
Type IV Secretion and Signal Transduction of Helicobacter pylori CagA through Interactions with Host Cell Receptors


{kind=link}
Abstract
:1. Introduction
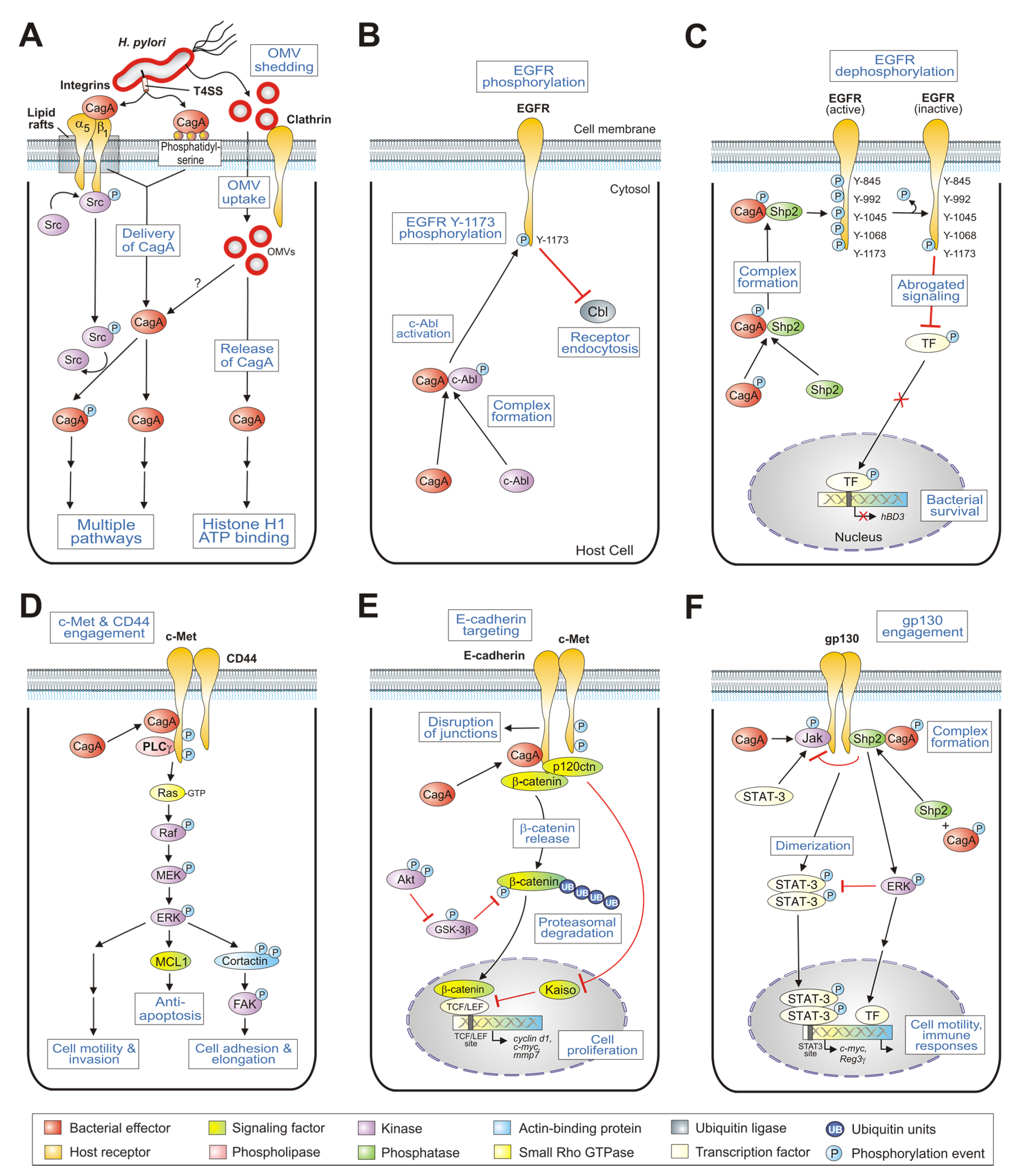
2. Type IV Secretion-Dependent Delivery of CagA via Integrin-β1
3. Internalization of CagA by Binding to Membrane-Associated Phosphatidylserine
4. Type IV Secretion-Independent Uptake of CagA through Outer Membrane Vesicles
5. Inhibition of EGFR Endocytosis by the Non-Receptor Kinase c-Abl and CagA
6. CagA Abrogates Human β-Defensin 3 Expression by the Dephosphorylation of EGFR
7. CagA Protein Targets the c-Met Receptor and Enhances the Motogenic Host Cell Response
8. CagA Associates with c-Met and CD44 Activating Host Cell Proliferation
9. CagA Targets E-Cadherin and Deregulates Catenin Signaling
10. CagA Associates with c-Met, E-Cadherin, and p120ctn with an Impact on Host Cell Invasion
11. CagA Determines gp130-Activated Shp2/ERK and JAK/STAT Signal Transduction
12. Summary
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Schreiber, S.; Konradt, M.; Groll, C.; Scheid, P.; Hanauer, G.; Werling, H.O.; Josenhans, C.; Suerbaum, S. The spatial orientation of Helicobacter pylori in the gastric mucus. Proc. Natl. Acad. Sci. USA 2004, 101, 5024–5029. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Y.; Sweeney, E.G.; Sigal, M.; Zhang, H.C.; Remington, S.J.; Cantrell, M.A.; Kuo, C.J.; Guillemin, K.; Amieva, M.R. Chemodetection and Destruction of Host Urea Allows Helicobacter pylori to Locate the Epithelium. Cell Host Microbe 2015, 18, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Salama, N.R.; Hartung, M.L.; Mueller, A. Life in the human stomach: Persistence strategies of the bacterial pathogen Helicobacter pylori. Nat. Rev. Microbiol. 2013, 11, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y.; Graham, D.Y. Helicobacter pylori virulence and cancer pathogenesis. Future Oncol. 2014, 10, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.; Peek, R.M., Jr. Pathobiology of Helicobacter pylori-Induced Gastric Cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Maixner, F.; Krause-Kyora, B.; Turaev, D.; Herbig, A.; Hoopmann, M.R.; Hallows, J.L.; Kusebauch, U.; Vigl, E.E.; Malfertheiner, P.; Megraud, F.; et al. The 5300-year-old Helicobacter pylori genome of the Iceman. Science 2016, 351, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Atherton, J.C.; Blaser, M.J. Coadaptation of Helicobacter pylori and humans: Ancient history, modern implications. J. Clin. Investig. 2009, 119, 2475–2487. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Blaser, M.J. The Role of CagA in the Gastric Biology of Helicobacter pylori. Cancer Res. 2016, 76, 4028–4031. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 (Internet); International Agency for Research on Cancer: Lyon, France, 2013. Available online: http://globocan.iarc.fr (accessed on 18 January 2017).
- Aspholm, M.; Kalia, A.; Ruhl, S.; Schedin, S.; Arnqvist, A.; Lindén, S.; Sjöström, R.; Gerhard, M.; Semino-Mora, C.; Dubois, A.; et al. Helicobacter pylori adhesion to carbohydrates. Methods Enzymol. 2006, 417, 293–339. [Google Scholar] [PubMed]
- Odenbreit, S.; Swoboda, K.; Barwig, I.; Ruhl, S.; Borén, T.; Koletzko, S.; Haas, R. Outer membrane protein expression profile in Helicobacter pylori clinical isolates. Infect. Immun. 2009, 77, 3782–3790. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Clyne, M.; Tegtmeyer, N. Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori. Cell Commun. Signal. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Belogolova, E.; Bauer, B.; Pompaiah, M.; Asakura, H.; Brinkman, V.; Ertl, C.; Bartfeld, S.; Nechitaylo, T.Y.; Haas, R.; Machuy, N.; et al. Helicobacter pylori outer membrane protein HopQ identified as a novel T4SS-associated virulence factor. Cell Microbiol. 2013, 15, 1896–1912. [Google Scholar] [PubMed]
- Oleastro, M.; Ménard, A. The Role of Helicobacter pylori Outer Membrane Proteins in Adherence and Pathogenesis. Biology (Basel) 2013, 2, 1110–1134. [Google Scholar] [CrossRef] [PubMed]
