
Venomics of Remipede Crustaceans Reveals Novel Peptide Diversity and Illuminates the Venom’s Biological Role



Abstract
:1. Introduction
2. Results and Discussion
2.1. The Effect of Transcriptome Sequencing Platforms and Assembly Strategies
2.1.1. Transcriptome Assembly Strategy
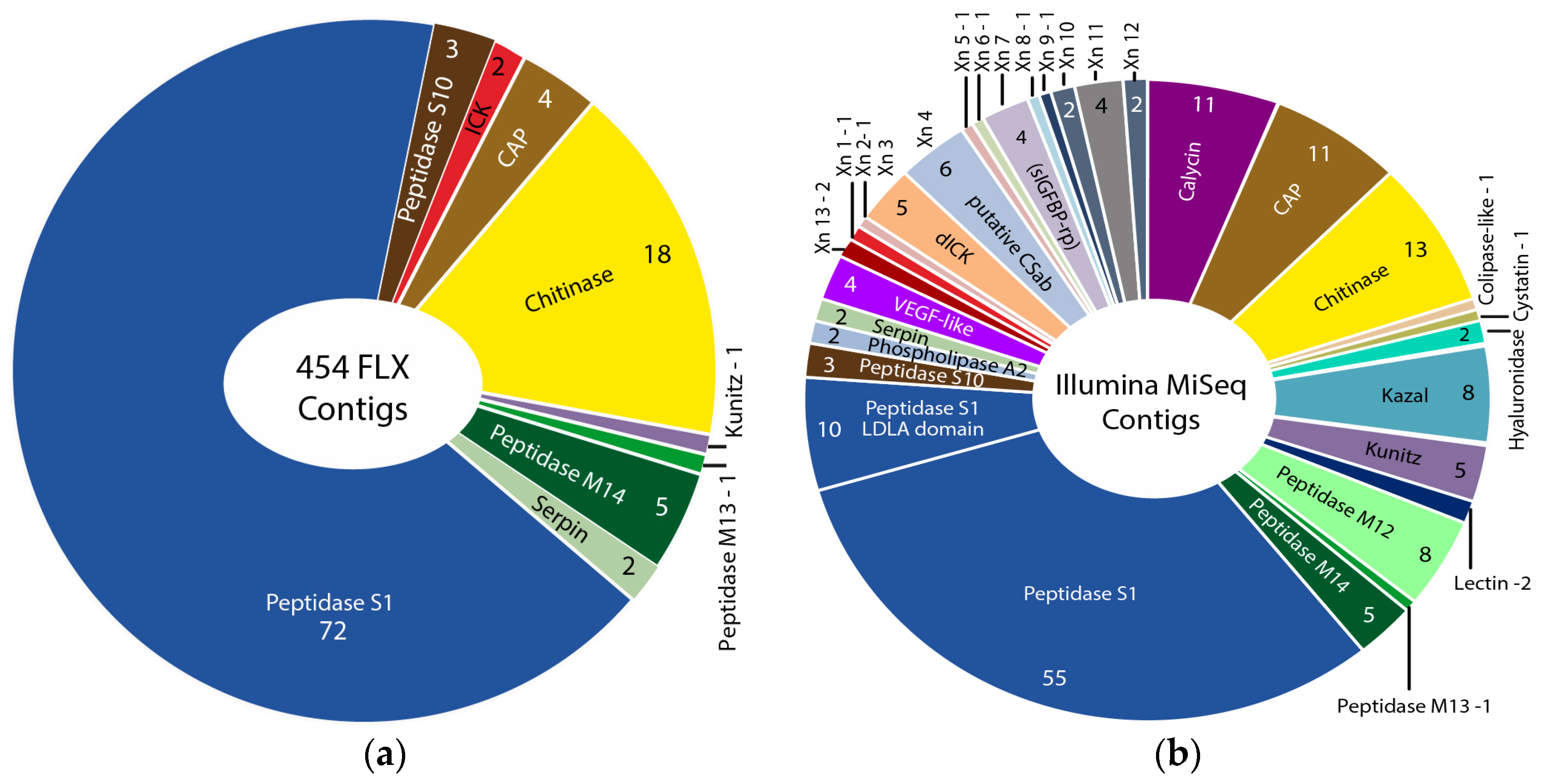
2.1.2. Comparison of 454 FLX and Illumina MiSeq and HiSeq Transcriptomes
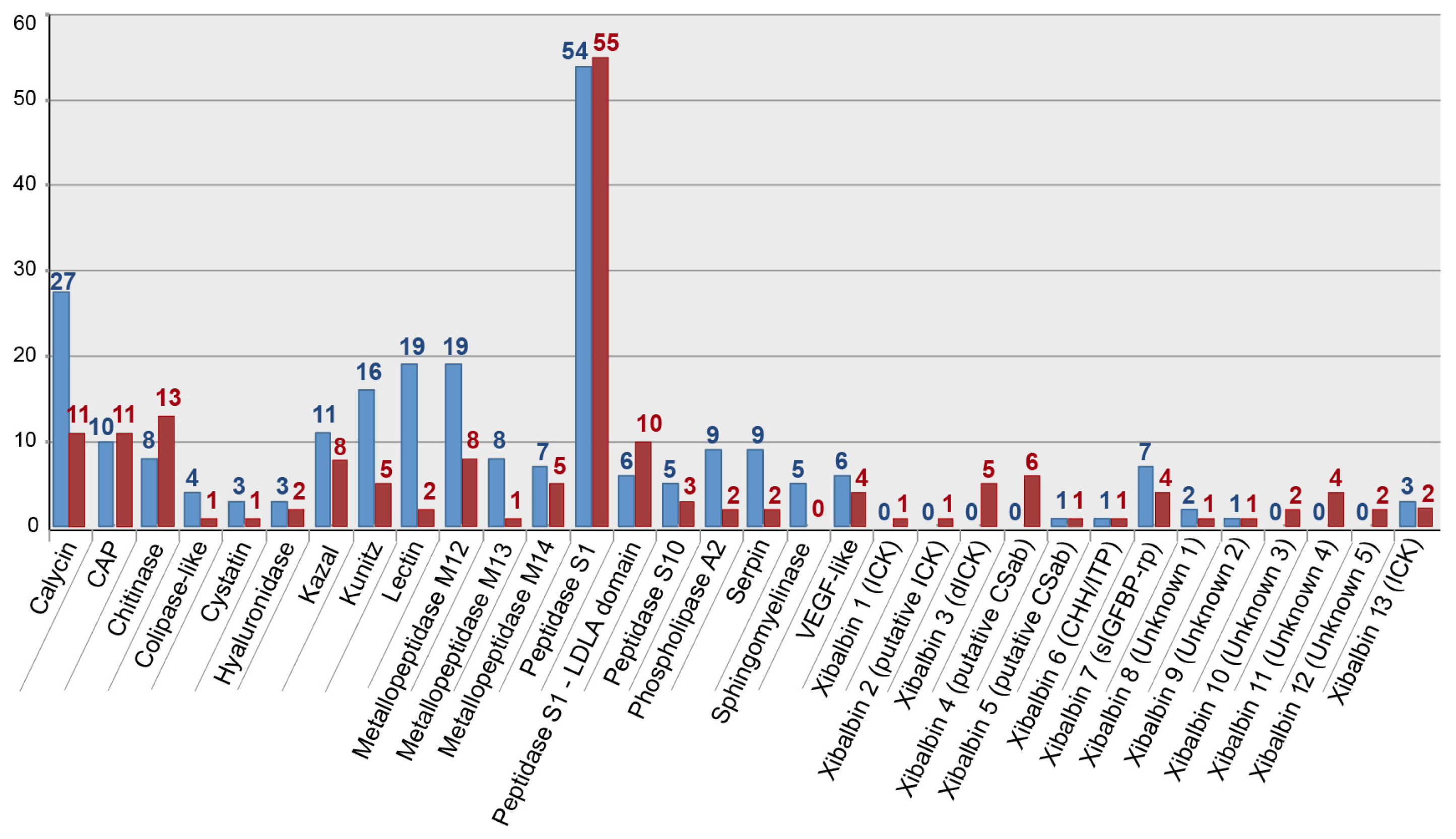
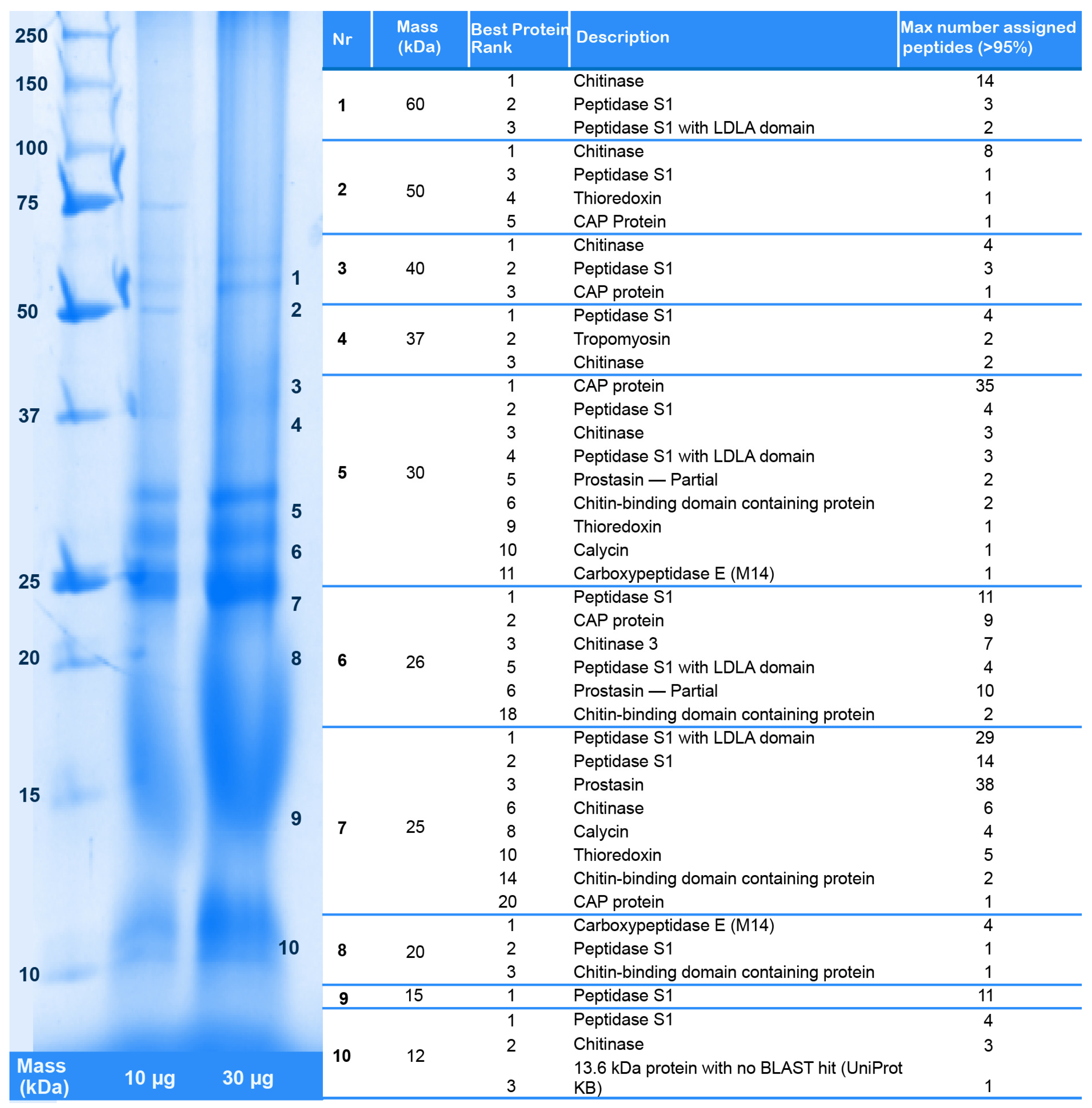
2.2. Revising the Venomic Profile of X. Tulumensis
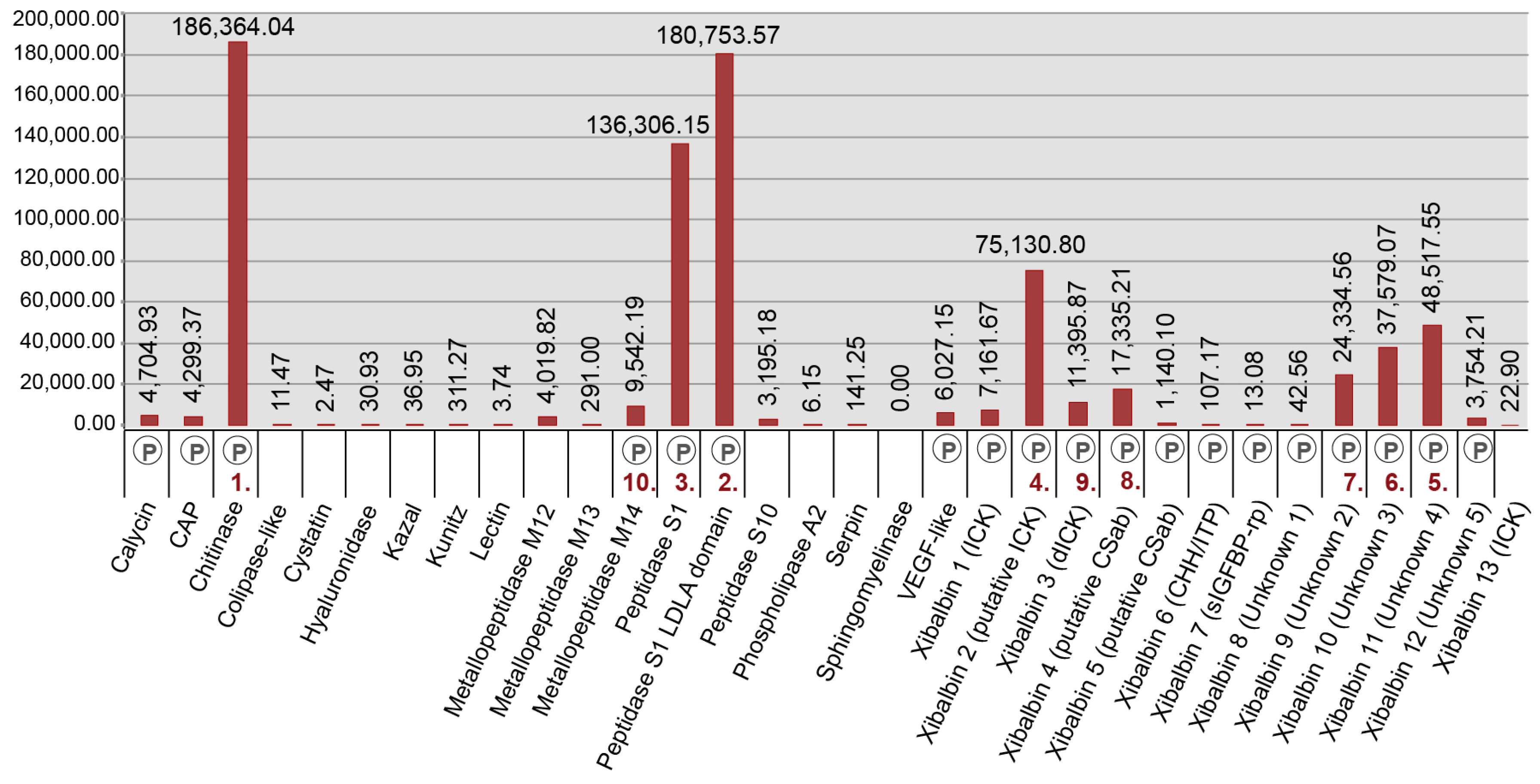
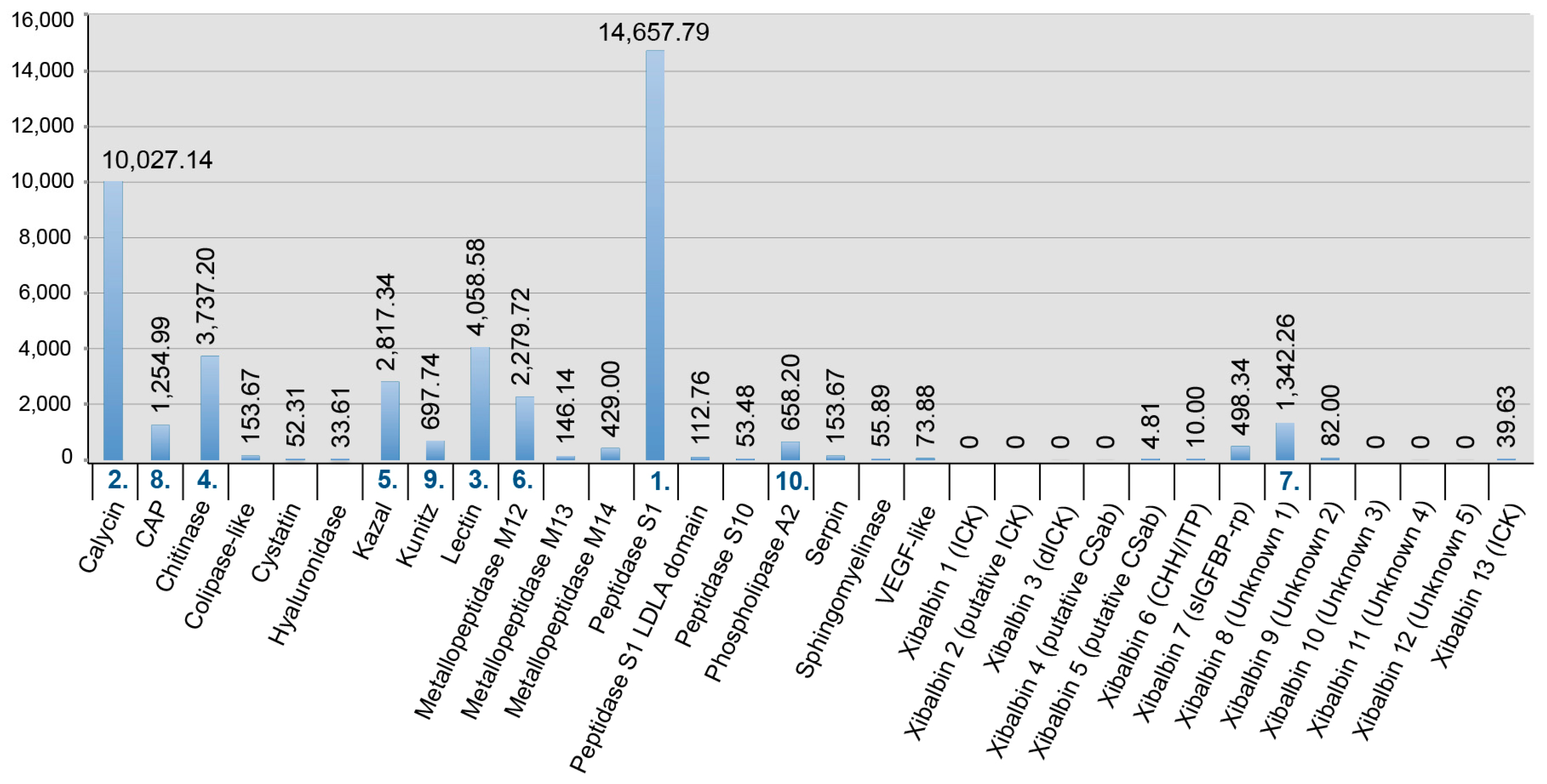
2.3. New Venomic Profile Provides Insights into Putative Function of Venom
2.3.1. Enzymes
2.3.2. Non-Enzymatic Proteins
2.3.3. Peptides
3. Conclusions
4. Materials and Methods
4.1. Species Collection, Dissection and Preservation
4.2. Identification of Putative Toxins via Transcriptomics
4.2.1. RNA Extraction and Library Construction
4.2.2. Pre-Processing of RNA Sequence Reads
4.2.3. Comparative Read Assembly Strategy Using Trinity and SOAPdenovo-Trans
4.2.4. Read Mapping to Assess Expression Levels of Identified Putative Toxin Contigs
4.2.5. Identification of Putative Venom Toxins in Secreted Proteins of Assembly and Selection of Optimal Assembly Strategy
4.2.6. Identification of Putative Venom Toxins in the Complete (Secreted and Non-Secreted Proteins) Assembled Data
4.3. Identification of Putative Toxins via Proteomics
4.4. Final Comparative Toxin Identification and Analyses
4.4.1. Sequence Alignments, Phylogenetic Tree and Network Reconstructions
4.4.2. Assessing the Evolution of ICK Sequences by Extensive Data Mining in NCBI and UniProt
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Neiber, M.T.; Hartke, T.R.; Stemme, T.; Bergmann, A.; Rust, J.; Iliffe, T.M.; Koenemann, S. Global biodiversity and phylogenetic evaluation of Remipedia (Crustacea). PLoS ONE 2011, 6, e19627. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J.; Meland, K.; Glenner, H.; Van Hengstum, P.J.; Iliffe, T.M. Xibalbanus cozumelensis, a new species of Remipedia (Crustacea) from Cozumel, Mexico, and a molecular phylogeny of Xibalbanus on the Yucatán Peninsula. Eur. J. Taxon. 2017, 316, 1–27. [Google Scholar]
- Yager, J. Remipedia, a new class of Crustacea from a marine cave in the Bahamas. J. Crustac. Biol. 1981, 1, 328–333. [Google Scholar] [CrossRef]
- Schram, F.R.; Hof, C.H.J. Fossils and interrelationships of major crustacean groups. In Arthropod Fossils and Phylogeny; Edgecombe, G.D., Ed.; Columbia University Press: New York, NY, USA, 1998; pp. 233–302. [Google Scholar]
- Martin, J.W.; Davis, G.E. An Updated Classification of the Recent Crustacea; Science Series; Natural History Museum Los Angeles County: Los Angeles, CA, USA, 2001; Volume 39, pp. 1–124. [Google Scholar]
- Von Reumont, B.M.; Jenner, R.A.; Wills, M.A.; Dell’Ampio, E.; Ebersberger, I.; Meyer, B.; Koenemann, S.; Iliffe, T.M.; Stamatakis, A.; Niehuis, O.; et al. Pancrustacean phylogeny in the light of new phylogenomic data: Support for Remipedia as the sister group of Hexapoda. Mol. Biol. Evol. 2012, 29, 1031–1045. [Google Scholar] [CrossRef] [PubMed]
