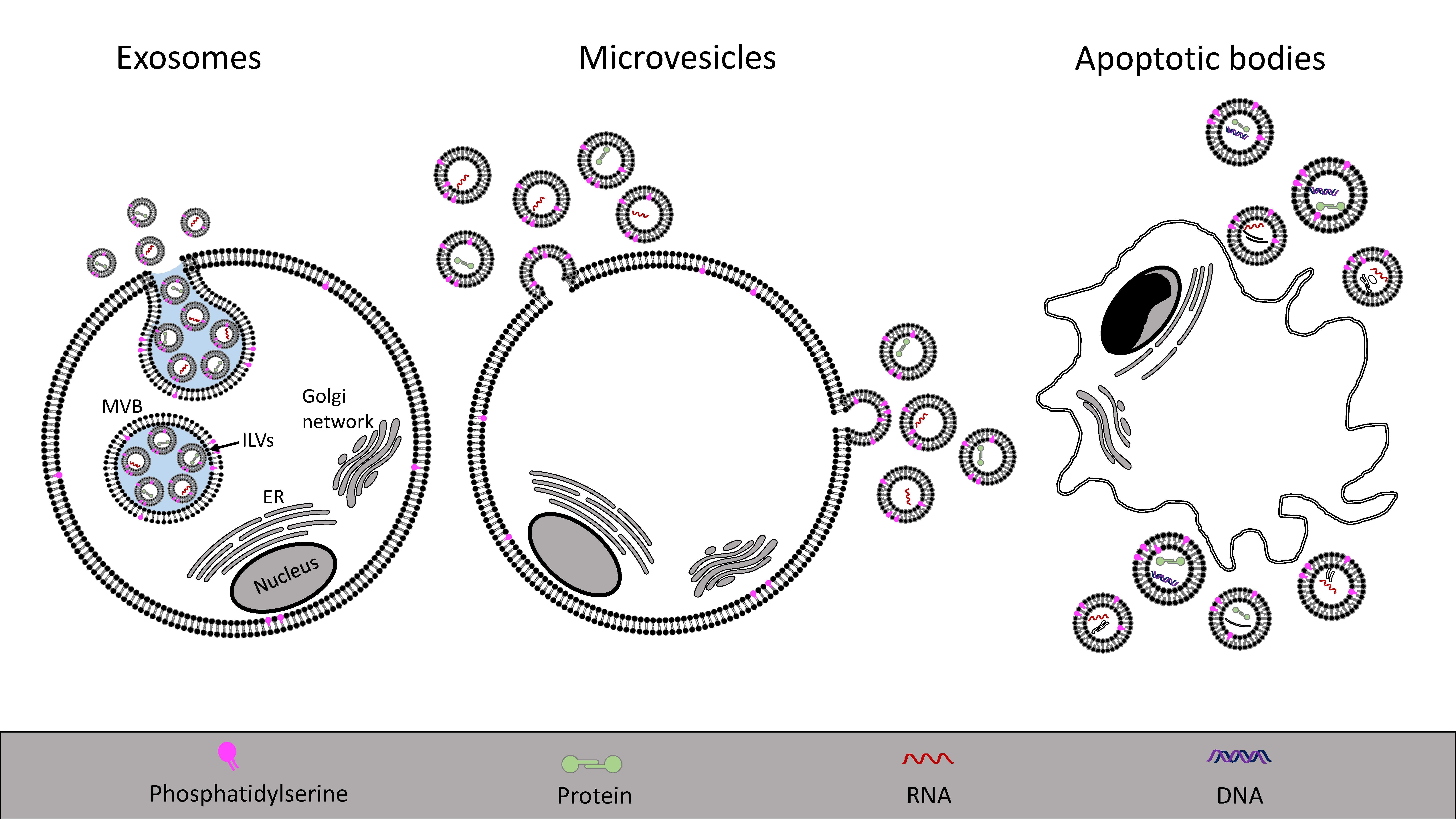
3. Characteristics of Extracellular Vesicles
3.1. Exosomes
3.2. Microvesicles
3.2.1. Microvesicle Formation
3.2.2. Microvesicle Uptake
3.2.3. The role of Microvesicles in Intercellular Communication
4. Microvesicles in Infectious Diseases
Microvesicles May Transfer Infectious Agents or Their Virulence Factors
5. Shiga Toxin-Induced Microvesicles in Laboratory Models
6. Microvesicles in the Pathogenesis of Hemolytic Uremic Syndrome
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Karpman, D.; Loos, S.; Tati, R.; Arvidsson, I. Haemolytic uraemic syndrome. J. Intern. Med. 2017, 281, 123–148. [Google Scholar] [CrossRef] [PubMed]
- Griffin, P.M.; Ostroff, S.M.; Tauxe, R.V.; Greene, K.D.; Wells, J.G.; Lewis, J.H.; Blake, P.A. Illnesses associated with Escherichia coli O157:H7 infections. A broad clinical spectrum. Ann. Intern. Med. 1988, 109, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.; Werber, D.; Cramer, J.P.; Askar, M.; Faber, M.; an der Heiden, M.; Bernard, H.; Fruth, A.; Prager, R.; Spode, A.; et al. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N. Engl. J. Med. 2011, 365, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- McKee, M.L.; O’Brien, A.D. Investigation of enterohemorrhagic Escherichia coli o157:H7 adherence characteristics and invasion potential reveals a new attachment pattern shared by intestinal E. coli. Infect. Immun. 1995, 63, 2070–2074. [Google Scholar] [PubMed]
- Kaper, J.B. The locus of enterocyte effacement pathogenicity island of Shiga toxin-producing Escherichia coli O157:H7 and other attaching and effacing E. coli. Jpn. J. Med. Sci. Biol. 1998, 51 (Suppl. 1), S101–S107. [Google Scholar] [CrossRef] [PubMed]
- Rogers, T.J.; Paton, J.C.; Wang, H.; Talbot, U.M.; Paton, A.W. Reduced virulence of an flic mutant of Shiga-toxigenic Escherichia coli O113:H21. Infect. Immun. 2006, 74, 1962–1966. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, A.R.; Sperandio, V. Inter-kingdom signaling: Chemical language between bacteria and host. Curr. Opin. Microbiol. 2009, 12, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Karmali, M.A.; Petric, M.; Lim, C.; Fleming, P.C.; Arbus, G.S.; Lior, H. The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J. Infect. Dis. 1985, 151, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Ståhl, A.L. Enterohemorrhagic Escherichia coli pathogenesis and the host response. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Loos, S.; Ahlenstiel, T.; Kranz, B.; Staude, H.; Pape, L.; Hartel, C.; Vester, U.; Buchtala, L.; Benz, K.; Hoppe, B.; et al. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: Presentation and short-term outcome in children. Clin. Infect. Dis. 2012, 55, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Schuller, S.; Frankel, G.; Phillips, A.D. Interaction of Shiga toxin from Escherichia coli with human intestinal epithelial cell lines and explants: Stx2 induces epithelial damage in organ culture. Cell Microbiol. 2004, 6, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Schuller, S.; Heuschkel, R.; Torrente, F.; Kaper, J.B.; Phillips, A.D. Shiga toxin binding in normal and inflamed human intestinal mucosa. Microbes Infect. 2007, 9, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Hurley, B.P.; Thorpe, C.M.; Acheson, D.W. Shiga toxin translocation across intestinal epithelial cells is enhanced by neutrophil transmigration. Infect. Immun. 2001, 69, 6148–6155. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, A.; Arondel, J.; Sansonetti, P.J. Role of Shiga toxin in the pathogenesis of bacillary dysentery, studied by using a tox- mutant of Shigella dysenteriae 1. Infect. Immun. 1988, 56, 3099–3109. [Google Scholar] [PubMed]
- Brigotti, M.; Tazzari, P.L.; Ravanelli, E.; Carnicelli, D.; Rocchi, L.; Arfilli, V.; Scavia, G.; Minelli, F.; Ricci, F.; Pagliaro, P.; et al. Clinical relevance of Shiga toxin concentrations in the blood of patients with hemolytic uremic syndrome. Pediatr. Infect. Dis. J. 2011, 30, 486–490. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Quinones, B.; Loo, M.T.; Loos, S.; Scavia, G.; Brigotti, M.; Levtchenko, E.; Monnens, L. Serum Shiga toxin 2 values in patients during acute phase of diarrhoea-associated haemolytic uraemic syndrome. Acta Paediatr. 2015, 104, e564–e568. [Google Scholar] [CrossRef] [PubMed]
- Te Loo, D.M.; van Hinsbergh, V.W.; van den Heuvel, L.P.; Monnens, L.A. Detection of verocytotoxin bound to circulating polymorphonuclear leukocytes of patients with hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2001, 12, 800–806. [Google Scholar] [PubMed]
- Ståhl, A.L.; Sartz, L.; Nelsson, A.; Békássy, Z.D.; Karpman, D. Shiga toxin and lipopolysaccharide induce platelet-leukocyte aggregates and tissue factor release, a thrombotic mechanism in hemolytic uremic syndrome. PLoS ONE 2009, 4, e6990. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, I.; Ståhl, A.L.; Hedström, M.M.; Kristoffersson, A.C.; Rylander, C.; Westman, J.S.; Storry, J.R.; Olsson, M.L.; Karpman, D. Shiga toxin-induced complement-mediated hemolysis and release of complement-coated red blood cell-derived microvesicles in hemolytic uremic syndrome. J. Immunol. 2015, 194, 2309–2318. [Google Scholar] [CrossRef] [PubMed]
- Bitzan, M.; Richardson, S.; Huang, C.; Boyd, B.; Petric, M.; Karmali, M.A. Evidence that verotoxins (Shiga-like toxins) from Escherichia coli bind to p blood group antigens of human erythrocytes in vitro. Infect. Immun. 1994, 62, 3337–3347. [Google Scholar] [PubMed]
- Ståhl, A.L.; Arvidsson, I.; Johansson, K.E.; Chromek, M.; Rebetz, J.; Loos, S.; Kristoffersson, A.C.; Békássy, Z.D.; Mörgelin, M.; Karpman, D. A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathog. 2015, 11, e1004619. [Google Scholar] [CrossRef] [PubMed]