- Wunder, C.; Churin, Y.; Winau, F.; Warnecke, D.; Vieth, M.; Lindner, B.; Zähringer, U.; Mollenkopf, H.J.; Heinz, E.; Meyer, T.F. Cholesterol glucosylation promotes immune evasion by Helicobacter pylori. Nat. Med. 2006, 12, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Hoy, B.; Löwer, M.; Weydig, C.; Carra, G.; Tegtmeyer, N.; Geppert, T.; Schröder, P.; Sewald, N.; Backert, S.; Schneider, G.; et al. Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep. 2010, 11, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Posselt, G.; Backert, S.; Wessler, S. The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun. Signal. 2013, 11. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.N.; Hartung, M.L.; Urban, S.; Kyburz, A.; Bahlmann, A.S.; Lind, J.; Backert, S.; Taube, C.; Müller, A. Helicobacter urease-induced activation of the TLR2/NLRP3/IL-18 axis protects against asthma. J. Clin. Investig. 2015, 125, 3297–3302. [Google Scholar] [CrossRef] [PubMed]
- Pyburn, T.M.; Foegeding, N.J.; González-Rivera, C.; McDonald, N.A.; Gould, K.L.; Cover, T.L.; Ohi, M.D. Structural organization of membrane-inserted hexamers formed by Helicobacter pylori VacA toxin. Mol. Microbiol. 2016, 102, 22–36. [Google Scholar]
- Hatakeyama, M. Helicobacter pylori CagA-a potential bacterial oncoprotein that functionally mimics the mammalian Gab family of adaptor proteins. Microbes Infect. 2003, 5, 143–150. [Google Scholar] [CrossRef]
- Backert, S.; Tegtmeyer, N.; Selbach, M. The versatility of Helicobacter pylori CagA effector protein functions: The master key hypothesis. Helicobacter 2010, 15, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Christie, P.J.; Whitaker, N.; González-Rivera, C. Mechanism and structure of the bacterial type IV secretion systems. Biochim. Biophys. Acta 2014, 1843, 1578–1591. [Google Scholar] [CrossRef] [PubMed]
- Frick-Cheng, A.E.; Pyburn, T.M.; Voss, B.J.; McDonald, W.H.; Ohi, M.D.; Cover, T.L. Molecular and Structural Analysis of the Helicobacter pylori cag Type IV Secretion System Core Complex. MBio 2016, 7. [Google Scholar] [CrossRef]
- Kwok, T.; Zabler, D.; Urman, S.; Rohde, M.; Hartig, R.; Wessler, S.; Misselwitz, R.; Berger, J.; Sewald, N.; König, W.; et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 2007, 449, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Rohde, M.; Püls, J.; Buhrdorf, R.; Fischer, W.; Haas, R. A novel sheathed surface organelle of the Helicobacter pylori cag type IV secretion system. Mol. Microbiol. 2003, 49, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Soto, L.F.; Kutter, S.; Sewald, X.; Ertl, C.; Weiss, E.; Kapp, U.; Rohde, M.; Pirch, T.; Jung, K.; Retta, S.F.; et al. Helicobacter pylori type IV secretion apparatus exploits beta1 integrin in a novel RGD-independent manner. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, C.L.; Gaddy, J.A.; Loh, J.T.; Johnson, E.M.; Hill, S.; Hennig, E.E.; McClain, M.S.; McDonald, W.H.; Cover, T.L. Helicobacter pylori exploits a unique repertoire of type IV secretion system components for pilus assembly at the bacteria-host cell interface. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Barden, S.; Lange, S.; Tegtmeyer, N.; Conradi, J.; Sewald, N.; Backert, S.; Niemann, H.H. A helical RGD motif promoting cell adhesion: Crystal structures of the Helicobacter pylori type IV secretion system pilus protein CagL. Structure 2013, 21, 1931–1941. [Google Scholar] [CrossRef] [PubMed]
- Javaheri, A.; Kruse, T.; Moonens, K.; Mejías-Luque, R.; Debraekeleer, A.; Asche, C.I.; Tegtmeyer, N.; Kalali, B.; Bach, N.C.; Sieber, S.A.; et al. Helicobacter pylori adhesin HopQ engages in a virulence-enhancing interaction with human CEACAMs. Nat. Microbiol. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Koniger, V.; Holsten, L.; Harrison, U.; Loell, E.; Busch, B.; Zhao, Q.; Bonsor, D.A.; Roth, A.; Kengmo-Tchoupa, A.; Smith, S.I.; et al. Helicobacter pylori exploits human CEACAMs via HopQ for adherence and translocation of CagA. Nat. Microbiol. 2016, 2. [Google Scholar] [CrossRef]
- Lai, C.H.; Chang, Y.C.; Du, S.Y.; Wang, H.J.; Kuo, C.H.; Fang, S.H.; Fu, H.W.; Lin, H.H.; Chiang, A.S.; Wang, W.C. Cholesterol depletion reduces Helicobacter pylori CagA translocation and CagA-induced responses in AGS cells. Infect. Immun. 2008, 76, 3293–3303. [Google Scholar] [CrossRef] [PubMed]