- Oakley, T.H.; Wolfe, J.M.; Lindgren, A.R.; Zaharoff, A.K. Phylotranscriptomics to bring the understudied into the fold: Monophyletic Ostracoda, fossil placement, and pancrustacean phylogeny. Mol. Biol. Evol. 2013, 30, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.; Donath, A.; Mayer, C.; Frandsen, P.; Ware, J.; Flouri, T.; Beutel, R. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Schwentner, M.; Combosch, D.J.; Nelson, J.P.; Giribet, G. A phylogenomic solution to the origin of insects by resolving crustacean-hexapod relationships. Curr. Biol. 2017, 27, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M.; Burmester, T. Remipedia and the evolution of hexapods. In Encyclopedia of Life Science; John Wiley & Sons, Ltd.: Chichester, UK, 2010; pp. 1–6. [Google Scholar]
- Von Reumont, B.M.; Campbell, L.I.; Jenner, R.A. Quo Vadis venomics? A roadmap to neglected venomous invertebrates. Toxins 2014, 6, 3488–3551. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M.; Blanke, A.; Richter, S.; Alvarez, F.; Bleidorn, C.; Jenner, R.A. The first venomous crustacean revealed by transcriptomics and functional morphology: Remipede venom glands express a unique toxin cocktail dominated by enzymes and a neurotoxin. Mol. Biol. Evol. 2014, 31, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Hoenemann, M.; Neiber, M.T.; Humphreys, W.F.; Iliffe, T.M.; Li, D.; Schram, F.R.; Koenemann, S. Phylogenetic analysis and systematic revision of Remipedia (Nectiopoda) from Bayesian analysis of molecular data. J. Crustac. Biol. 2013, 33, 603–619. [Google Scholar] [CrossRef]
- Stemme, T.; Iliffe, T.M.; Bicker, G. Olfactory pathway in Xibalbanus tulumensis: Remipedian hemiellipsoid body as homologue of hexapod mushroom body. Cell Tissue Res. 2016, 363, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Kohlhage, K.; Yager, J. An analysis of swimming in remipede crustaceans. Philos. Trans. R. Soc. Lond. B 1994, 346, 213–221. [Google Scholar] [CrossRef]
- Fanenbruck, M.; Harzsch, S.; Wägele, J.W. The brain of the Remipedia (Crustacea) and an alternative hypothesis on their phylogenetic relationships. Proc. Natl. Acad. Sci. USA 2004, 101, 3868–3873. [Google Scholar] [CrossRef] [PubMed]
- Undheim, E.; Fry, B.; King, G. Centipede venom: Recent discoveries and current state of knowledge. Toxins 2015, 7, 679–704. [Google Scholar] [CrossRef] [PubMed]
- Kuhn-Nentwig, L.; Stöcklin, R.; Nentwig, W. Venom composition and strategies in spiders. Adv. Insect Physiol. 2011, 40, 1–86. [Google Scholar]
- Liu, Z.; Chen, S.; Zhou, Y.; Xie, C.; Zhu, B.; Zhu, H.; Liu, S.; Wang, W.; Chen, H.; Ji, Y. Deciphering the venomic transcriptome of killer-wasp Vespa velutina. Sci. Rep. 2015, 5, 9454. [Google Scholar] [CrossRef] [PubMed]
- Touchard, A.; Aili, S.R.; Fox, E.G.; Escoubas, P.; Orivel, J.; Nicholson, G.M.; Dejean, A. The biochemical toxin arsenal from ant venoms. Toxins 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Dias, N.B.; de Souza, B.M.; Gomes, P.C.; Brigatte, P.; Palma, M.S. Peptidome profiling of venom from the social wasp Polybia paulista. Toxicon 2015, 107, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.O. The Sting of the Wild; Johns Hopkins University Press: Baltimore, MD, USA, 2016. [Google Scholar]