- Te Loo, D.M.; Monnens, L.A.; van Der Velden, T.J.; Vermeer, M.A.; Preyers, F.; Demacker, P.N.; van Den Heuvel, L.P.; van Hinsbergh, V.W. Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood 2000, 95, 3396–3402. [Google Scholar] [PubMed]
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Dilks, J.R.; Richardson, J.; Alden, E.; Patel-Hett, S.R.; Battinelli, E.; Klement, G.L.; Sola-Visner, M.; Italiano, J.E., Jr. Megakaryocyte-derived microparticles: Direct visualization and distinction from platelet-derived microparticles. Blood 2009, 113, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A. Leukocyte-derived microparticles in vascular homeostasis. Circ. Res. 2012, 110, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Rubin, O.; Canellini, G.; Delobel, J.; Lion, N.; Tissot, J.D. Red blood cell microparticles: Clinical relevance. Transfus. Med. Hemother. 2012, 39, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Chironi, G.N.; Boulanger, C.M.; Simon, A.; Dignat-George, F.; Freyssinet, J.M.; Tedgui, A. Endothelial microparticles in diseases. Cell Tissue Res. 2009, 335, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Turco, A.E.; Lam, W.; Rule, A.D.; Denic, A.; Lieske, J.C.; Miller, V.M.; Larson, J.J.; Kremers, W.K.; Jayachandran, M. Specific renal parenchymal-derived urinary extracellular vesicles identify age-associated structural changes in living donor kidneys. J. Extracell. Vesicles 2016, 5, 29642. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G.; Deregibus, M.C.; Bruno, S.; Cantaluppi, V.; Biancone, L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010, 78, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Abid Hussein, M.N.; Boing, A.N.; Sturk, A.; Hau, C.M.; Nieuwland, R. Inhibition of microparticle release triggers endothelial cell apoptosis and detachment. Thromb. Haemost. 2007, 98, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Pap, E. The role of microvesicles in malignancies. Adv. Exp. Med. Biol. 2011, 714, 183–199. [Google Scholar] [PubMed]
- Karpman, D.; Ståhl, A.L.; Arvidsson, I. Extracellular vesicles in renal disease. Nat. Rev. Nephrol. 2017, 13, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.; Vicencio, J.M.; Yellon, D.M.; Davidson, S.M. Microvesicles and exosomes: New players in metabolic and cardiovascular disease. J. Endocrinol. 2016, 228, R57–R71. [Google Scholar] [CrossRef] [PubMed]
- Distler, J.H.; Huber, L.C.; Gay, S.; Distler, O.; Pisetsky, D.S. Microparticles as mediators of cellular cross-talk in inflammatory disease. Autoimmunity 2006, 39, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Waldor, M.K.; Friedman, D.I. Phage regulatory circuits and virulence gene expression. Curr. Opin. Microbiol. 2005, 8, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.E.; Boodhoo, A.; Tyrrell, G.J.; Brunton, J.L.; Read, R.J. Crystal structure of the cell-binding b oligomer of verotoxin-1 from E. coli. Nature 1992, 355, 748–750. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, C.A.; Law, H.; Richardson, S.; Petric, M.; Brunton, J.L.; De Grandis, S.; Karmali, M. Glycolipid binding of purified and recombinant Escherichia coli produced verotoxin in vitro. J. Biol. Chem. 1987, 262, 8834–8839. [Google Scholar] [PubMed]
- Lindberg, A.A.; Brown, J.E.; Strömberg, N.; Westling-Ryd, M.; Schultz, J.E.; Karlsson, K.A. Identification of the carbohydrate receptor for Shiga toxin produced by Shigella dysenteriae type 1. J. Biol. Chem. 1987, 262, 1779–1785. [Google Scholar] [PubMed]
- Nakajima, H.; Kiyokawa, N.; Katagiri, Y.U.; Taguchi, T.; Suzuki, T.; Sekino, T.; Mimori, K.; Ebata, T.; Saito, M.; Nakao, H.; et al. Kinetic analysis of binding between Shiga toxin and receptor glycolipid gb3cer by surface plasmon resonance. J. Biol. Chem. 2001, 276, 42915–42922. [Google Scholar] [CrossRef] [PubMed]
- Kovbasnjuk, O.; Edidin, M.; Donowitz, M. Role of lipid rafts in Shiga toxin 1 interaction with the apical surface of caco-2 cells. J. Cell Sci. 2001, 114, 4025–4031. [Google Scholar] [PubMed]
- Romer, W.; Berland, L.; Chambon, V.; Gaus, K.; Windschiegl, B.; Tenza, D.; Aly, M.R.; Fraisier, V.; Florent, J.C.; Perrais, D.; et al. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature 2007, 450, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Torgersen, M.L.; Lauvrak, S.U.; Sandvig, K. The a-subunit of surface-bound Shiga toxin stimulates clathrin-dependent uptake of the toxin. FEBS J. 2005, 272, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Skotland, T.; van Deurs, B.; Klokk, T.I. Retrograde transport of protein toxins through the golgi apparatus. Histochem. Cell Biol. 2013, 140, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Romer, W.; Pontani, L.L.; Sorre, B.; Rentero, C.; Berland, L.; Chambon, V.; Lamaze, C.; Bassereau, P.; Sykes, C.; Gaus, K.; et al. Actin dynamics drive membrane reorganization and scission in clathrin-independent endocytosis. Cell 2010, 140, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Parton, R.G.; Bassereau, P.; Mayor, S. Building endocytic pits without clathrin. Nat. Rev. Mol. Cell Biol. 2015, 16, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Malyukova, I.; Murray, K.F.; Zhu, C.; Boedeker, E.; Kane, A.; Patterson, K.; Peterson, J.R.; Donowitz, M.; Kovbasnjuk, O. Macropinocytosis in Shiga toxin 1 uptake by human intestinal epithelial cells and transcellular transcytosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G78–G92. [Google Scholar] [CrossRef] [PubMed]