- Lind, J.; Backert, S.; Pfleiderer, K.; Berg, D.E.; Yamaoka, Y.; Sticht, H.; Tegtmeyer, N. Systematic analysis of phosphotyrosine antibodies recognizing single phosphorylated EPIYA-motifs in CagA of Western-type Helicobacter pylori strains. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Lind, J.; Backert, S.; Hoffmann, R.; Eichler, J.; Yamaoka, Y.; Perez-Perez, G.I.; Torres, J.; Sticht, H.; Tegtmeyer, N. Systematic analysis of phosphotyrosine antibodies recognizing single phosphorylated EPIYA-motifs in CagA of East Asian-type Helicobacter pylori strains. BMC Microbiol. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Selbach, M.; Moese, S.; Hauck, C.R.; Meyer, T.F.; Backert, S. Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J. Biol. Chem. 2002, 277, 6775–6778. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.; Bagnoli, F.; Halenbeck, R.; Rappuoli, R.; Fantl, W.J.; Covacci, A. c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol. Microbiol. 2002, 43, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Poppe, M.; Feller, S.M.; Römer, G.; Wessler, S. Phosphorylation of Helicobacter pylori CagA by c-Abl leads to cell motility. Oncogene 2007, 26, 3462–3472. [Google Scholar] [CrossRef] [PubMed]
- Tammer, I.; Brandt, S.; Hartig, R.; König, W.; Backert, S. Activation of Abl by Helicobacter pylori: A novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology 2007, 132, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; Tegtmeyer, N.; Brandt, S.; Yamaoka, Y.; De Poire, E.; Sgouras, D.; Wessler, S.; Torres, J.; Smolka, A.; Backert, S. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J. Clin. Investig. 2012, 122, 1553–1566. [Google Scholar] [CrossRef] [PubMed]
- El-Etr, S.H.; Mueller, A.; Tompkins, L.S.; Falkow, S.; Merrell, D.S. Phosphorylation-independent effects of CagA during interaction between Helicobacter pylori and T84 polarized monolayers. J. Infect. Dis. 2004, 190, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Glowinski, F.; Holland, C.; Thiede, B.; Jungblut, P.R.; Meyer, T.F. Analysis of T4SS-induced signaling by H. pylori using quantitative phosphoproteomics. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.S.; Tegtmeyer, N.; Traube, L.; Jindal, S.; Perez-Perez, G.; Sticht, H.; Backert, S.; Blaser, M.J. A specific A/T polymorphism in Western tyrosine phosphorylation B-motifs regulates Helicobacter pylori CagA epithelial cell interactions. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, N.; Yuasa, H.; Tanaka, S.; Sawa, H.; Miura, M.; Matsui, A.; Higashi, H.; Musashi, M.; Iwabuchi, K.; Suzuki, M.; et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. USA 2008, 105, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Longhurst, C.M.; Jennings, L.K. Integrin-mediated signal transduction. Cell Mol. Life Sci. 1998, 54, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Horton, E.R.; Humphries, J.D.; James, J.; Jones, M.C.; Askari, J.A.; Humphries, M.J. The integrin adhesome network at a glance. J. Cell Sci. 2016, 129, 4159–4163. [Google Scholar] [CrossRef] [PubMed]
- Conradi, J.; Huber, S.; Gaus, K.; Mertink, F.; Royo Gracia, S.; Strijowski, U.; Backert, S.; Sewald, N. Cyclic RGD peptides interfere with binding of the Helicobacter pylori protein CagL to integrins αVβ3 and α5β1. Amino Acids 2012, 43, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Conradi, J.; Tegtmeyer, N.; Woźna, M.; Wissbrock, M.; Michalek, C.; Gagell, C.; Cover, T.L.; Frank, R.; Sewald, N.; Backert, S. An RGD helper sequence in CagL of Helicobacter pylori assists in interactions with integrins and injection of CagA. Front. Cell Infect. Microbiol. 2012, 2, 70. [Google Scholar] [CrossRef] [PubMed]