- King, G.F.; Hardy, M.C. Spider-venom peptides: Structure, pharmacology, and potential for control of insect pests. Annu. Rev. Entomol. 2013, 58, 475–496. [Google Scholar] [CrossRef] [PubMed]
- Luna-Ramírez, K.; Quintero-Hernández, V.; Juárez-González, V.R.; Possani, L.D. Whole transcriptome of the venom gland from Urodacus yaschenkoi scorpion. PLoS ONE 2015, 10, e0127883. [Google Scholar] [CrossRef] [PubMed]
- Mathe-Hubert, H.; Colinet, D.; Deleury, E.; Belghazi, M.; Ravallec, M.; Poulain, J.; Dossat, C.; Poirie, M.; Gatti, J.L. Comparative venomics of Psyttalia lounsburyi and P. concolor, two olive fruit fly parasitoids: A hypothetical role for a GH1 beta-glucosidase. Sci. Rep. 2016, 6, 35873. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.; Von Reumont, B.M.; Erhart, J.; Chagas, A.C.; Ribeiro, J.M.C.; Kotsyfakis, M. De novo Ixodes ricinus salivary gland transcriptome analysis using two next-generation sequencing methodologies. FASEB J. 2013, 27, 4745–4756. [Google Scholar] [CrossRef] [PubMed]
- Rokyta, D.R.; Lemmon, A.R.; Margres, M.J.; Aronow, K. The venom-gland transcriptome of the Eastern diamondback rattlesnake (Crotalus adamanteus). BMC Genom. 2012, 13, 312. [Google Scholar] [CrossRef] [PubMed]
- Rokyta, D.R.; Wray, K.P.; Lemmon, A.R.; Lemmon, E.M.; Caudle, S.B. A high-throughput venom-gland transcriptome for the Eastern diamondback rattlesnake (Crotalus adamanteus) and evidence for pervasive positive selection across toxin classes. Toxicon 2011, 57, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Barghi, N.; Concepcion, G.P.; Olivera, B.M.; Lluisma, A.O. High conopeptide diversity in Conus tribblei revealed through analysis of venom duct transcriptome using two high-throughput sequencing platforms. Mar. Biotechnol. 2015, 17, 81–98. [Google Scholar] [CrossRef] [PubMed]
- King, G.K.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Undheim, E.A.B.; Jones, A.; Clauser, K.R.; Holland, J.W.; Pineda, S.S.; King, G.F.; Fry, B.G. Clawing through evolution: Toxin diversification and convergence in the ancient lineage Chilopoda (centipedes). Mol. Biol. Evol. 2014, 31, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Macrander, J.; Broe, M.; Daly, M. Multi-copy venom genes hidden in de novo transcriptome assemblies, a cautionary tale with the snakelocks sea anemone Anemonia sulcata (Pennant, 1977). Toxicon 2015, 108, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, U.C.D.; Candido, D.M.; Dorce, V.A.C.; Junqueira-De-Azevedo, I.D.L.M. The transcriptome recipe for the venom cocktail of Tityus bahiensis scorpion. Toxicon 2015, 95, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Rokyta, D.R.; Ward, M.J. Venom-gland transcriptomics and venom proteomics of the black-back scorpion (Hadrurus spadix) reveal detectability challenges and an unexplored realm of animal toxin diversity. Toxicon 2017, 128, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Cornet, V.; Henry, J.; Corre, E.; Le Corguille, G.; Zanuttini, B.; Zatylny-Gaudin, C. Dual role of the cuttlefish salivary proteome in defense and predation. J. Proteom. 2014, 108, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Whitelaw, B.L.; Strugnell, J.M.; Faou, P.; da Fonseca, R.A.; Hall, N.E.; Norman, M.; Finn, J.; Cooke, I.R. Combined transcriptomic and proteomic analysis of the posterior salivary gland from the Southern blue-ringed octopus and the Southern sand octopus. J. Proteome Res. 2016, 15, 3284–3297. [Google Scholar] [CrossRef] [PubMed]