- Schuller, S. Shiga toxin interaction with human intestinal epithelium. Toxins (Basel) 2011, 3, 626–639. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Garred, O.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; van Deurs, B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 1992, 358, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Garred, O.; van Deurs, B.; Sandvig, K. Furin-induced cleavage and activation of Shiga toxin. J Biol. Chem. 1995, 270, 10817–10821. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K. Shiga toxins. Toxicon 2001, 39, 1629–1635. [Google Scholar] [CrossRef]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a vero toxin (vt2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA n-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Håkansson, A.; Perez, M.T.; Isaksson, C.; Carlemalm, E.; Caprioli, A.; Svanborg, C. Apoptosis of renal cortical cells in the hemolytic-uremic syndrome: In vivo and in vitro studies. Infect. Immun. 1998, 66, 636–644. [Google Scholar] [PubMed]
- Burlaka, I.; Liu, X.L.; Rebetz, J.; Arvidsson, I.; Yang, L.; Brismar, H.; Karpman, D.; Aperia, A. Ouabain protects against Shiga toxin-triggered apoptosis by reversing the imbalance between bax and bcl-xl. J. Am. Soc. Nephrol. 2013, 24, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. Activation of cell stress response pathways by Shiga toxins. Cell. Microbiol. 2012, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Koo, S.; Jeong, D.G.; Tesh, V.L. Shiga toxins as multi-functional proteins: Induction of host cellular stress responses, role in pathogenesis and therapeutic applications. Toxins (Basel) 2016, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Békássy, Z.D.; Calderon Toledo, C.; Leoj, G.; Kristoffersson, A.; Leopold, S.R.; Perez, M.T.; Karpman, D. Intestinal damage in enterohemorrhagic Escherichia coli infection. Pediatr. Nephrol. 2011, 26, 2059–2071. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, C.M.; Hurley, B.P.; Lincicome, L.L.; Jacewicz, M.S.; Keusch, G.T.; Acheson, D.W. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect. Immun. 1999, 67, 5985–5993. [Google Scholar] [PubMed]
- Thorpe, C.M.; Smith, W.E.; Hurley, B.P.; Acheson, D.W. Shiga toxins induce, superinduce, and stabilize a variety of c-x-c chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. Infect. Immun. 2001, 69, 6140–6147. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, C.; Natori, Y.; Zeng, X.T.; Ohmura, M.; Yamasaki, S.; Takeda, Y.; Natori, Y. Induction of cytokines in a human colon epithelial cell line by Shiga toxin 1 (stx1) and stx2 but not by non-toxic mutant stx1 which lacks n-glycosidase activity. FEBS Lett. 1999, 442, 231–234. [Google Scholar] [CrossRef]
- Tesh, V.L.; Ramegowda, B.; Samuel, J.E. Purified Shiga-like toxins induce expression of proinflammatory cytokines from murine peritoneal macrophages. Infect. Immun. 1994, 62, 5085–5094. [Google Scholar] [PubMed]
- Karpman, D.; Connell, H.; Svensson, M.; Scheutz, F.; Alm, P.; Svanborg, C. The role of lipopolysaccharide and Shiga-like toxin in a mouse model of Escherichia coli O157:H7 infection. J. Infect. Dis. 1997, 175, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Calderon Toledo, C.; Rogers, T.J.; Svensson, M.; Tati, R.; Fischer, H.; Svanborg, C.; Karpman, D. Shiga toxin-mediated disease in myd88-deficient mice infected with Escherichia coli O157:H7. Am. J. Pathol. 2008, 173, 1428–1439. [Google Scholar] [CrossRef] [PubMed]
- Robson, W.L.; Fick, G.H.; Wilson, P.C. Prognostic factors in typical postdiarrhea hemolytic-uremic syndrome. Child Nephrol. Urol. 1988, 9, 203–207. [Google Scholar] [PubMed]
- Tazzari, P.L.; Ricci, F.; Carnicelli, D.; Caprioli, A.; Tozzi, A.E.; Rizzoni, G.; Conte, R.; Brigotti, M. Flow cytometry detection of Shiga toxins in the blood from children with hemolytic uremic syndrome. Cytometry B. Clin. Cytom. 2004, 61, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Van Setten, P.A.; Monnens, L.A.; Verstraten, R.G.; van den Heuvel, L.P.; van Hinsbergh, V.W. Effects of verocytotoxin-1 on nonadherent human monocytes: Binding characteristics, protein synthesis, and induction of cytokine release. Blood 1996, 88, 174–183. [Google Scholar] [PubMed]
- Guessous, F.; Marcinkiewicz, M.; Polanowska-Grabowska, R.; Keepers, T.R.; Obrig, T.; Gear, A.R. Shiga toxin 2 and lipopolysaccharide cause monocytic thp-1 cells to release factors which activate platelet function. Thromb. Haemost. 2005, 94, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Higuchi, T.; Takada, K.; Oida, K.; Horie, S.; Ishii, H. Verotoxin-1 stimulation of macrophage-like THP-1 cells up-regulates tissue factor expression through activation of c-yes tyrosine kinase: Possible signal transduction in tissue factor up-regulation. Biochim. Biophys. Acta 2006, 1762, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Buelli, S.; Morigi, M. Shiga toxin-associated hemolytic uremic syndrome: Pathophysiology of endothelial dysfunction. Pediatr. Nephrol. 2010, 25, 2231–2240. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Manea, M.; Vaziri-Sani, F.; Ståhl, A.L.; Kristoffersson, A.C. Platelet activation in hemolytic uremic syndrome. Semin. Thromb. Hemost. 2006, 32, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Fong, J.S.; Kaplan, B.S. Impairment of platelet aggregation in hemolytic uremic syndrome: Evidence for platelet “exhaustion”. Blood 1982, 60, 564–570. [Google Scholar] [PubMed]
- Appiani, A.C.; Edefonti, A.; Bettinelli, A.; Cossu, M.M.; Paracchini, M.L.; Rossi, E. The relationship between plasma levels of the factor viii complex and platelet release products (beta-thromboglobulin and platelet factor 4) in children with the hemolytic-uremic syndrome. Clin. Nephrol. 1982, 17, 195–199. [Google Scholar] [PubMed]
- Katayama, M.; Handa, M.; Araki, Y.; Ambo, H.; Kawai, Y.; Watanabe, K.; Ikeda, Y. Soluble p-selectin is present in normal circulation and its plasma level is elevated in patients with thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome. Br. J. Haematol. 1993, 84, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Cooling, L.L.; Walker, K.E.; Gille, T.; Koerner, T.A. Shiga toxin binds human platelets via globotriaosylceramide (pk antigen) and a novel platelet glycosphingolipid. Infect. Immun. 1998, 66, 4355–4366. [Google Scholar] [PubMed]