- Kaplan-Türköz, B.; Jiménez-Soto, L.F.; Dian, C.; Ertl, C.; Remaut, H.; Louche, A.; Tosi, T.; Haas, R.; Terradot, L. Structural insights into Helicobacter pylori oncoprotein CagA interaction with β1 integrin. Proc. Natl. Acad. Sci. USA 2012, 109, 14640–14645. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Senda, M.; Morohashi, H.; Higashi, H.; Horio, M.; Kashiba, Y.; Nagase, L.; Sasaya, D.; Shimizu, T.; Venugopalan, N.; et al. Tertiary structure-function analysis reveals the pathogenic signaling potentiation mechanism of Helicobacter pylori oncogenic effector CagA. Cell Host Microbe 2012, 12, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, G.R. The type III secretion injectisome. Nat. Rev. Microbiol. 2006, 4, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Murata-Kamiya, N.; Kikuchi, K.; Hayashi, T.; Higashi, H.; Hatakeyama, M. Helicobacter pylori exploits host membrane phosphatidylserine for delivery, localization, and pathophysiological action of the CagA oncoprotein. Cell Host Microbe 2010, 7, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Scheffzek, K.; Welti, S. Pleckstrin homology (PH) like domains—Versatile modules in protein-protein interaction platforms. FEBS Lett. 2012, 586, 2662–2673. [Google Scholar] [CrossRef] [PubMed]
- Roujeinikova, A. Phospholipid binding residues of eukaryotic membrane-remodelling F-BAR domain proteins are conserved in Helicobacter pylori CagA. BMC Res. Notes 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Kumar Pachathundikandi, S.; Brandt, S.; Madassery, J.; Backert, S. Induction of TLR-2 and TLR-5 expression by Helicobacter pylori switches cagPAI-dependent signalling leading to the secretion of IL-8 and TNF-α. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Bauer, B.; Moese, S.; Bartfeld, S.; Meyer, T.F.; Selbach, M. Analysis of cell type-specific responses mediated by the type IV secretion system of Helicobacter pylori. Infect. Immun. 2005, 73, 4643–4652. [Google Scholar] [CrossRef] [PubMed]
- Schwechheimer, C.; Kuehn, M.J. Outer-membrane vesicles from Gram-negative bacteria: Biogenesis and functions. Nat. Rev. Microbiol. 2015, 13, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Pathirana, R.D.; Kaparakis-Liaskos, M. Bacterial membrane vesicles: Biogenesis, immune regulation and pathogenesis. Cell. Microbiol. 2016, 18, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Fiocca, R.; Necchi, V.; Sommi, P.; Ricci, V.; Telford, J.; Cover, T.L.; Solcia, E. Release of Helicobacter pylori vacuolating cytotoxin by both a specific secretion pathway and budding of outer membrane vesicles. Uptake of released toxin and vesicles by gastric epithelium. J. Pathol. 1999, 188, 220–226. [Google Scholar] [CrossRef]
- Heczko, U.; Smith, V.C.; Meloche, M.R.; Buchan, A.M.; Finlay, B.B. Characteristics of Helicobacter pylori attachment to human primary antral epithelial cells. Microbes Infect. 2000, 2, 1669–1676. [Google Scholar] [CrossRef]
- Keenan, J.; Day, T.; Neal, S.; Cook, B.; Perez-Perez, G.; Allardyce, R.; Bagshaw, P. A role for the bacterial outer membrane in the pathogenesis of Helicobacter pylori infection. FEMS Microbiol. Lett. 2000, 182, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Ismail, S.; Hampton, M.B.; Keenan, J.I. Helicobacter pylori outer membrane vesicles modulate proliferation and interleukin-8 production by gastric epithelial cells. Infect. Immun. 2003, 71, 5670–5675. [Google Scholar] [CrossRef] [PubMed]
- Hynes, S.O.; Keenan, J.I.; Ferris, J.A.; Annuk, H.; Moran, A.P. Lewis epitopes on outer membrane vesicles of relevance to Helicobacter pylori pathogenesis. Helicobacter 2005, 10, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Ricci, V.; Chiozzi, V.; Necchi, V.; Oldani, A.; Romano, M.; Solcia, E.; Ventura, U. Free-soluble and outer membrane vesicle-associated VacA from Helicobacter pylori: Two forms of release, a different activity. Biochem. Biophys. Res. Commun. 2005, 337, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G.; Torres, L.; Espinosa, M.; Fierros-Zarate, G.; Maldonado, V.; Meléndez-Zajgla, J. External membrane vesicles from Helicobacter pylori induce apoptosis in gastric epithelial cells. FEMS Microbiol. Lett. 2006, 260, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Keenan, J.I.; Davis, K.A.; Beaugie, C.R.; McGovern, J.J.; Moran, A.P. Alterations in Helicobacter pylori outer membrane and outer membrane vesicle-associated lipopolysaccharides under iron-limiting growth conditions. Innate Immun. 2008, 14, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Chitcholtan, K.; Hampton, M.B.; Keenan, J.I. Outer membrane vesicles enhance the carcinogenic potential of Helicobacter pylori. Carcinogenesis 2008, 29, 2400–2405. [Google Scholar] [CrossRef] [PubMed]