- Mackessy, S.P.; Saviola, A.J. Understanding biological roles of venoms among the Caenophidia: The importance of rear-fanged snakes. Integr. Comp. Biol. 2016, 56, 1004–1021. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G. Venomous Reptiles & Their Toxins. Evolution, Pathophysiology & Biodiversity; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Sanggaard, K.W.; Dyrlund, T.F.; Thomsen, L.R.; Nielsen, T.A.; Brøndum, L.; Wang, T.; Thøgersen, I.B.; Enghild, J.J. Characterization of the gila monster (Heloderma suspectum suspectum) venom proteome. J. Proteom. 2015, 117, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pilson, M.E.Q.; Taylor, P.B. Hole drilling by octopus. Science 1961, 134, 1366–1368. [Google Scholar] [CrossRef] [PubMed]
- Runham, N.W.; Bailey, C.J.; Carr, M.; Evans, C.A.; Malham, S. Hole drilling in crab and gastropod shells by Eledone cirrhosa (Lamarck, 1798). Sci. Mar. 1997, 61, 67–76. [Google Scholar]
- Grisley, M.S.; Boyle, P.R.; Key, L.N. Eye puncture as a route of entry for saliva during predation on crabs by the octopus Eledone cirrhosa (Lamarck). J. Exp. Mar. Biol. Ecol. 1996, 202, 225–237. [Google Scholar] [CrossRef]
- Grisley, M.S. Separation and partial characterization of salivary enzymes expressed during prey handling in the octopus Eledone cirrhosa. Comp. Biochem. Physiol. 1993, 105B, 183–192. [Google Scholar] [CrossRef]
- Grisley, M.S.; Boyle, P.R. Bioassay and proteolytic activity of digestive enzymes from octopus saliva. Comp. Biochem. Physiol. 1987, 88, 1117–1123. [Google Scholar] [CrossRef]
- Nixon, M. Is there external digestion by Octopus? J. Zool. 1984, 202, 441–447. [Google Scholar] [CrossRef]
- Fry, B.G.; Roelants, K.; Norman, J.A. Tentacles of venom: Toxic protein convergence in the Kingdom Animalia. J. Mol. Evol. 2009, 68, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Ruder, T.; Sunagar, K.; Undheim, E.A.B.; Ali, S.A.; Wai, T.-C.; Low, D.H.W.; Jackson, T.N.W.; King, G.F.; Antunes, A.; Fry, B.G. Molecular phylogeny and evolution of the proteins encoded by coleoid (cuttlefish, octopus, and squid) posterior venom glands. J. Mol. Evol. 2013, 76, 192–204. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, D.C.; Aerts, M.; Brunain, M.; Desjardins, C.A.; Jacobs, F.J.; Werren, J.H.; Devreese, B. Insights into the venom composition of the ectoparasitoid wasp Nasonia vitripennis from bioinformatic and proteomic studies. Insect Mol. Biol. 2010, 19, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Fang, Q.; Wang, L.; Liu, J.; Zhu, Y.; Wang, F.; Li, F.; Werren, J.H.; Ye, G. Insights into the venom composition and evolution of an endoparasitoid wasp by combining proteomic and transcriptomic analyses. Sci. Rep. 2016, 6, 19604. [Google Scholar] [CrossRef] [PubMed]