- Ståhl, A.L.; Svensson, M.; Mörgelin, M.; Svanborg, C.; Tarr, P.I.; Mooney, J.C.; Watkins, S.L.; Johnson, R.; Karpman, D. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood 2006, 108, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.A.; Polanowska-Grabowska, R.K.; Fujii, J.; Obrig, T.; Gear, A.R. Shiga toxin binds to activated platelets. J. Thromb. Haemost. 2004, 2, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Papadopoulou, D.; Nilsson, K.; Sjögren, A.C.; Mikaelsson, C.; Lethagen, S. Platelet activation by Shiga toxin and circulatory factors as a pathogenetic mechanism in the hemolytic uremic syndrome. Blood 2001, 97, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.L.; Sartz, L.; Karpman, D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood 2011, 117, 5503–5513. [Google Scholar] [CrossRef] [PubMed]
- Polley, M.J.; Nachman, R.L. Human complement in thrombin-mediated platelet function: Uptake of the C5b-9 complex. J. Exp. Med. 1979, 150, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Chandler, W.L.; Jelacic, S.; Boster, D.R.; Ciol, M.A.; Williams, G.D.; Watkins, S.L.; Igarashi, T.; Tarr, P.I. Prothrombotic coagulation abnormalities preceding the hemolytic-uremic syndrome. N. Engl. J. Med. 2002, 346, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Polley, M.J.; Nachman, R. The human complement system in thrombin-mediated platelet function. J. Exp. Med. 1978, 147, 1713–1726. [Google Scholar] [CrossRef] [PubMed]
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of C5a in the absence of C3: A new complement activation pathway. Nat. Med. 2006, 12, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Tati, R. Complement contributes to the pathogenesis of Shiga toxin-associated hemolytic uremic syndrome. Kidney Int. 2016, 90, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.M.; Marotta, S. Therapeutic complement inhibition in complement-mediated hemolytic anemias: Past, present and future. Semin. Immunol. 2016, 28, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Spitalnik, P.F.; Spitalnik, S.L. The P blood group system: Biochemical, serological, and clinical aspects. Transfus. Med. Rev. 1995, 9, 110–122. [Google Scholar] [CrossRef]
- Karpman, D. Management of Shiga toxin-associated Escherichia coli-induced haemolytic uraemic syndrome: Randomized clinical trials are needed. Nephrol. Dial. Transplant. 2012, 27, 3669–3674. [Google Scholar] [CrossRef] [PubMed]
- Geelen, J.M.; van der Velden, T.J.; van den Heuvel, L.P.; Monnens, L.A. Interactions of Shiga-like toxin with human peripheral blood monocytes. Pediatr. Nephrol. 2007, 22, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Psotka, M.A.; Obata, F.; Kolling, G.L.; Gross, L.K.; Saleem, M.A.; Satchell, S.C.; Mathieson, P.W.; Obrig, T.G. Shiga toxin 2 targets the murine renal collecting duct epithelium. Infect. Immun. 2009, 77, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Van de Kar, N.C.; Monnens, L.A.; Karmali, M.A.; van Hinsbergh, V.W. Tumor necrosis factor and interleukin-1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: Implications for the pathogenesis of the hemolytic uremic syndrome. Blood 1992, 80, 2755–2764. [Google Scholar] [PubMed]
- Stone, M.K.; Kolling, G.L.; Lindner, M.H.; Obrig, T.G. P38 mitogen-activated protein kinase mediates lipopolysaccharide and tumor necrosis factor alpha induction of Shiga toxin 2 sensitivity in human umbilical vein endothelial cells. Infect. Immun. 2008, 76, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Louise, C.B.; Obrig, T.G. Specific interaction of Escherichia coli O157:H7-derived Shiga-like toxin ii with human renal endothelial cells. J. Infect. Dis. 1995, 172, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Guessous, F.; Marcinkiewicz, M.; Polanowska-Grabowska, R.; Kongkhum, S.; Heatherly, D.; Obrig, T.; Gear, A.R. Shiga toxin 2 and lipopolysaccharide induce human microvascular endothelial cells to release chemokines and factors that stimulate platelet function. Infect. Immun. 2005, 73, 8306–8316. [Google Scholar] [CrossRef] [PubMed]
- Nevard, C.H.; Jurd, K.M.; Lane, D.A.; Philippou, H.; Haycock, G.B.; Hunt, B.J. Activation of coagulation and fibrinolysis in childhood diarrhoea-associated haemolytic uraemic syndrome. Thromb. Haemost. 1997, 78, 1450–1455. [Google Scholar] [PubMed]
- Van Geet, C.; Proesmans, W.; Arnout, J.; Vermylen, J.; Declerck, P.J. Activation of both coagulation and fibrinolysis in childhood hemolytic uremic syndrome. Kidney Int. 1998, 54, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Van de Kar, N.C.; van Hinsbergh, V.W.; Brommer, E.J.; Monnens, L.A. The fibrinolytic system in the hemolytic uremic syndrome: In vivo and in vitro studies. Pediatr. Res. 1994, 36, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Sappino, A.P.; Huarte, J.; Vassalli, J.D.; Belin, D. Sites of synthesis of urokinase and tissue-type plasminogen activators in the murine kidney. J. Clin. Investig. 1991, 87, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Hertel, B.; Emden, S.H.; Beneke, J.; Menne, J.; Haller, H.; von Vietinghoff, S. Microparticle generation and leucocyte death in Shiga toxin-mediated hus. Nephrol. Dial. Transplant. 2012, 27, 2768–2775. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Gyorgy, B.; Szabo, T.G.; Pasztoi, M.; Pal, Z.; Misjak, P.; Aradi, B.; Laszlo, V.; Pallinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life. Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [PubMed]