- Mullaney, E.; Brown, P.A.; Smith, S.M.; Botting, C.H.; Yamaoka, Y.Y.; Terres, A.M.; Kelleher, D.P.; Windle, H.J. Proteomic and functional characterization of the outer membrane vesicles from the gastric pathogen Helicobacter pylori. Proteomics Clin. Appl. 2009, 3, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Kaparakis, M.; Turnbull, L.; Carneiro, L.; Firth, S.; Coleman, H.A.; Parkington, H.C.; Le Bourhis, L.; Karrar, A.; Viala, J.; Mak, J.; et al. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell Microbiol. 2010, 12, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, A.; Vallström, A.; Petzold, K.; Tegtmeyer, N.; Schleucher, J.; Carlsson, S.; Haas, R.; Backert, S.; Wai, S.N.; Gröbner, G.; et al. Biochemical and functional characterization of Helicobacter pylori vesicles. Mol. Microbiol. 2010, 77, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Letley, D.; Rhead, J.; Atherton, J.; Robinson, K. Helicobacter pylori membrane vesicles stimulate innate pro- and anti-inflammatory responses and induce apoptosis in Jurkat T cells. Infect. Immun. 2014, 82, 1372–1381. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.H.; Rho da, J.; Jeon, J.I.; Kim, Y.J.; Woo, H.A.; Kim, N.; Kim, J.M. Crude Preparations of Helicobacter pylori Outer Membrane Vesicles Induce Upregulation of Heme Oxygenase-1 via Activating Akt-Nrf2 and mTOR-IκB Kinase-NF-κB Pathways in Dendritic Cells. Infect. Immun. 2016, 84, 2162–2174. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.; Chitcholtan, K.; Hampton, M.B.; Keenan, J.I. Uptake of Helicobacter pylori outer membrane vesicles by gastric epithelial cells. Infect. Immun. 2010, 78, 5054–5061. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, A.; Nygård Skalman, L.; Obi, I.; Lundmark, R.; Arnqvist, A. Uptake of Helicobacter pylori vesicles is facilitated by clathrin-dependent and clathrin-independent endocytic pathways. MBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, E.J.; Krachler, A.M. Mechanisms of outer membrane vesicle entry into host cells. Cell Microbiol. 2016, 18, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Turkina, M.V.; Olofsson, A.; Magnusson, K.E.; Arnqvist, A.; Vikström, E. Helicobacter pylori vesicles carrying CagA localize in the vicinity of cell-cell contacts and induce histone H1 binding to ATP in epithelial cells. FEMS Microbiol. Lett. 2015, 362. [Google Scholar] [CrossRef] [PubMed]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signaling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Holowka, D.; Baird, B. Mechanisms of epidermal growth factor receptor signaling as characterized by patterned ligand activation and mutational analysis. Biochim. Biophys. Acta 2016. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.H.; Yoon, J.H.; Choi, S.S.; Ashktorab, H.; Smoot, D.T.; Song, K.Y.; Nam, S.W.; Lee, J.Y.; Park, C.H.; Park, W.S. The effect of Helicobacter pylori CagA on the HER-2 copy number and expression in gastric cancer. Gene 2014, 546, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, C.; Camargo, M.C.; Leite, M.; Fuentes-Pananá, E.M.; Rabkin, C.S.; Machado, J.C. Pathogenesis of Gastric Cancer: Genetics and Molecular Classification. In Molecular Pathogenesis and Signal Transduction by Helicobacter pylori Research; Tegtmeyer, N., Backert, S., Eds.; Springer: Heidelberg, Germany, 2017; pp. 277–304. [Google Scholar]
- Coyle, W.J.; Sedlack, R.E.; Nemec, R.; Peterson, R.; Duntemann, T.; Murphy, M.; Lawson, J.M. Eradication of Helicobacter pylori normalizes elevated mucosal levels of epidermal growth factor and its receptor. Am. J. Gastroenterol. 1999, 94, 2885–2889. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.C.; Wang, W.P.; So, W.H.; Shin, V.Y.; Wong, W.M.; Fung, F.M.; Liu, E.S.; Hiu, W.M.; Lam, S.K.; Cho, C.H. Epidermal growth factor and its receptor in chronic active gastritis and gastroduodenal ulcer before and after Helicobacter pylori eradication. Aliment. Pharmacol. Ther. 2001, 15, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Wallasch, C.; Crabtree, J.E.; Bevec, D.; Robinson, P.A.; Wagner, H.; Ullrich, A. Helicobacter pylori-stimulated EGF receptor transactivation requires metalloprotease cleavage of HB-EGF. Biochem. Biophys. Res. Commun. 2002, 295, 695–701. [Google Scholar] [CrossRef]