- Veiga, A.B.G.; Ribeiro, J.M.C.; Guimarães, J.A.; Francischetti, I.M.B. A catalog for the transcripts from the venomous structures of the caterpillar Lonomia obliqua: Identification of the proteins potentially involved in the coagulation disorder and hemorrhagic syndrome. Gene 2005, 355, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Haney, R.A.; Ayoub, N.A.; Clarke, T.H.; Hayashi, C.Y.; Garb, J.E. Dramatic expansion of the black widow toxin arsenal uncovered by multi-tissue transcriptomics and venom proteomics. BMC Genom. 2014, 15, 366. [Google Scholar] [CrossRef] [PubMed]
- Haney, R.A.; Clarke, T.H.; Gadgil, R.; Fitzpatrick, R.; Hayashi, C.Y.; Ayoub, N.A.; Garb, J.E. Effects of gene duplication, positive selection and shifts in gene expression on the evolution of the venom gland transcriptome in widow spiders. Genome Biol. Evol. 2016, 8, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Santiago, P.B.; Assumpção, T.C.F.; Nunes de Araújo, C.; Marques Dourado Bastos, I.; Neves, D.; Garcia da Silva, I.; Charneau, S.; Queiroz, R.M.L.; Raiol, T.; de Araújo Oliveira, J.V.; et al. A deep insight into the sialome of Rhodnius neglectus, a cector of Chagas disease. PLoS Negl. Trop. Dis. 2016, 10, e0004581. [Google Scholar] [CrossRef] [PubMed]
- Dantuma, N.P.; Potters, M.; De Winther, M.P.J.; Tensen, C.P.; Kooiman, F.P.; Bogerd, J.; van der Horst, D.J. An insect homolog of the vertebrate very low density lipoprotein receptor mediates endocytosis of lipophorins. J. Lipid Res. 1999, 40, 973–978. [Google Scholar] [PubMed]
- Parra-Peralbo, E.; Culi, J. Drosophila lipophorin receptors mediate the uptake of neutral lipids in oocytes and imaginal disc cells by an endocytosis-independent mechanism. PLoS Genet. 2011, 7, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodenburg, K.W.; van der Horst, D.J. Lipoprotein-mediated lipid transport in insects: Analogy to the mammalian lipid carrier system and novel concepts for the functioning of LDL receptor family members. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2005, 1736, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Muhlia-Almazán, A.; Sánchez-Paz, A.; García-Carreño, F.L. Invertebrate trypsins: A review. J. Comp. Physiol. B 2008, 178, 655–672. [Google Scholar] [CrossRef] [PubMed]
- Yepiz-Plascencia, G.; Vargas-Albores, F.; Higuera-Ciapara, I. Penaeid shrimp hemolymph lipoproteins. Aquaculture 2000, 191, 177–189. [Google Scholar] [CrossRef]
- Koenemann, S.; Iliffe, T.M. Class Remipedia Yager, 1981. In Treatise on Zoology—Anatomy, Taxonomy, Biology. The Crustacea; von Vaupel Klein, J.C., Charmantier-Daures, M., Schram, F.R., Eds.; Brill: Leiden, The Netherlands, 2013; Volume 4, pp. 125–177. [Google Scholar]
- Fernandes-Pedrosa, M.; Junqueira-De-Azevedo, I.; Gonçalves-De-Andrade, R.; Kobashi, L.; Almeida, D.; Ho, P.; Tambourgi, D. Transcriptome analysis of Loxosceles laeta (Araneae, Sicariidae) spider venomous gland using expressed sequence tags. BMC Genom. 2008, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Baek, J.H.; Yoon, K.A. Differential properties of venom peptides and proteins in solitary vs. social hunting wasps. Toxins 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Moreau, S.; Asgari, S. Venom proteins from parasitoid wasps and their biological functions. Toxins 2015, 7, 2385–2412. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, P.G.; Beckmann, A.; Warnken, U.; Schnolzer, M.; Schuler, A.; Bornberg-Bauer, E.; Holstein, T.W.; Ozbek, S. Proteome of Hydra nematocyst. J. Biol. Chem. 2012, 287, 9672–9681. [Google Scholar] [CrossRef] [PubMed]
