
Spider Neurotoxins, Short Linear Cationic Peptides and Venom Protein Classification Improved by an Automated Competition between Exhaustive Profile HMM Classifiers

Abstract
:1. Introduction
2. Results
2.1. New Classification System and Exhaustive Classifiers for Spider Venom Components
2.2. Spider Peptide Classification is Improved
2.3. Hmmcompete, an Add-On to the HMMER3 Suite
2.3.1. Synopsis
2.3.2. Description
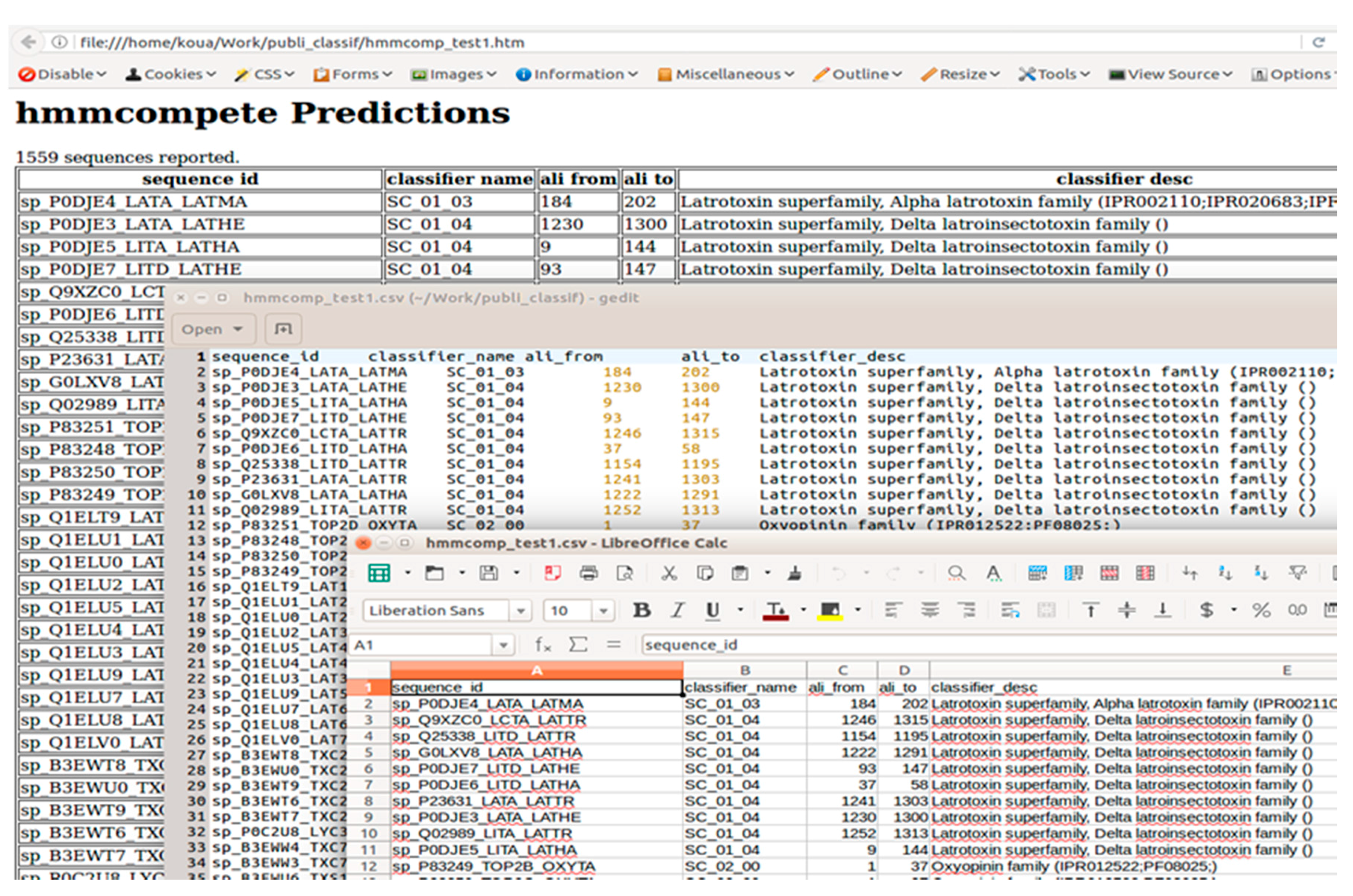
2.3.3. Output Description
- sequence_id: the first part of target sequence FASTA header from the > sign to the first space.