- Saha, A.; Backert, S.; Hammond, C.E.; Gooz, M.; Smolka, A.J. Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase alpha subunit. Gastroenterology 2010, 139, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Keates, S.; Sougioultzis, S.; Keates, A.C.; Zhao, D.; Peek, R.M., Jr.; Shaw, L.M.; Kelly, C.P. Cag+ H. pylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J. Biol. Chem. 2001, 276, 48127–48134. [Google Scholar] [PubMed]
- Tegtmeyer, N.; Zabler, D.; Schmidt, D.; Hartig, R.; Brandt, S.; Backert, S. Importance of EGF receptor, HER2/Neu and Erk1/2 kinase signalling for host cell elongation and scattering induced by the Helicobacter pylori CagA protein: Antagonistic effects of the vacuolating cytotoxin VacA. Cell Microbiol. 2009, 11, 488–505. [Google Scholar] [CrossRef] [PubMed]
- Mimuro, H.; Suzuki, T.; Nagai, S.; Rieder, G.; Suzuki, M.; Nagai, T.; Fujita, Y.; Nagamatsu, K.; Ishijima, N.; Koyasu, S.; et al. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe 2007, 2, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, R.; Asim, M.; Piazuelo, M.B.; Yan, F.; Barry, D.P.; Sierra, J.C.; Delgado, A.G.; Hill, S.; Casero, R.A., Jr.; Bravo, L.E.; et al. Activation of EGFR and ERBB2 by Helicobacter pylori results in survival of gastric epithelial cells with DNA damage. Gastroenterology 2014, 146, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.E.; Beeson, C.; Suarez, G.; Peek, R.M., Jr.; Backert, S.; Smolka, A.J. Helicobacter pylori virulence factors affecting gastric proton pump expression and acid secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G193–G201. [Google Scholar] [CrossRef] [PubMed]
- Tanos, B.; Pendergast, A.M. Abl tyrosine kinase regulates endocytosis of the epidermal growth factor receptor. J. Biol. Chem. 2006, 281, 32714–32723. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, A.; Goh, L.K. Endocytosis and intracellular trafficking of ErbBs. Exp. Cell Res. 2009, 315, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Bauer, B.; Bartfeld, S.; Meyer, T.F. H. pylori selectively blocks EGFR endocytosis via the non-receptor kinase c-Abl and CagA. Cell Microbiol. 2009, 11, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Bauer, B.; Pang, E.; Holland, C.; Kessler, M.; Bartfeld, S.; Meyer, T.F. The H. pylori virulence effector CagA abrogates human β-defensin 3 expression via inactivation of EGFR signaling. Cell Host Microbe 2012, 11, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, J.S.; Zaidi, S.F.; Zhou, Y.; Sakurai, H.; Sugiyama, T. Novel epidermal growth factor receptor pathway mediates release of human β-defensin 3 from Helicobacter pylori-infected gastric epithelial cells. Pathog. Dis. 2016, 74. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Bauer, B.; Wex, T.; Kuester, D.; Meyer, T.; Malfertheiner, P. Differential expression of human beta defensin 2 and 3 in gastric mucosa of Helicobacter pylori-infected individuals. Helicobacter 2013, 18, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signaling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Churin, Y.; Al-Ghoul, L.; Kepp, O.; Meyer, T.F.; Birchmeier, W.; Naumann, M. Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. J. Cell Biol. 2003, 161, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Mimuro, H.; Suzuki, T.; Tanaka, J.; Asahi, M.; Haas, R.; Sasakawa, C. Grb2 is a key mediator of H. pylori CagA protein activities. Mol. Cell 2002, 10, 745–755. [Google Scholar] [CrossRef]
