Microfluidic Device for Screening for Target Cell-Specific Binding Molecules by Using Adherent Cells

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microfluidic Device for Screening for Target Cell-Specific Binding Molecules
2.1.1. Fabrication Process of the Microfluidic Device
2.1.2. Design of the Microfluidic Device
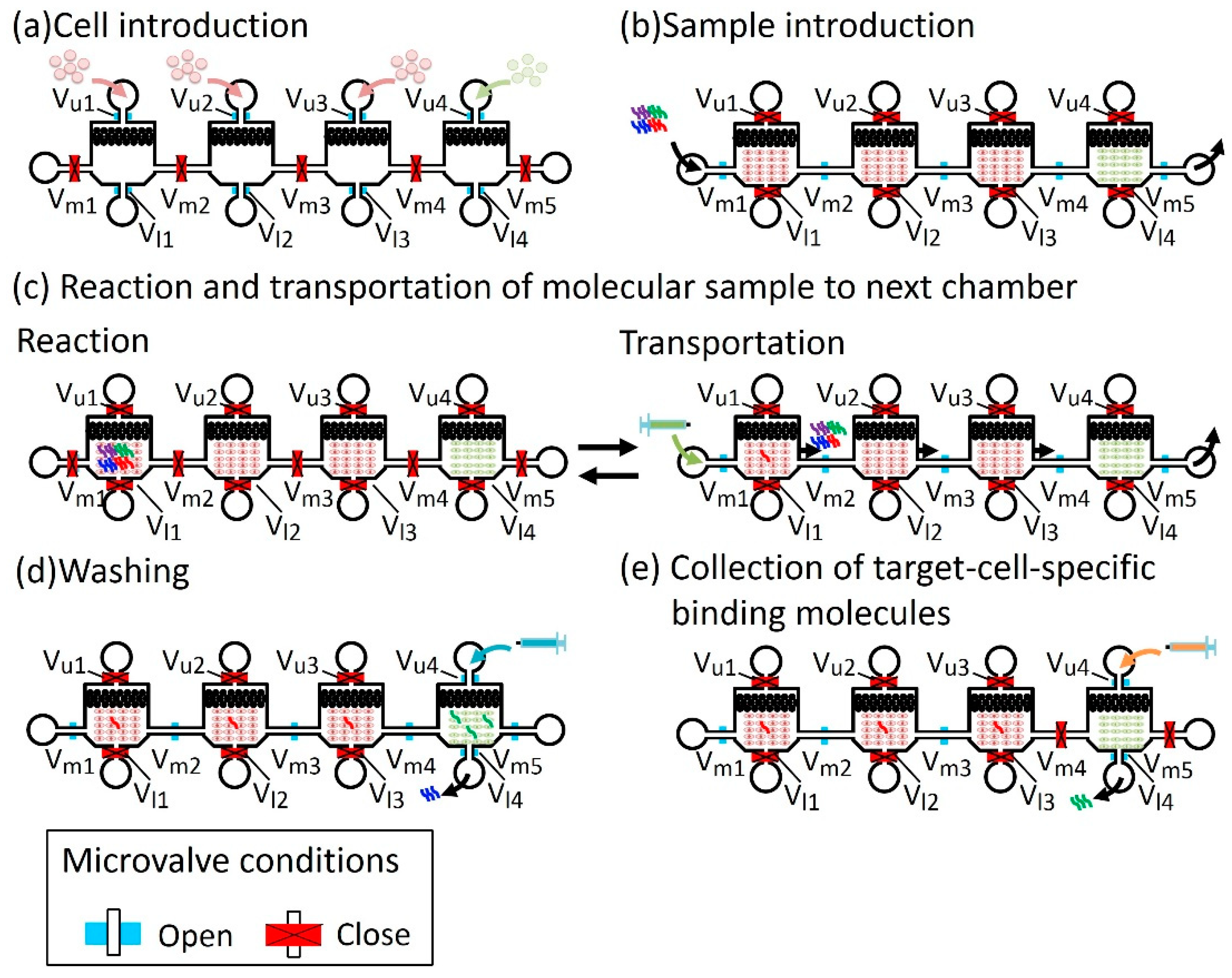
2.1.3. Operation of the Microfluidic Device
2.1.4. Cells
2.1.5. Reagent
2.2. Experimental Procedure
2.2.1. Experimental Setup
2.2.2. Filtering Non-Specific antibodies (Abs)
2.2.3. Collecting Target-Specific Antibodies (Abs)
3. Results and Discussion
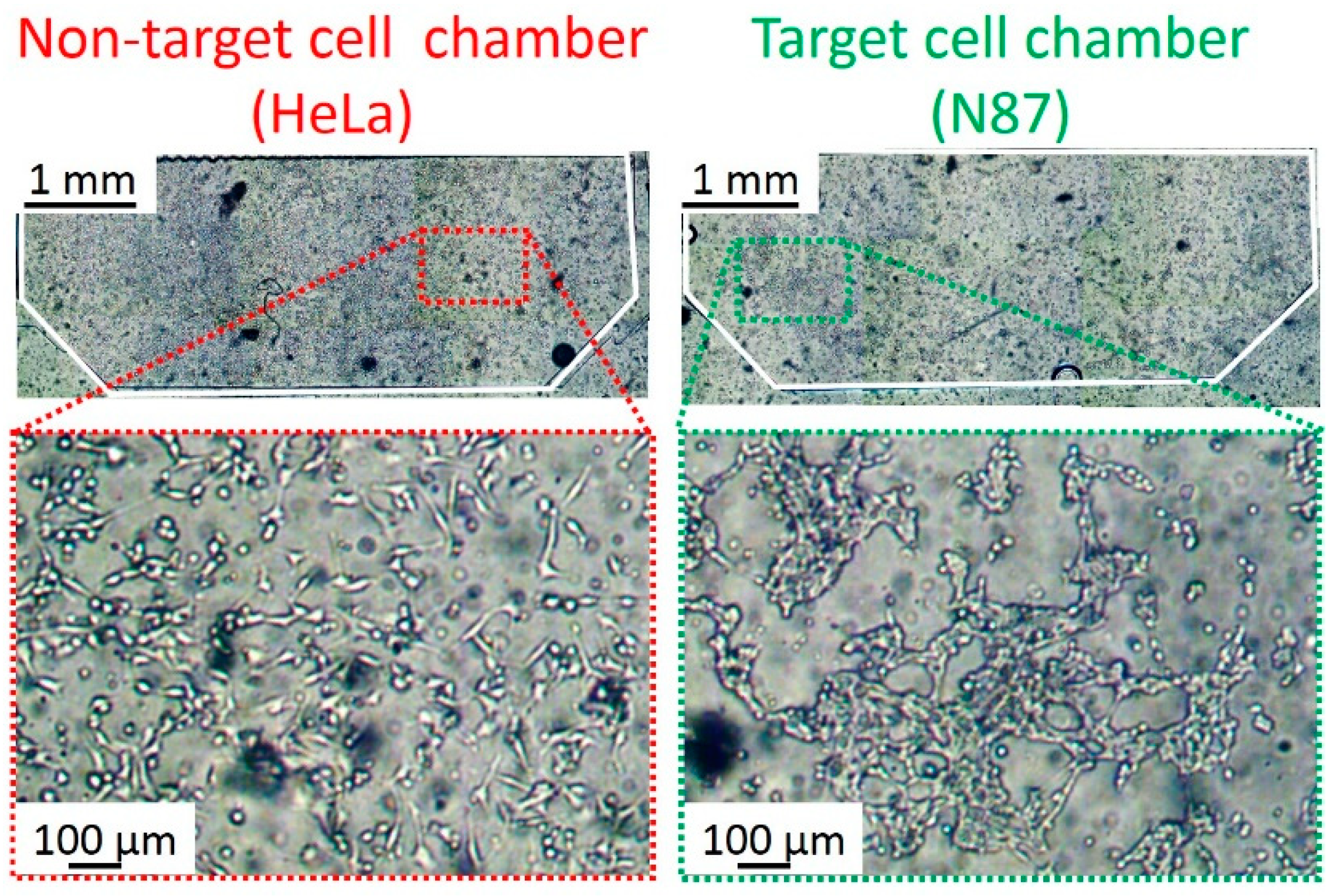
3.1. Results of Filtering Non-Specific Antibodies (Abs)
3.2. Results of Collecting Target-Specific Antibodies (Abs)
3.3. Future Prospects on the Development of the Device
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- DeVita, V.T., Jr.; Chu, E. A History of Cancer Chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, T.M.; Cullis, P.R. Drug Delivery Systems: Entering the Mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef] [PubMed]
- Boekhout, A.H.; Beijnen, J.H.; Schellens, J.H. Trastuzumab. Oncologist 2011, 16, 800–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danhier, F.; Breton, A.L.; Préat, V. RGD-Based Strategies to Target Alpha(v) Beta (3) Integrin in Cancer Therapy and Diagnosis. Mol. Pharmaceutics 2012, 9, 2916–2973. [Google Scholar] [CrossRef] [PubMed]
- Henri, J.L.; Macdonald, J.; Strom, M.; Duan, W.; Shigdar, S. Aptamers as potential therapeutic agents for ovarian cancer. Biochimie 2018, 145, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Ishima, Y.; Chen, D.; Fang, J.; Maeda, H.; Minomo, A.; Kragh-Hansen, U.; Kai, T.; Maruyama, T.; Otagiri, M. S-Nitrosated Human Serum Albumin Dimer is not only a Novel Anti-Tumor Drug but also a Potentiator for Anti-Tumor Drugs with Augmented EPR Effects. Bioconjugate Chem. 2012, 23, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Wanga, J.; Liu, Y.; Teesalu, T.; Sugahara, K.N.; Kotamrajua, V.R.; Adams, J.D.; Ferguson, B.S.; Gong, Q.; Oh, S.S.; Csordas, A.T.; et al. Selection of phage-displayed peptides on live adherent cells in microfluidic channels. Proc. Natl. Acad. Sci. 2011, 108, 6909–6914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, L.-Y.; Wang, C.-H.; Hsu, K.-F.; Chou, C.-Y.; Lee, G.-B. An on-chip Cell-SELEX process for automatic selection of high-affinity aptamers specific to different histologically classified ovarian cancer cells. Lab Chip 2014, 14, 4017–4028. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.-J.; Wu, H.-W.; Hung, L.-Y.; Liu, C.-A.; Chang, H.-Y.; Wang, K.; Lee, G.-B. An integrated microfluidic system for screening of phage-displayed peptides specific to colon cancer cells and colon cancer stem cells. Biomicrofluidics 2015, 9, 054121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, C.-H.; Hsieh, I.-S.; Hung, L.-Y.; Lin, H.-I.; Shiesh, S.-C.; Chen, Y.-L.; Lee, G.-B. An automatic microfluidic system for rapid screening of cancer stem-like cell-specific aptamers. Microfluid. Nanofluid. 2013, 14, 753–765. [Google Scholar] [CrossRef]
- Wang, C.-H.; Weng, C.-H.; Che, Y.-J.; Wang, K.; Lee, G.-B. Cancer Cell-Specific Oligopeptides Selected by an Integrated Microfluidic System from a Phage Display Library for Ovarian Cancer Diagnosis. Theranostics 2015, 5, 431–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminaga, M.; Ishida, T.; Kadonosono, T.; Kizaka-Kondoh, S.; Omata, T. Uniform Cell Distribution Achieved by Using Cell Deformation in a Micropillar Array. Micromachines 2015, 6, 409–422. [Google Scholar] [CrossRef] [Green Version]
- Kaminaga, M.; Ishida, T.; Omata, T. Fabrication of Pneumatic Microvalve for Tall Microchannel Using Inclined Lithography. Micromachines 2016, 7, 224. [Google Scholar] [CrossRef] [PubMed]