- Grisley, M.S.; Boyle, P.R. Chitinase, a new enzyme in octopus saliva. Comp. Biochem. Physiol. 1990, 95B, 311–316. [Google Scholar] [CrossRef]
- Schram, F.R.; Lewis, C.A. Functional morphology of feeding in the Nectiopoda. In Crustacean Issues 6. Functional Morphology of Feeding and Grooming in Crustacea; Felgenhauer, B.E., Watling, L., Thistle, A.B., Eds.; Balkema: Rotterdam, The Netherlands, 1989; pp. 115–122. [Google Scholar]
- Junqueira-de-Azevedo, I.L.M.; Campos, P.F.; Ching, A.T.C.; Mackessy, S.P. Colubrid venom composition: An -omics perspective. Toxins 2016, 8, 230. [Google Scholar] [CrossRef] [PubMed]
- Whittington, C.M.; Papenfuss, A.T.; Locke, D.P.; Mardis, E.R.; Wilson, R.K.; Abubucker, S.; Mitreva, M.; Wong, E.S.W.; Hsu, A.L.; Kuchel, P.W.; et al. Novel venom gene discovery in the platypus. Genome Biol. Evol. 2010, 11, R95. [Google Scholar] [CrossRef] [PubMed]
- Mcgivern, J.J.; Wray, K.P.; Margres, M.J.; Couch, M.E.; Mackessy, S.P.; Rokyta, D.R. RNA-seq and high-definition mass spectrometry reveal the complex and divergent venoms of two rear-fanged colubrid snakes. BMC Genom. 2014, 15, 1061. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Vidal, N.; van der Weerd, L.; Kochva, E.; Renjifo, C. Evolution and diversification of the Toxifera reptile venom system. J. Proteom. 2009, 72, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Pinto, J.R.A.; Fox, E.G.P.; Saidemberg, D.M.; Santos, L.D.; da Silva Menegasso, A.R.; Costa-Manso, E.; Machado, E.A.; Bueno, O.C.; Palma, M.S. Proteomic view of the venom from the fire ant Solenopsis invicta Buren. J. Proteome Res. 2012, 11, 4643–4653. [Google Scholar] [CrossRef] [PubMed]
- Casewell, N.R.; Visser, J.C.; Baumann, K.; Dobson, J.; Han, H.; Kuruppu, S.; Morgan, M.; Romilio, A.; Weisbecker, V.; Ali, S.A.; et al. The evolution of fangs, venom, and mimicry systems in blenny fishes. Curr. Biol. 2017, 27, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Undheim, E.A.; Mobli, M.; King, G.F. Toxin structures as evolutionary tools: Using conserved 3D folds to study the evolution of rapidly evolving peptides. Bioessays 2016, 38, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Bohlen, C.J.; Priel, A.; Zhou, S.; King, D.; Siemens, J.; Julius, D. A bivalent tarantula toxin activates the capsaicin receptor, TRPV1, by targeting the outer pore domain. Cell 2010, 141, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Chassagnon, I.R.; McCarthy, C.A.; Chin, Y.K.-Y.; Pineda, S.S.; Keramidas, A.; Mobli, M.; Pham, V.; De Silva, T.M.; Lynch, J.W.; Widdop, R.E.; et al. Potent neuroprotection after stroke afforded by a double-knot spider-venom peptide that inhibits acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. USA 2017, 114, 3750–3755. [Google Scholar] [CrossRef] [PubMed]