- Brandt, S.; Kwok, T.; Hartig, R.; König, W.; Backert, S. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9300–9305. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeyer, N.; Wittelsberger, R.; Hartig, R.; Wessler, S.; Martinez-Quiles, N.; Backert, S. Serine phosphorylation of cortactin controls focal adhesion kinase activity and cell scattering induced by Helicobacter pylori. Cell Host Microbe 2011, 9, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signaling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Wroblewski, L.E.; Piazuelo, M.B.; Chaturvedi, R.; Schumacher, M.; Aihara, E.; Feng, R.; Noto, J.M.; Delgado, A.; Israel, D.A.; Zavros, Y.; et al. Helicobacter pylori targets cancer-associated apical-junctional constituents in gastroids and gastric epithelial cells. Gut 2015, 64, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Schlaermann, P.; Toelle, B.; Berger, H.; Schmidt, S.C.; Glanemann, M.; Ordemann, J.; Bartfeld, S.; Mollenkopf, H.J.; Meyer, T.F. A novel human gastric primary cell culture system for modelling Helicobacter pylori infection in vitro. Gut 2016, 65, 202–213. [Google Scholar] [CrossRef] [PubMed]
- McCracken, K.W.; Catá, E.M.; Crawford, C.M.; Sinagoga, K.L.; Schumacher, M.; Rockich, B.E.; Tsai, Y.H.; Mayhew, C.N.; Spence, J.R.; Zavros, Y.; et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 2014, 516, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Bertaux-Skeirik, N.; Feng, R.; Schumacher, M.A.; Li, J.; Mahe, M.M.; Engevik, A.C.; Javier, J.E.; Peek, R.M., Jr.; Ottemann, K.; Orian-Rousseau, V.; et al. CD44 plays a functional role in Helicobacter pylori-induced epithelial cell proliferation. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.M.; Leite, M.; Seruca, R.; Figueiredo, C. Adherens junctions as targets of microorganisms: A focus on Helicobacter pylori. FEBS Lett. 2013, 587, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Braga, V. Spatial integration of E-cadherin adhesion, signaling and the epithelial cytoskeleton. Curr. Opin. Cell Biol. 2016, 42, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Murata-Kamiya, N.; Kurashima, Y.; Teishikata, Y.; Yamahashi, Y.; Saito, Y.; Higashi, H.; Aburatani, H.; Akiyama, T.; Peek, R.M., Jr.; Azuma, T.; et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 2007, 26, 4617–4626. [Google Scholar] [CrossRef] [PubMed]
- Kurashima, Y.; Murata-Kamiya, N.; Kikuchi, K.; Higashi, H.; Azuma, T.; Kondo, S.; Hatakeyama, M. Deregulation of beta-catenin signal by Helicobacter pylori CagA requires the CagA-multimerization sequence. Int. J. Cancer 2008, 122, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Shun, C.T.; Wu, M.S.; Lin, M.T.; Chang, M.C.; Lin, J.T.; Chuang, S.M. Immunohistochemical evaluation of cadherin and catenin expression in early gastric carcinomas: Correlation with clinicopathologic characteristics and Helicobacter pylori infection. Oncology 2001, 60, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Yong, X.; Tang, B.; Xiao, Y.F.; Xie, R.; Qin, Y.; Luo, G.; Hu, C.J.; Dong, H.; Yang, S.M. Helicobacter pylori upregulates Nanog and Oct4 via Wnt/β-catenin signaling pathway to promote cancer stem cell-like properties in human gastric cancer. Cancer Lett. 2016, 374, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.T.; Israel, D.A.; Washington, M.K.; Krishna, U.; Fox, J.G.; Rogers, A.B.; Neish, A.S.; Collier-Hyams, L.; Perez-Perez, G.I.; Hatakeyama, M.; et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc. Natl. Acad. Sci. USA 2005, 102, 10646–10651. [Google Scholar] [CrossRef] [PubMed]
- Ogden, S.R.; Wroblewski, L.E.; Weydig, C.; Romero-Gallo, J.; O’Brien, D.P.; Israel, D.A.; Krishna, U.S.; Fingleton, B.; Reynolds, A.B.; Wessler, S.; et al. p120 and Kaiso regulate Helicobacter pylori-induced expression of matrix metalloproteinase-7. Mol. Biol. Cell 2008, 19, 4110–4121. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, O.; Bozko, P.M.; Naumann, M. Helicobacter pylori suppresses glycogen synthase kinase 3beta to promote beta-catenin activity. J. Biol. Chem. 2008, 283, 29367–29374. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.J.; Costa, A.C.; Costa, A.M.; Henriques, L.; Suriano, G.; Atherton, J.C.; Machado, J.C.; Carneiro, F.; Seruca, R.; Mareel, M.; et al. Helicobacter pylori induces gastric epithelial cell invasion in a c-Met and type IV secretion system-dependent manner. J. Biol. Chem. 2006, 281, 34888–34896. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.J.; Costa, A.M.; Costa, A.C.; Ferreira, R.M.; Sampaio, P.; Machado, J.C.; Seruca, R.; Mareel, M.; Figueiredo, C. CagA associates with c-Met, E-cadherin, and p120-catenin in a multiproteic complex that suppresses Helicobacter pylori-induced cell-invasive phenotype. J. Infect. Dis. 2009, 200, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Hoy, B.; Geppert, T.; Boehm, M.; Reisen, F.; Plattner, P.; Gadermaier, G.; Sewald, N.; Ferreira, F.; Briza, P.; Schneider, G.; et al. Distinct roles of secreted HtrA proteases from gram-negative pathogens in cleaving the junctional protein and tumor suppressor E-cadherin. J. Biol. Chem. 2012, 287, 10115–10120. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Thiem, S.; Nguyen, P.M.; Eissmann, M.; Putoczki, T.L. Epithelial gp130/Stat3 functions: An intestinal signaling node in health and disease. Semin. Immunol. 2014, 26, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, T.; Ishihara, K.; Atsumi, T.; Nishida, K.; Kaneko, Y.; Miyata, T.; Itoh, S.; Narimatsu, M.; Maeda, H.; Fukada, T.; et al. Dissection of signaling cascades through gp130 in vivo: Reciprocal roles for STAT3- and SHP2-mediated signals in immune responses. Immunity 2000, 12, 95–105. [Google Scholar] [CrossRef]
- Jackson, C.B.; Judd, L.M.; Menheniott, T.R.; Kronborg, I.; Dow, C.; Yeomans, N.D.; Boussioutas, A.; Robb, L.; Giraud, A.S. Augmented gp130-mediated cytokine signaling accompanies human gastric cancer progression. J. Pathol. 2007, 213, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Bronte-Tinkew, D.M.; Terebiznik, M.; Franco, A.; Ang, M.; Ahn, D.; Mimuro, H.; Sasakawa, C.; Ropeleski, M.J.; Peek, R.M., Jr.; Jones, N.L. Helicobacter pylori cytotoxin-associated gene A activates the signal transducer and activator of transcription 3 pathway in vitro and in vivo. Cancer Res. 2009, 69, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.O.; Kim, J.H.; Choi, Y.J.; Pillinger, M.H.; Kim, S.Y.; Blaser, M.J.; Lee, Y.C. Helicobacter pylori CagA phosphorylation status determines the gp130-activated SHP2/ERK and JAK/STAT signal transduction pathways in gastric epithelial cells. J. Biol. Chem. 2010, 285, 16042–16050. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Kalantzis, A.; Jackson, C.B.; O’Connor, L.; Murata-Kamiya, N.; Hatakeyama, M.; Judd, L.M.; Giraud, A.S.; Menheniott, T.R. Helicobacter pylori CagA triggers expression of the bactericidal lectin REG3γ via gastric STAT3 activation. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Moese, S.; Selbach, M.; Zimny-Arndt, U.; Jungblut, P.R.; Meyer, T.F.; Backert, S. Identification of a tyrosine-phosphorylated 35 kDa carboxy-terminal fragment (p35CagA) of the Helicobacter pylori CagA protein in phagocytic cells: Processing or breakage? Proteomics 2001, 1, 618–629. [Google Scholar] [CrossRef]
- Odenbreit, S.; Gebert, B.; Fischer, W.; Püls, J.; Haas, R. Interaction of H. pylori with professional phagocytes: Role of the cag pathogenicity island and translocation, phosphorylation and processing of CagA. Cell Microbiol. 2001, 3, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Krisch, L.M.; Posselt, G.; Hammerl, P.; Wessler, S. CagA Phosphorylation in Helicobacter pylori-Infected B Cells Is Mediated by the Nonreceptor Tyrosine Kinases of the Src and Abl Families. Infect. Immun. 2016, 84, 2671–2680. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Backert, S.; Tegtmeyer, N. Type IV Secretion and Signal Transduction of Helicobacter pylori CagA through Interactions with Host Cell Receptors. Toxins 2017, 9, 115. https://doi.org/10.3390/toxins9040115
Backert S, Tegtmeyer N. Type IV Secretion and Signal Transduction of Helicobacter pylori CagA through Interactions with Host Cell Receptors. Toxins. 2017; 9(4):115. https://doi.org/10.3390/toxins9040115
Chicago/Turabian StyleBackert, Steffen, and Nicole Tegtmeyer. 2017. "Type IV Secretion and Signal Transduction of Helicobacter pylori CagA through Interactions with Host Cell Receptors" Toxins 9, no. 4: 115. https://doi.org/10.3390/toxins9040115
APA StyleBackert, S., & Tegtmeyer, N. (2017). Type IV Secretion and Signal Transduction of Helicobacter pylori CagA through Interactions with Host Cell Receptors. Toxins, 9(4), 115. https://doi.org/10.3390/toxins9040115