- classifier_name: name of the profile giving the higher bit score for the considered peptide sequence.
- ali_from: the position in the target sequence at which the best hit starts.
- ali_to: the position in the target sequence at which the best hit ends.
- target_region: display region of the target sequence that matched the best model hit (region going from ali_from to ali_to). Only available if the --pepreg option is set.
- classifier_desc: description line (DESC) from the best classifier when available in the concerned HMM entry of the <hmmDb>. Only available if the --desc (-d) option is set.
- matches_count: number of models from the <hmmDb> producing a valuable match for the target peptide sequence. Only available if the --altpred option is set.
- matches_position: a list of all HMMs in <hmmDb> producing a match. Each hit is described by giving some useful details formatted as (ali_from-ali_to#classifier_name#bit_score). Only available if the --altpred option is set.
2.3.4. hmmcompete Use Example
3. Discussion
3.1. Precision and Evolution of the Proposed Classification System
3.2. hmmcompete Allows Taking Advantage of New Classifiers
4. Conclusions
5. Materials and Methods
5.1. Data Acquisition
5.2. Model Construction
5.3. Profile HMM-Based Classification
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maggio, F.; Sollod, B.L.; Tedford, H.W.; Herzig, V.; King, G.F. Insect Pharmacology: Channels, Receptors, Toxins and Enzymes; Gilbert, L.I., Gill, S.S., Eds.; Academic Press: Oxford, UK, 2010; pp. 101–123. [Google Scholar]
- Bohlen, C.J.; Julius, D. Receptor-targeting mechanisms of pain-causing toxins: How ow? Toxicon 2012, 60, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Saez, N.J.; Senff, S.; Jensen, J.E.; Er, S.Y.; Herzig, V.; Rash, L.D.; King, G.F. Spider-venom peptides as therapeutics. Toxins 2010, 2, 2851–2871. [Google Scholar] [CrossRef] [PubMed]
- Windley, M.J.; Herzig, V.; Dziemborowicz, S.A.; Hardy, M.C.; King, G.F.; Nicholson, G.M. Spider-venom peptides as bioinsecticides. Toxins 2012, 4, 191–227. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Lau, C.H.Y.; Herzig, V.; Ikonomopoulou, M.P.; Rash, L.D.; King, G.F. Venoms to Drugs: Therapeutic Applications of Spider-Venom Peptides; King, G.F., Ed.; Royal Society of Chemistry: Cambridge, UK, 2015; pp. 221–244. [Google Scholar]
- Samy, R.P.; Stiles, B.G.; Franco, O.L.; Sethi, G.; Lim, L.H.K. Animal venoms as antimicrobial agents. Biochem. Pharmacol. 2017, 134, 127–138. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium/ToxProt Project. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Herzig, V.; Wood, D.L.A.; Newell, F.; Chaumeil, P.A.; Kaas, Q.; Binford, G.J.; Nicholson, G.M.; Gorse, D.; King, G.F. ArachnoServer 2.0, an updated online resource for spider toxin sequences and structures. Nucleic Acids Res. 2011, 39, D653–D657. [Google Scholar] [CrossRef] [PubMed]