- Vassilevski, A.A.; Fedorova, I.M.; Maleeva, E.E.; Korolkova, Y.V.; Efimova, S.S.; Samsonova, O.V.; Schagina, L.V.; Feofanov, A.V.; Magazanik, L.G.; Grishin, E.V. Novel class of spider toxin: Active principle from the yellow sac spider Cheiracanthium punctorium venom is a unique two-domain polypeptide. J. Biol. Chem. 2010, 285, 32293–32302. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Stensvåg, K.; Haug, T.; Sperstad, S.V.; Rekdal, O.; Indrevoll, B.; Styrvold, O.B. Arasin 1, a proline-arginine-rich antimicrobial peptide isolated from the spider crab, Hyas araneus. Dev. Comp. Immunol. 2008, 32, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Kokryakov, V.N.; Harwig, S.S.; Panyutich, E.A.; Shevchenko, A.A.; Aleshina, G.M.; Shamova, O.V.; Korneva, H.A.; Lehrer, R.I. Protegrins: Leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993, 327, 231–236. [Google Scholar] [CrossRef]
- Paulsen, V.S.; Blencke, H.-M.; Benincasa, M.; Haug, T.; Eksteen, J.J.; Styrvold, O.B.; Scocchi, M.; Stensvåg, K. Structure-activity relationships of the antimicrobial peptide arasin 1—And mode of action studies of the N-terminal, proline-rich region. PLoS ONE 2013, 8, e53326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolov, Y.; Mirzabekov, T.; Martin, D.W.; Lehrer, R.I.; Kagan, B.L. Membrane channel formation by antimicrobial protegrins. Biochim. Biophys. Acta 1999, 1420, 23–29. [Google Scholar] [CrossRef]
- Chen, J.; Falla, T.J.; Liu, H.; Hurst, M.A.; Fujii, C.A.; Mosca, D.A.; Embree, J.R.; Loury, D.J.; Radel, P.A.; Cheng Chang, C.; et al. Development of protegrins for the treatment and prevention of oral mucositis: Structure-activity relationships of synthetic protegrin analogues. Biopolymers 2000, 55, 88–98. [Google Scholar] [CrossRef]
- Ebberink, R.H.M.; Smit, A.B.; Van Minnen, J. The insulin family: Evolution of structure and function in vertebrates and invertebrates. Biol. Bull. 1989, 117, 176–182. [Google Scholar] [CrossRef]
- Montagné, N.; Desdevises, Y.; Soyez, D.; Toullec, J.-Y. Molecular evolution of the crustacean hyperglycemic hormone family in ecdysozoans. BMC Evol. Biol. 2010, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Safavi-Hemami, H.; Lu, A.; Li, Q.; Fedosov, A.E.; Biggs, J.; Showers Corneli, P.; Seger, J.; Yandell, M.; Olivera, B.M. Venom insulins of cone snails diversify rapidly and track prey taxa. Mol. Biol. Evol. 2016, 33, 2924–2934. [Google Scholar] [CrossRef] [PubMed]
- Undheim, E.A.B.; Grimm, L.L.; Low, C.-F.; Morgenstern, D.; Herzig, V.; Zobel-Thropp, P.; Pineda, S.S.; Habib, R.; Dziemborowicz, S.; Fry, B.G.; et al. Weaponization of a hormone: Convergent recruitment of hyperglycemic hormone into the venom of arthropod predators. Structure 2015, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics. Available online: www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 April 2016).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Natural History Museum’s Data Portal. Available online: http://data.nhm.ac.uk/dataset/supplementary-data-remipede-toxins-paper (accessed on 10 July 2017).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- UniVec. Available online: ftp.ncbi.nlm.nih.gov/pub/UniVec/ (accessed on 1 April 2016).
- Emvec. Available online: ftp.ebi.ac.uk/pub/databases/emvec (accessed on 1 April 2016).