- Erdbrügger, U.; Lannigan, J. Analytical challenges of extracellular vesicle detection: A comparison of different techniques. Cytometry A. 2016, 89, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Buzas, E.I.; Bemis, L.T.; Bora, A.; Lasser, C.; Lotvall, J.; Nolte-’t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.S.; Kim, D.K.; Kim, Y.K.; Gho, Y.S. Proteomics of extracellular vesicles: Exosomes and ectosomes. Mass Spectrom. Rev. 2015, 34, 474–490. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; Coumans, F.A.; Grootemaat, A.E.; Gardiner, C.; Sargent, I.L.; Harrison, P.; Sturk, A.; van Leeuwen, T.G.; Nieuwland, R. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J. Thromb. Haemost. 2014, 12, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [PubMed]
- Kowal, J.; Tkach, M.; Thery, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Thery, C.; Raposo, G. Analysis of escrt functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [PubMed]
- Hanson, P.I.; Roth, R.; Lin, Y.; Heuser, J.E. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J. Cell Biol. 2008, 180, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Escola, J.M.; Kleijmeer, M.J.; Stoorvogel, W.; Griffith, J.M.; Yoshie, O.; Geuze, H.J. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human b-lymphocytes. J. Biol. Chem. 1998, 273, 20121–20127. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Boussac, M.; Veron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.; Santos, S.G.; Campbell, E.C.; Nimmo, A.M.; Botting, C.; Prescott, A.; Antoniou, A.N.; Powis, S.J. Novel MHC class I structures on exosomes. J. Immunol. 2009, 183, 1884–1891. [Google Scholar] [CrossRef] [PubMed]
- Haraszti, R.A.; Didiot, M.C.; Sapp, E.; Leszyk, J.; Shaffer, S.A.; Rockwell, H.E.; Gao, F.; Narain, N.R.; DiFiglia, M.; Kiebish, M.A.; et al. High-resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. J. Extracell. Vesicles 2016, 5, 32570. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Xu, Q. Functions and application of exosomes. Acta Pol. Pharm. 2014, 71, 537–543. [Google Scholar] [PubMed]
- Little, K.M.; Smalley, D.M.; Harthun, N.L.; Ley, K. The plasma microparticle proteome. Semin. Thromb. Hemost. 2010, 36, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Barry, O.P.; Pratico, D.; Lawson, J.A.; FitzGerald, G.A. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J. Clin. Investig. 1997, 99, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Sapet, C.; Simoncini, S.; Loriod, B.; Puthier, D.; Sampol, J.; Nguyen, C.; Dignat-George, F.; Anfosso, F. Thrombin-induced endothelial microparticle generation: Identification of a novel pathway involving rock-II activation by caspase-2. Blood 2006, 108, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, J.; Miekus, K.; Kucia, M.; Zhang, J.; Reca, R.; Dvorak, P.; Ratajczak, M.Z. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: Evidence for horizontal transfer of mRNA and protein delivery. Leukemia 2006, 20, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2016, 8, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Pomatto, M.A.C.; Gai, C.; Bussolati, B.; Camussi, G. Extracellular vesicles in renal pathophysiology. Front. Mol. Biosci. 2017, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Whitlow, M.B.; Nussenzweig, V. Membrane vesiculation protects erythrocytes from destruction by complement. J. Immunol. 1991, 147, 2638–2642. [Google Scholar] [PubMed]
- Willekens, F.L.; Werre, J.M.; Groenen-Dopp, Y.A.; Roerdinkholder-Stoelwinder, B.; de Pauw, B.; Bosman, G.J. Erythrocyte vesiculation: A self-protective mechanism? Br. J. Haematol. 2008, 141, 549–556. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Boucrot, E. Membrane curvature at a glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, J.J.; Jy, W.; Mauro, L.M.; Soderland, C.; Horstman, L.L.; Ahn, Y.S. Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis. Thromb. Res. 2003, 109, 175–180. [Google Scholar] [CrossRef]
- Nomura, S.; Tandon, N.N.; Nakamura, T.; Cone, J.; Fukuhara, S.; Kambayashi, J. High-shear-stress-induced activation of platelets and microparticles enhances expression of cell adhesion molecules in THP-1 and endothelial cells. Atherosclerosis 2001, 158, 277–287. [Google Scholar] [CrossRef]
- Bernimoulin, M.; Waters, E.K.; Foy, M.; Steele, B.M.; Sullivan, M.; Falet, H.; Walsh, M.T.; Barteneva, N.; Geng, J.G.; Hartwig, J.H.; et al. Differential stimulation of monocytic cells results in distinct populations of microparticles. J. Thromb. Haemost. 2009, 7, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.J.; Gibbons, E.; Bailey, R.W.; Fairbourn, J.; Nguyen, T.; Smith, S.K.; Best, K.B.; Nelson, J.; Judd, A.M.; Bell, J.D. The influence of membrane physical properties on microvesicle release in human erythrocytes. PMC Biophys. 2009, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol. 2009, 19, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.C.; Tan, S.S.; Yeo, R.W.; Choo, A.B.; Reiner, A.T.; Su, Y.; Shen, Y.; Fu, Z.; Alexander, L.; Sze, S.K.; et al. MSC secretes at least 3 EV types each with a unique permutation of membrane lipid, protein and RNA. J. Extracell. Vesicles 2016, 5, 29828. [Google Scholar] [CrossRef] [PubMed]
- Biro, E.; Akkerman, J.W.; Hoek, F.J.; Gorter, G.; Pronk, L.M.; Sturk, A.; Nieuwland, R. The phospholipid composition and cholesterol content of platelet-derived microparticles: A comparison with platelet membrane fractions. J. Thromb. Haemost. 2005, 3, 2754–2763. [Google Scholar] [CrossRef] [PubMed]
- Sinauridze, E.I.; Kireev, D.A.; Popenko, N.Y.; Pichugin, A.V.; Panteleev, M.A.; Krymskaya, O.V.; Ataullakhanov, F.I. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb. Haemost. 2007, 97, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Seigneuret, M.; Zachowski, A.; Hermann, A.; Devaux, P.F. Asymmetric lipid fluidity in human erythrocyte membrane: New spin-label evidence. Biochemistry 1984, 23, 4271–4275. [Google Scholar] [CrossRef] [PubMed]