- Nowak, J.E.; Chatterjee, M.; Mohapatra, S.; Dryde, S.C.; Tainsky, M.A. Direct production and purification of T7 phage display cloned proteins selected and analyzed on microarrays. BioTechniques 2006, 40, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Kurosawa, O. A Three-dimensional Cell Culture Method with a Micromesh Sheet and Its Application to Hepatic Cells. Tissue Eng. Part C: Methods 2018, 24. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Cell Lines | Expressing Surface Molecules | ||
|---|---|---|---|
| Target cells | N87 cell lines | HER2 (Target molecules) | Integrin αvβ5 |
| Non-target cells | HeLa cell lines | - | Integrin αvβ5 |
| Name of Molecules | Binding Cells | Fluorescent Dye | ||
|---|---|---|---|---|
| Target cell-specific binding molecules | Anti-HER2 antibody (Target-specific Ab) | N87 cells (Target cells) | - | AF 488 (Green fluorescence) |
| Non-target cell-binding molecules | Anti-inegrin antibody (Non-specific Ab) | N87 cells (Target cells) | HeLa cells (Non-target cells) | AF 555 (Red fluorescence) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaminaga, M.; Ishida, T.; Kadonosono, T.; Kizaka-Kondoh, S.; Omata, T. Microfluidic Device for Screening for Target Cell-Specific Binding Molecules by Using Adherent Cells. Micromachines 2019, 10, 41. https://doi.org/10.3390/mi10010041
Kaminaga M, Ishida T, Kadonosono T, Kizaka-Kondoh S, Omata T. Microfluidic Device for Screening for Target Cell-Specific Binding Molecules by Using Adherent Cells. Micromachines. 2019; 10(1):41. https://doi.org/10.3390/mi10010041
Chicago/Turabian StyleKaminaga, Maho, Tadashi Ishida, Tetsuya Kadonosono, Shinae Kizaka-Kondoh, and Toru Omata. 2019. "Microfluidic Device for Screening for Target Cell-Specific Binding Molecules by Using Adherent Cells" Micromachines 10, no. 1: 41. https://doi.org/10.3390/mi10010041
APA StyleKaminaga, M., Ishida, T., Kadonosono, T., Kizaka-Kondoh, S., & Omata, T. (2019). Microfluidic Device for Screening for Target Cell-Specific Binding Molecules by Using Adherent Cells. Micromachines, 10(1), 41. https://doi.org/10.3390/mi10010041