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.-Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017—Beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Oldrati, V.; Koua, D.; Allard, P.M.; Hulo, N.; Arrell, M.; Nentwig, W.; Lisacek, F.; Wolfender, J.L.; Kuhn-Nentwig, L.; Stöcklin, R. Peptidomic and transcriptomic profiling of four distinct spider venoms. PLoS ONE 2017, 12, e0172966. [Google Scholar] [CrossRef] [PubMed]
- Naamati, G.; Askenazi, M.; Linial, M. ClanTox: A classifier of short animal toxins. Nucleic Acids Res. 2009, 37, W363–W368. [Google Scholar] [CrossRef] [PubMed]
- Lavergne, V.; Alewood, P.F.; Mobli, M.; King, G.F. Venoms to Drugs: The Structural Universe of Disulfide-Rich Venom Peptides; King, G.F., Ed.; Royal Society of Chemistry: Cambridge, UK, 2015; pp. 37–79. [Google Scholar]
- Koua, D.; Brauer, A.; Laht, S.; Kaplinski, L.; Favreau, P.; Remm, M.; Lisacek, F.; Stöcklin, R. ConoDictor: A tool for prediction of conopeptide superfamilies. Nucleic Acids Res. 2012, 40, W238–W241. [Google Scholar] [CrossRef] [PubMed]
- Laht, S.; Koua, D.; Kaplinski, L.; Remm, M.; Stöcklin, R. Identification and classification of conopeptides using hidden Markov models. Biochim. Biophys. Acta 2012, 1824, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Durbin, R.; Eddy, S.R.; Krogh, A.; Mitchison, G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform. 2010, 11, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| InterPro Signature Combination | Pfam HMM | ToxProt First Classification Level (TPL1) * | Total of Annotated Sequences in ToxProt |
|---|---|---|---|
| IPR000737; IPR011052 | PF00299 | Protease inhibitor I7 (squash-type serine protease inhibitor) family | 1 |
| IPR002110; IPR020683 | PF00023; PF12796 | Latrotoxin superfamily | 4 |
| IPR002110; IPR020683; IPR013829 | PF00023; PF12796; PF13606 | Latrotoxin superfamily | 3 |
| IPR002223 | PF00014 | Venom Kunitz-type family | 39 |
| IPR002223; IPR020901 | PF00014 | Venom Kunitz-type family | 8 |
| IPR003614 | 1 | ||
| IPR004169 | Plectoxin superfamily (16) | 18 | |
| Spider toxin CSTX superfamily (1) | |||
| No class (1) | |||
| IPR004214 | PF02950 | Spider toxin Tx2 family (1) | 2 |
| Huwentoxin-1 family (1) | |||
| IPR005853; IPR013605 | PF08396 | Omega-agatoxin superfamily | 13 |
| IPR007733; IPR027300 | PF05039 | Spider agouti family | 1 |
| IPR008017 | PF05353 | Delta-atracotoxin family | 7 |
| IPR008197 | Spider wap-1 family(17) | 21 | |
| Spider wap-2 family (4) | |||
| IPR009243 | Beta/delta-agatoxin family | 12 | |
| IPR009243; IPR004169 | Beta/delta-agatoxin family | 2 | |
| IPR009415 | PF06357 | Shiva superfamily | 13 |
| IPR009415; IPR018071 | PF06357 | Shiva superfamily (14) | 15 |
| No class (1) | |||
| IPR011142 | Spider toxin CSTX superfamily | 6 | |
| IPR011696 | PF07740 | Huwentoxin-1 family | 114 |
| IPR011696; IPR013140 | PF07740 | Huwentoxin-1 family | 119 |
| IPR011696; IPR016191 | PF07740 | Huwentoxin-1 family | 4 |
| IPR012499 | PF07945 | Shiva superfamily | 7 |
| IPR012522 | PF08025 | Oxyopinin-2 family | 4 |
| IPR012625 | PF08089 | Huwentoxin-2 family | 79 |
| IPR012625; IPR012627 | PF08092 | Magi-1 superfamily | 1 |
| IPR012626 | PF08091 | Insecticidal toxin ABC family | 5 |
| IPR012627 | PF08092 | Magi-1 superfamily | 82 |