- Manno, S.; Takakuwa, Y.; Mohandas, N. Identification of a functional role for lipid asymmetry in biological membranes: Phosphatidylserine-skeletal protein interactions modulate membrane stability. Proc. Natl. Acad. Sci. USA 2002, 99, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Daleke, D.L. Regulation of transbilayer plasma membrane phospholipid asymmetry. J. Lipid Res. 2003, 44, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Kim, A.; David, T.; Palmer, D.; Jin, T.; Tien, J.; Huang, F.; Cheng, T.; Coughlin, S.R.; Jan, Y.N.; et al. Tmem16f forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 2012, 151, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Morel, O.; Jesel, L.; Freyssinet, J.M.; Toti, F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Farsad, K.; De Camilli, P. Mechanisms of membrane deformation. Curr. Opin. Cell Biol. 2003, 15, 372–381. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podbilewicz, B. Virus and cell fusion mechanisms. Annu. Rev. Cell Dev. Biol. 2014, 30, 111–139. [Google Scholar] [CrossRef] [PubMed]
- Prada, I.; Meldolesi, J. Binding and fusion of extracellular vesicles to the plasma membrane of their cell targets. Int. J. Mol. Sci. 2016, 17, 1296. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.L.; Breakefield, X.O.; Weaver, A.M. Extracellular vesicles: Unique intercellular delivery vehicles. Trends Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef] [PubMed]
- French, K.C.; Antonyak, M.A.; Cerione, R.A. Extracellular vesicle docking at the cellular port: Extracellular vesicle binding and uptake. Semin. Cell Dev. Biol. 2017, 67, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Pluskota, E.; Woody, N.M.; Szpak, D.; Ballantyne, C.M.; Soloviev, D.A.; Simon, D.I.; Plow, E.F. Expression, activation, and function of integrin alphaMbeta2 (Mac-1) on neutrophil-derived microparticles. Blood 2008, 112, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Falati, S.; Liu, Q.; Gross, P.; Merrill-Skoloff, G.; Chou, J.; Vandendries, E.; Celi, A.; Croce, K.; Furie, B.C.; Furie, B. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle p-selectin glycoprotein ligand 1 and platelet p-selectin. J. Exp. Med. 2003, 197, 1585–1598. [Google Scholar] [CrossRef] [PubMed]
- Piccin, A.; Murphy, W.G.; Smith, O.P. Circulating microparticles: Pathophysiology and clinical implications. Blood Rev. 2007, 21, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Kuhne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef] [PubMed]
- Losche, W.; Scholz, T.; Temmler, U.; Oberle, V.; Claus, R.A. Platelet-derived microvesicles transfer tissue factor to monocytes but not to neutrophils. Platelets 2004, 15, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Deregibus, M.C.; Cantaluppi, V.; Calogero, R.; Lo Iacono, M.; Tetta, C.; Biancone, L.; Bruno, S.; Bussolati, B.; Camussi, G. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mrna. Blood 2007, 110, 2440–2448. [Google Scholar] [CrossRef] [PubMed]
- Mause, S.F.; Weber, C. Microparticles: Protagonists of a novel communication network for intercellular information exchange. Circ. Res. 2010, 107, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Collino, F.; Bruno, S.; Incarnato, D.; Dettori, D.; Neri, F.; Provero, P.; Pomatto, M.; Oliviero, S.; Tetta, C.; Quesenberry, P.J.; et al. AKI recovery induced by mesenchymal stromal cell-derived extracellular vesicles carrying micrornas. J. Am. Soc. Nephrol. 2015, 26, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Mack, M.; Kleinschmidt, A.; Bruhl, H.; Klier, C.; Nelson, P.J.; Cihak, J.; Plachy, J.; Stangassinger, M.; Erfle, V.; Schlondorff, D. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: A mechanism for cellular human immunodeficiency virus 1 infection. Nat. Med. 2000, 6, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Rozmyslowicz, T.; Majka, M.; Kijowski, J.; Murphy, S.L.; Conover, D.O.; Poncz, M.; Ratajczak, J.; Gaulton, G.N.; Ratajczak, M.Z. Platelet- and megakaryocyte-derived microparticles transfer CXCR4 receptor to CXCR4-null cells and make them susceptible to infection by X4-HIV. AIDS 2003, 17, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Kerbel, R.S.; Allison, A.C.; Rak, J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 3794–3799. [Google Scholar] [CrossRef] [PubMed]
- Kahn, R.; Mossberg, M.; Ståhl, A.L.; Johansson, K.; Lopatko Lindman, I.; Heijl, C.; Segelmark, M.; Mörgelin, M.; Leeb-Lundberg, L.M.; Karpman, D. Microvesicle transfer of kinin B1-receptors is a novel inflammatory mechanism in vasculitis. Kidney Int. 2017, 91, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Baj-Krzyworzeka, M.; Majka, M.; Pratico, D.; Ratajczak, J.; Vilaire, G.; Kijowski, J.; Reca, R.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Platelet-derived microparticles stimulate proliferation, survival, adhesion, and chemotaxis of hematopoietic cells. Exp. Hematol. 2002, 30, 450–459. [Google Scholar] [CrossRef]
- Salanova, B.; Choi, M.; Rolle, S.; Wellner, M.; Luft, F.C.; Kettritz, R. Beta2-integrins and acquired glycoprotein IIb/IIIa (gpIIb/IIIa) receptors cooperate in NF-kappaB activation of human neutrophils. J. Biol. Chem. 2007, 282, 27960–27969. [Google Scholar] [CrossRef] [PubMed]
- Taraboletti, G.; D’Ascenzo, S.; Borsotti, P.; Giavazzi, R.; Pavan, A.; Dolo, V. Shedding of the matrix metalloproteinases mmp-2, mmp-9, and mt1-mmp as membrane vesicle-associated components by endothelial cells. Am. J. Pathol. 2002, 160, 673–680. [Google Scholar] [CrossRef]