- Hoffmann, S.; Otto, C.; Kurtz, S.; Sharma, C.M.; Khaitovich, P.; Vogel, J.; Stadler, P.F.; Hackermüller, J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009, 5, e1000502. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.A.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using Tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M.; Campbell, L.I.; Richter, S.; Hering, L.; Sykes, D.; Hetmank, J.; Jenner, R.A.; Bleidorn, C. A polychaete’s powerful punch: Venom gland transcriptomics of Glycera reveals a complex cocktail of toxin homologs. Genome Biol. Evol. 2014, 6, 2406–2423. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; Heijne, G.V.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; Arnold, R.; Sebastian-Leon, P.; Martin-Rodriguez, S.; Tischler, P.; Jehl, M.A.; Dopazo, J.; Rattei, T.; Conesa, A. B2G-FAR, a species-centered GO annotation repository. Bioinformatics 2011, 27, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Han, M.V.; Zmasek, C.M. phyloXML: XML for evolutionary biology and comparative genomics. Bioinformatics 2009, 10, 356. [Google Scholar] [CrossRef] [PubMed]
- Kloepper, T.H.; Huson, D.H. Drawing explicit phylogenetic networks and their integration into SplitsTree. BMC Evol. Biol. 2008, 8, 22. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly Strategy | Raw Data | Clipped Data | Cleaned Data | Number of Secreted Proteins |
|---|---|---|---|---|
| “Raw” Contigs | Contigs > 137 nuc | Trimmomatic Seqclean | BLASTX vs. UniProt (SL0243) | |
| Venom gland transcriptome (library: 27,421,129 reads, paired end) | ||||
| SOAPdenovo-Trans (k-mer 31) | 294,931 | 177,668 | 176,408 | - |
| SOAPdenovo-Trans (k-mer 47) | 247,453 | 198,075 | 197,240 | 1799 1 |
| SOAPdenovo-Trans (k-mer 65) | 203.964 | 179,168 | 178,835 | - |
| Trinity (read length > 101) | 191,255 | 166,309 | 165,333 | 1943 1 |
| Body transcriptome (library: 9,165,598 reads, paired end) | ||||
| SOAPdenovo-Trans (k-mer 47) | 151,399 | 123,676 | 123,241 | 2376 1 |
| Trinity (read length > 101) | 203,113 | 161,511 | 161,100 | 3004 1 |
| Peptide Family | Structural Fold | Scaffold | Predicted Mature Length | Contigs VG | FPKM VG |
|---|---|---|---|---|---|
| Xibalbin 1 | ICK | xCx6Cx6CCx4CxCx6CxCx | 49 | 1 | 7138.77 |
| Xibalbin 2 | Putative ICK | Cx5Cx5Cx6Cx7CCx4CxCx8CxCx | 50 | 1 | 75,130.80 |
| Xibalbin 3 | Double ICK | xCx6Cx6CCx4Cx9–11C [x4] Cx6Cx6CCx4Cx9Cx | 74–79 | 2 | 11,366.77 |
| Xibalbin 4 | Putative CSαβ | xCx12Cx3Cx5Cx5Cx5CxCxC | 61 | 1 | 17,073.49 |
| Xibalbin 5 | Putative CSαβ | xCx14Cx8CxCx3Cx17CxCx4Cx10CCx | 87 | 1 | 1140.10 |
| Xibalbin 6 | ITP/CHH | xCx15Cx2Cx12Cx3Cx8Cx | 82 | 1 | 107.17 |
| Xibalbin 7 | sIGFBP-rp | Cx2Cx4Cx5Cx8Cx4Cx7Cx9Cx5CxCx2Cx2Cx6Cx4Cx | 77 | 2 | 1.96 |
| Xibalbin 8 | Unknown | xCx10Cx28CCx4Cx7Cx10Cx21Cx | 105 | 1 | 42.56 |
| Xibalbin 9 | Unknown | xCx5CxCx2Cx21Cx3Cx3Cx2Cx>50 | 114 | 1 | 24,334.56 |
| Xibalbin 10 | Unknown | xCxCx4CxCx | 21 | 2 | 37,579.07 |
| Xibalbin 11 | Unknown | No cysteines; two P-rich domains | 18/32 | 2 | 46,289.60 |
| Xibalbin 12 | Unknown | No cysteines; multiple ‘SIFQK’/‘FIFPK’ domains | 5 | 2 | 0 1 |
| Pancrustacean Group | Major Group | Species | TSA | SRA | No Matching Sequence |
|---|---|---|---|---|---|
| Major crustacean lineages | Thecostraca (Cirripedia) | Tetraclita japonica (OA) | SRR426837 | x | |
| Copepoda | Calanus finmarchicus | x | |||
| Tigriopus californicus | x | ||||
| Caligus rogercresseyi (OA) | SRR1232138 | ||||
| Caligus rogercresseyi (bad data) | x | ||||
| Lepeophtheirus salmonis (OA) | ERR262962 | ||||
| Lepeophtheirus salmonis (bad data) | x | x | |||
| Branchiura | Argulus siamensis (OA) | SRR514120 | |||
| Argulus foliaceus (OA) | SRR3183279 | ||||
| Decapoda | Procambarus clarkii | x | |||
| Astacus astacus | x | x | |||
| Carcinus maenas | x | ||||
| Eriocheir sinsensis | x | ||||
| Euphausia crystallorophias (OA) | ERR264582 | ||||
| Amphipoda | Ligia exotica (OA) | DRR054553 | |||
| Asellus aquaticus | x | ||||
| Armadillidium vulgare (OA) | SRR1324800 | ||||
| Hyalella azteca | x | ||||
| Branchiopoda | Triops newberryi | x | |||
| Early hexapod lineages | Protura | Acerentomon sp. AD-2013 | x | ||
| Diplura | Campodea augens | x | |||
| Occasjapyx japonicus | x | ||||
| Collembola | Tetrodontophora bielanensis | x | |||
| Anurida maritima | x | ||||
| Folsomia candida | x | x | |||
| Sminthurus viridis | x | ||||
| Pogonognathellus sp. AD-2013 | x | ||||
| Archaeognatha | Machilis hrabei | x | |||
| Zygentoma | Thermobia domestica | x | |||
| Odonata | Calopteryx splendens | x | x |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Von Reumont, B.M.; Undheim, E.A.B.; Jauss, R.-T.; Jenner, R.A. Venomics of Remipede Crustaceans Reveals Novel Peptide Diversity and Illuminates the Venom’s Biological Role. Toxins 2017, 9, 234. https://doi.org/10.3390/toxins9080234
Von Reumont BM, Undheim EAB, Jauss R-T, Jenner RA. Venomics of Remipede Crustaceans Reveals Novel Peptide Diversity and Illuminates the Venom’s Biological Role. Toxins. 2017; 9(8):234. https://doi.org/10.3390/toxins9080234
Chicago/Turabian StyleVon Reumont, Björn M., Eivind A. B. Undheim, Robin-Tobias Jauss, and Ronald A. Jenner. 2017. "Venomics of Remipede Crustaceans Reveals Novel Peptide Diversity and Illuminates the Venom’s Biological Role" Toxins 9, no. 8: 234. https://doi.org/10.3390/toxins9080234
APA StyleVon Reumont, B. M., Undheim, E. A. B., Jauss, R. -T., & Jenner, R. A. (2017). Venomics of Remipede Crustaceans Reveals Novel Peptide Diversity and Illuminates the Venom’s Biological Role. Toxins, 9(8), 234. https://doi.org/10.3390/toxins9080234