| IPR012628 | PF08093 | Magi-5 family | 3 |
| IPR012633 | PF08115 | Spider toxin SFI family | 10 |
| IPR012634 | PF08116 | Spider neurotoxin 21C2 family | 4 |
| IPR013139; IPR012628 | PF08093 | Omega-atracotoxin type 2 family | 5 |
| IPR013605 | PF08396 | Omega-agatoxin superfamily | 14 |
| IPR016328; IPR009243 | Beta/delta-agatoxin family | 13 | |
| IPR017946 | Arthropod phospholipase D family | 199 | |
| IPR017946; IPR000909 | Arthropod phospholipase D family | 2 | |
| IPR018802 | PF10279 | Latarcin superfamily | 11 |
| IPR019553 | PF10530 | Plectoxin superfamily (1) | 62 |
| Spider toxin CSTX superfamily (6) | |||
| U6-lycotoxin family(10) | |||
| U7-lycotoxin family (11) | |||
| U8-lycotoxin family (28) | |||
| U11-lycotoxin family (6) | |||
| IPR019553; IPR004169 | PF10530 | U10-lycotoxin family | 5 |
| IPR019553; IPR011142 | PF10530 | Spider toxin CSTX superfamily | 104 |
| IPR020683 | Latrotoxin superfamily | 1 | |
| IPR020683; IPR007094 | Latrotoxin superfamily | 1 | |
| IPR023569 | PF06607 | AVIT (prokineticin) family | 9 |
| IPR024079; IPR001506; IPR006026 | PF01400 | Peptidase M12A family | 1 |
| IPR027300 | Plectoxin superfamily (5) | 6 | |
| No class (1) | |||
| IPR027300; IPR004169 | No class | 1 | |
| IPR034035; IPR024079; IPR001506; IPR006026 | PF01400 | Peptidase M12A family | 4 |
| ToxProt Family | Number of Sequences |
|---|---|
| Aptotoxin family | 4 |
| Arthropod phospholipase D family | 13 |
| AVIT (prokineticin) family | 1 |
| Bradykinin-related peptide family | 5 |
| Cupiennin family | 43 |
| Cytoinsectotoxin family | 20 |
| Helical arthropod-neuropeptide-derived (HAND) family | 3 |
| Huwentoxin-1 family | 12 |
| HWTX-LSTX family | 2 |
| Insecticidal toxin DTX family | 3 |
| JZTX-72 family | 3 |
| Latrotoxin superfamily | 1 |
| Litx family | 3 |
| Magi-1 superfamily | 1 |
| Omega-agatoxin superfamily | 2 |
| Omega-lycotoxin family | 7 |
| Phrixotoxin family | 13 |
| Plectoxin superfamily | 7 |
| Shiva superfamily | 2 |
| Spider agouti family | 9 |
| Spider LiTx3-related peptide family | 2 |
| Spider toxin CSTX superfamily | 6 |
| Spider toxin Tx2 family | 7 |
| Spider toxin Tx3-6 family | 7 |
| Spider wap-1 family | 1 |
| U12-lycotoxin family | 6 |
| U2-agatoxin family | 24 |
| Venom metalloproteinase (M12B) family | 1 |
| No family name | 114 |
| Toxin Type (Level 1) | Classifiers * (Level 2 and 3) | Discriminative ToxProt Annotation | InterPro Signatures | Pfam HMMs |
|---|---|---|---|---|
| Spider Cationic peptides (SC) 21 profile HMMs | SC_01_00 | Cytoinsectotoxin family | ||
| SC_02_00 | Oxyopinin family | IPR012522 | PF08025 | |
| SC_03_00 to SC_03_07 | Latarcin superfamily | IPR018802 | PF10279 | |
| SC_04_01 to SC_04_10 | Cupiennin family | |||
| CsTx-16 ** | ||||
| SC_05_00 | Bradykinin-related peptide family | |||
| Spider Neurotoxin (SN) 170 profile HMM | SN_01_00 | U2-agatoxin family | ||
| SN_02_00 to SN_02_09 | Plectoxin superfamily | IPR004169 | ||
| CsTx-19 **, CsTx-28,34,36 *** | ||||
| SN_03_01 to SN_03_06 | Spider toxin Tx2 family | IPR004214 | PF02950 | |