- Barry, O.P.; Pratico, D.; Savani, R.C.; FitzGerald, G.A. Modulation of monocyte-endothelial cell interactions by platelet microparticles. J. Clin. Investig. 1998, 102, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Mossberg, M.; Ståhl, A.L.; Kahn, R.; Kristoffersson, A.C.; Tati, R.; Heijl, C.; Segelmark, M.; Leeb-Lundberg, L.M.F.; Karpman, D. C1-inhibitor decreases the release of vasculitis-like chemotactic endothelial microvesicles. J. Am. Soc. Nephrol. 2017, 28, 2472–2481. [Google Scholar] [CrossRef] [PubMed]
- Mause, S.F.; von Hundelshausen, P.; Zernecke, A.; Koenen, R.R.; Weber, C. Platelet microparticles: A transcellular delivery system for rantes promoting monocyte recruitment on endothelium. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Biancone, L.; Bruno, S.; Deregibus, M.C.; Tetta, C.; Camussi, G. Therapeutic potential of mesenchymal stem cell-derived microvesicles. Nephrol. Dial. Transplant. 2012, 27, 3037–3042. [Google Scholar] [CrossRef] [PubMed]
- Gasser, O.; Schifferli, J.A. Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood 2004, 104, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Bevers, E.M.; Williamson, P.L. Getting to the outer leaflet: Physiology of phosphatidylserine exposure at the plasma membrane. Physiol. Rev. 2016, 96, 605–645. [Google Scholar] [CrossRef] [PubMed]
- Sims, P.J.; Wiedmer, T.; Esmon, C.T.; Weiss, H.J.; Shattil, S.J. Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane. Studies in scott syndrome: An isolated defect in platelet procoagulant activity. J. Biol. Chem. 1989, 264, 17049–17057. [Google Scholar] [PubMed]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.L.; Vaziri-Sani, F.; Heinen, S.; Kristoffersson, A.C.; Gydell, K.H.; Raafat, R.; Gutierrez, A.; Beringer, O.; Zipfel, P.F.; Karpman, D. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood 2008, 111, 5307–5315. [Google Scholar] [CrossRef] [PubMed]
- Diamant, M.; Nieuwland, R.; Pablo, R.F.; Sturk, A.; Smit, J.W.; Radder, J.K. Elevated numbers of tissue-factor exposing microparticles correlate with components of the metabolic syndrome in uncomplicated type 2 diabetes mellitus. Circulation 2002, 106, 2442–2447. [Google Scholar] [CrossRef] [PubMed]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; Lopez, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Rubin, O.; Delobel, J.; Prudent, M.; Lion, N.; Kohl, K.; Tucker, E.I.; Tissot, J.D.; Angelillo-Scherrer, A. Red blood cell-derived microparticles isolated from blood units initiate and propagate thrombin generation. Transfusion 2013, 53, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Nieuwland, R.; Berckmans, R.J.; McGregor, S.; Boing, A.N.; Romijn, F.P.; Westendorp, R.G.; Hack, C.E.; Sturk, A. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood 2000, 95, 930–935. [Google Scholar] [PubMed]
- Fujimi, S.; Ogura, H.; Tanaka, H.; Koh, T.; Hosotsubo, H.; Nakamori, Y.; Kuwagata, Y.; Shimazu, T.; Sugimoto, H. Activated polymorphonuclear leukocytes enhance production of leukocyte microparticles with increased adhesion molecules in patients with sepsis. J. Trauma 2002, 52, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Tokes-Fuzesi, M.; Woth, G.; Ernyey, B.; Vermes, I.; Muhl, D.; Bogar, L.; Kovacs, G.L. Microparticles and acute renal dysfunction in septic patients. J. Crit. Care 2013, 28, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Mostefai, H.A.; Meziani, F.; Mastronardi, M.L.; Agouni, A.; Heymes, C.; Sargentini, C.; Asfar, P.; Martinez, M.C.; Andriantsitohaina, R. Circulating microparticles from patients with septic shock exert protective role in vascular function. Am. J. Respir. Crit. Care Med. 2008, 178, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Woei, A.J.F.J.; van der Starre, W.E.; Tesselaar, M.E.; Garcia Rodriguez, P.; van Nieuwkoop, C.; Bertina, R.M.; van Dissel, J.T.; Osanto, S. Procoagulant tissue factor activity on microparticles is associated with disease severity and bacteremia in febrile urinary tract infections. Thromb. Res. 2014, 133, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Delabranche, X.; Boisrame-Helms, J.; Asfar, P.; Berger, A.; Mootien, Y.; Lavigne, T.; Grunebaum, L.; Lanza, F.; Gachet, C.; Freyssinet, J.M.; et al. Microparticles are new biomarkers of septic shock-induced disseminated intravascular coagulopathy. Intensive Care Med. 2013, 39, 1695–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, A.O.; Jy, W.; Chirinos, J.A.; Valdivia, M.A.; Velasquez, H.S.; Jimenez, J.J.; Horstman, L.L.; Kett, D.H.; Schein, R.M.; Ahn, Y.S. Levels of endothelial and platelet microparticles and their interactions with leukocytes negatively correlate with organ dysfunction and predict mortality in severe sepsis. Crit. Care Med. 2005, 33, 2540–2546. [Google Scholar] [CrossRef] [PubMed]
- Mortaza, S.; Martinez, M.C.; Baron-Menguy, C.; Burban, M.; de la Bourdonnaye, M.; Fizanne, L.; Pierrot, M.; Cales, P.; Henrion, D.; Andriantsitohaina, R.; et al. Detrimental hemodynamic and inflammatory effects of microparticles originating from septic rats. Crit. Care Med. 2009, 37, 2045–2050. [Google Scholar] [CrossRef] [PubMed]
- Meziani, F.; Delabranche, X.; Asfar, P.; Toti, F. Bench-to-bedside review: Circulating microparticles--a new player in sepsis? Crit. Care 2010, 14, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camussi, G.; Cantaluppi, V.; Deregibus, M.C.; Gatti, E.; Tetta, C. Role of microvesicles in acute kidney injury. Contrib. Nephrol. 2011, 174, 191–199. [Google Scholar] [PubMed]