| SN_04_00 to SN_04_04 | Omega-agatoxin superfamily | IPR005853; IPR013605 | PF08396 | |
| SN_05_00 to SN_05_06 | Spider agouti family | IPR007733; IPR027300 | PF05039 | |
| SN_06_00 | Delta-atracotoxin family | IPR008017 | PF05353 | |
| SN_07_00 to SN_07_04 | Beta/delta agatoxin family | IPR009243 | ||
| SN_08_01 to SN_08_02 | Shiva superfamily, Omega-toxin family | IPR009415; IPR018071 | PF06357 | |
| SN_09_00 | Spider toxin Tx3-6 family | |||
| SN_10_00 to SN_10_67 | Huwentoxin-1 family | IPR011696 | PF07740 | |
| SN_11_00 | Shiva superfamily, Kappa toxin family | IPR012499 | PF07945 | |
| SN_12_01 to SN_12_08 | Huwentoxin-2 family | IPR012625 | PF08089 | |
| SN_13_00 to SN_13_03 | Insecticidal toxin ABC family | IPR012626 | PF08091 | |
| SN_14_00 to SN_14_09 | Magi-1 superfamily | IPR012627 | PF08092 | |
| SN_15_01 to SN_15_02 | Magi-5 family | IPR012628 | PF08093 | |
| SN_16_00 | Spider toxin SFI family | IPR012633 | PF08115 | |
| SN_17_00 | Spider neurotoxin 21C2 family | IPR012634 | PF08116 | |
| SN_18_00 | AVIT (prokineticin) family | IPR023569 | PF06607 | |
| SN_19_00 to SN_19_12 | Spider toxin CsTx superfamily | IPR019553; IPR011142 | PF10530 | |
| SN_20_00 | CsTx-20 ** | |||
| SN_21_00 | Aptotoxin_family | |||
| SN_22_00 | Helical arthropod neuropeptide derived (HAND) family | |||
| SN_23_00 | Double-knot toxin subfamily | |||
| SN_24_00 | OAIP 4 subfamily | |||
| SN_25_00 | HWTX-LSTX family | |||
| SN_26_00 | Insecticidal toxin DTX family | |||
| SN_27_00 | JZTX-72 family | |||
| SN_28_00 | Litx family | |||
| SN_29_00 | Omega lycotoxin family | |||
| SN_30_00 | Phrixotoxin family | |||
| SN_31_00 | U12-lycotoxin family | |||
| SN_32_00 to SN_32_02 | MIT-like AcTx family ** | IPR020202 | PF17556 | |
| CsTx-21 **, CsTx-22 *** | ||||
| SN_33_00 | CsTx-26 *** | |||
| SN_34_00 | CsTx-29 *** | |||
| SN_35_00 | CsTx-35 *** | |||
| SN_36_00 | Huwentoxin type 10 ** | |||
| SN_37_00 | CsTx-37 ** | |||
| SN_38_00 | CsTx-38 ** | |||
| SN_39_00 | Spider LiTx3 related peptide family | |||
| SN_40_00 | Spiderine ** | |||
| Venom Proteins (VP) 28 profile HMMs | VP_01_00 | Protease inhibitor I7 (squash type serine protease inhibitor) family | IPR000737; IPR011052 | PF00299 |
| VP_02_00 | Peptidase M12A family | IPR024079; IPR001506; | PF01400 | |
| VP_03_01 to VP_03_02 | Arthropod phospholipase D family | IPR017946 | ||
| VP_04_00 | Venom metalloproteinase (M12B) family | |||
| VP_05_00 | Hyaluronidase ** | IPR018155 | ||
| VP_06_00 | Arthropod Phospholipase A2 ** | IPR001211 | ||
| VP_07_00 | Angiotensin-converting Enzyme ** | IPR033591 | ||
| VP_08_00 | Peptidylglycine alpha-amidating monooxygenase ** | IPR000720 | ||
| VP_09_00 | Signal peptidase ** | IPR001733 | ||
| VP_10_00 | Venom serine protease *** | IPR001314 | ||
| VP_11_01 to VP_11_02 | Spider WAP family | IPR008197 | ||
| VP_12_01 to VP_12_04 | Venom Kunitz-type family | IPR002223; IPR020901 | PF00014 | |
| VP_13_01 to VP_13_02 | Cysteine-rich secretory protein | IPR014044; | ||
| IPR002413 | ||||
| VP_14_00 | Thyroglobulin-like protein ** | IPR000716 | ||
| VP_15_00 | Leucine rich peptide ** | IPR032675 | ||
| VP_16_00 | Protein disulfide-isomerase ** | IPR005792 | ||
| VP_17_00 | Tachylectin 5A ** | IPR002181 | ||
| VP_18_00 | Cystatin ** | IPR027214 | ||
| VP_19_01 to VP_19_04 | Latrotoxin superfamily | IPR002110; IPR020683 | PF00023 |
| Option Name | Description |
|---|---|
| --hmm <hmmDbPath> | The profile database to be used for sequence classification. HMMER3 profiles. This is a mandatory argument. |
| -i or --in <seqFastaDb> | The sequence database to be classified. In FASTA format. This is a mandatory argument. |
| -h or --help | Help. Print a brief reminder of command line usage and available options. hmmcompete help page will also be displayed if the command is executed without any argument. |
| -v or --version | Print hmmcompete version and exit. |
| -o <file_path> or --out <file_path> | Direct the main tabular output to a file <f> instead of the default stdout. |
| -d or --desc | Display profile description in the main output when the description is present in the profile. Default: ‘Off’. |
| --altpred | Display number of alternative profile HMM matching a sequence, as well as a summarized description of each alternative match. This description includes positions of the query sequence matching the profile, as well as the produced score. Default: ‘Off’. |
| --allseq | Also report sequences not matched by any model. Default: Off, i.e., only query sequences matched by a profile in hmmDB are reported by default. |
| --pepreg | Display region of the target sequence that matched the reported profileHMM. Default: ‘Off’. |
| --hsout <file_path> | Save an output file similar to that of hmmsearch with the --domtblout option. Will only report the best prediction/classification where available. Sequences not matched by any model are not reported. Alternative profile HMM matches are also ignored. |
| --htmout <file_path> | Save an HTML version of the output. May be useful for web integration. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koua, D.; Kuhn-Nentwig, L. Spider Neurotoxins, Short Linear Cationic Peptides and Venom Protein Classification Improved by an Automated Competition between Exhaustive Profile HMM Classifiers. Toxins 2017, 9, 245. https://doi.org/10.3390/toxins9080245
Koua D, Kuhn-Nentwig L. Spider Neurotoxins, Short Linear Cationic Peptides and Venom Protein Classification Improved by an Automated Competition between Exhaustive Profile HMM Classifiers. Toxins. 2017; 9(8):245. https://doi.org/10.3390/toxins9080245
Chicago/Turabian StyleKoua, Dominique, and Lucia Kuhn-Nentwig. 2017. "Spider Neurotoxins, Short Linear Cationic Peptides and Venom Protein Classification Improved by an Automated Competition between Exhaustive Profile HMM Classifiers" Toxins 9, no. 8: 245. https://doi.org/10.3390/toxins9080245
APA StyleKoua, D., & Kuhn-Nentwig, L. (2017). Spider Neurotoxins, Short Linear Cationic Peptides and Venom Protein Classification Improved by an Automated Competition between Exhaustive Profile HMM Classifiers. Toxins, 9(8), 245. https://doi.org/10.3390/toxins9080245