- Timar, C.I.; Lorincz, A.M.; Csepanyi-Komi, R.; Valyi-Nagy, A.; Nagy, G.; Buzas, E.I.; Ivanyi, Z.; Kittel, A.; Powell, D.W.; McLeish, K.R.; et al. Antibacterial effect of microvesicles released from human neutrophilic granulocytes. Blood 2013, 121, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Oehmcke, S.; Westman, J.; Malmström, J.; Mörgelin, M.; Olin, A.I.; Kreikemeyer, B.; Herwald, H. A novel role for pro-coagulant microvesicles in the early host defense against streptococcus pyogenes. PLoS Pathog. 2013, 9, e1003529. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, L.; Qu, Y.; Wang, Y.; Lewis, C.J.; Cobb, B.A.; Takatsu, K.; Boom, W.H.; Dubyak, G.R.; Harding, C.V. Mycobacterium tuberculosis synergizes with ATP to induce release of microvesicles and exosomes containing major histocompatibility complex class ii molecules capable of antigen presentation. Infect. Immun. 2010, 78, 5116–5125. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D. The cell biology of HIV-1 virion genesis. Cell Host Microbe 2009, 5, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H. ESCRTs are everywhere. EMBO J. 2015, 34, 2398–2407. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Useros, N.; Naranjo-Gomez, M.; Archer, J.; Hatch, S.C.; Erkizia, I.; Blanco, J.; Borras, F.E.; Puertas, M.C.; Connor, J.H.; Fernandez-Figueras, M.T.; et al. Capture and transfer of HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood 2009, 113, 2732–2741. [Google Scholar] [CrossRef] [PubMed]
- Sims, B.; Farrow, A.L.; Williams, S.D.; Bansal, A.; Krendelchtchikov, A.; Gu, L.; Matthews, Q.L. Role of tim-4 in exosome-dependent entry of HIV-1 into human immune cells. Int. J. Nanomed. 2017, 12, 4823–4833. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr.; Raab-Traub, N. Microvesicles and viral infection. J. Virol. 2011, 85, 12844–12854. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.D.; Maier, C.L.; Pober, J.S. Cytomegalovirus-infected human endothelial cells can stimulate allogeneic CD4+ memory t cells by releasing antigenic exosomes. J. Immunol. 2009, 182, 1548–1559. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Silva, M.R.; Cabrera-Cabrera, F.; das Neves, R.F.; Souto-Padron, T.; de Souza, W.; Cayota, A. Gene expression changes induced by Trypanosoma cruzi shed microvesicles in mammalian host cells: Relevance of trna-derived halves. Biomed. Res. Int. 2014, 2014, 305239. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Silva, M.R.; das Neves, R.F.; Cabrera-Cabrera, F.; Sanguinetti, J.; Medeiros, L.C.; Robello, C.; Naya, H.; Fernandez-Calero, T.; Souto-Padron, T.; de Souza, W.; et al. Extracellular vesicles shed by Trypanosoma cruzi are linked to small RNA pathways, life cycle regulation, and susceptibility to infection of mammalian cells. Parasitol. Res. 2014, 113, 285–304. [Google Scholar] [CrossRef] [PubMed]
- Prados-Rosales, R.; Baena, A.; Martinez, L.R.; Luque-Garcia, J.; Kalscheuer, R.; Veeraraghavan, U.; Camara, C.; Nosanchuk, J.D.; Besra, G.S.; Chen, B.; et al. Mycobacteria release active membrane vesicles that modulate immune responses in a TLR2-dependent manner in mice. J. Clin. Investig. 2011, 121, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Ruter, C.; Bauwens, A.; Greune, L.; Jarosch, K.A.; Steil, D.; Zhang, W.; He, X.; Lloubes, R.; Fruth, A.; et al. Host cell interactions of outer membrane vesicle-associated virulence factors of enterohemorrhagic Escherichia coli O157: Intracellular delivery, trafficking and mechanisms of cell injury. PLoS Pathog. 2017, 13, e1006159. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Cleary, T.G.; Hernandez, J.D.; Abboud, H.E. Shiga toxin 1 elicits diverse biologic responses in mesangial cells. Kidney Int. 1998, 54, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Buelli, S.; Pezzotta, A.; Corna, D.; Perico, L.; Tomasoni, S.; Rottoli, D.; Rizzo, P.; Conti, D.; Thurman, J.M.; et al. Shiga toxin promotes podocyte injury in experimental hemolytic uremic syndrome via activation of the alternative pathway of complement. J. Am. Soc. Nephrol. 2014, 25, 1786–1798. [Google Scholar] [CrossRef] [PubMed]
- Polley, M.J.; Nachman, R.L. Human platelet activation by C3a and C3a des-arg. J. Exp. Med. 1983, 158, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, I.; Rebetz, J.; Loos, S.; Herthelius, M.; Kristoffersson, A.C.; Englund, E.; Chromek, M.; Karpman, D. Early terminal complement blockade and C6 deficiency are protective in enterohemorrhagic Escherichia coli-infected mice. J. Immunol. 2016, 197, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Galbusera, M.; Gastoldi, S.; Locatelli, M.; Buelli, S.; Pezzotta, A.; Pagani, C.; Noris, M.; Gobbi, M.; Stravalaci, M.; et al. Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J. Immunol. 2011, 187, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, M.; Kang, Y.; Tan, Y.S.; Pavlov, V.I.; Liu, B.; Boyle, D.C.; Kushak, R.I.; Skjoedt, M.O.; Grabowski, E.F.; Taira, Y.; et al. Human mannose-binding lectin inhibitor prevents Shiga toxin-induced renal injury. Kidney Int. 2016, 90, 774–782. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Extracellular Vesicle | Origin | Size | Content | References |
|---|---|---|---|---|
| Exosome | Intraluminal vesicle within multivesicular bodies | 30–100 nm | mRNA, miRNA, proteins, lipids | [97] |
| Microvesicle | Shedding from the plasma membrane with cellular content | 100–1000 nm | mRNA, miRNA, proteins, lipids | [29] |
| Apoptotic body | Cellular breakdown and shrinkage during apoptosis | 1–5 μm | Organelles, proteins, DNA, RNAs, lipids | [97] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villysson, A.; Tontanahal, A.; Karpman, D. Microvesicle Involvement in Shiga Toxin-Associated Infection. Toxins 2017, 9, 376. https://doi.org/10.3390/toxins9110376
Villysson A, Tontanahal A, Karpman D. Microvesicle Involvement in Shiga Toxin-Associated Infection. Toxins. 2017; 9(11):376. https://doi.org/10.3390/toxins9110376
Chicago/Turabian StyleVillysson, Annie, Ashmita Tontanahal, and Diana Karpman. 2017. "Microvesicle Involvement in Shiga Toxin-Associated Infection" Toxins 9, no. 11: 376. https://doi.org/10.3390/toxins9110376
APA StyleVillysson, A., Tontanahal, A., & Karpman, D. (2017). Microvesicle Involvement in Shiga Toxin-Associated Infection. Toxins, 9(11), 376. https://doi.org/10.3390/toxins9110376