Catalytic Hydrolysis of Phosphate Monoester by Supramolecular Complexes Formed by the Self-Assembly of a Hydrophobic Bis(Zn2+-cyclen) Complex, Copper, and Barbital Units That Are Functionalized with Amino Acids in a Two-Phase Solvent System

 and
and 
Abstract
: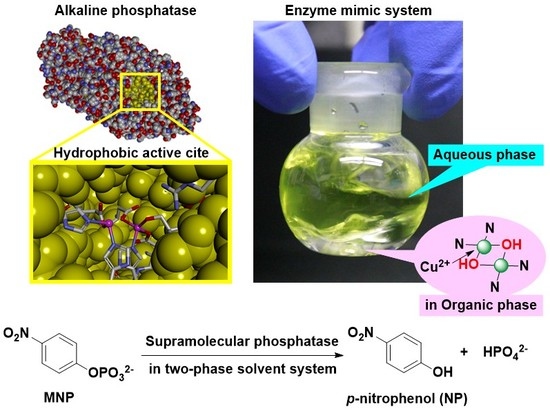
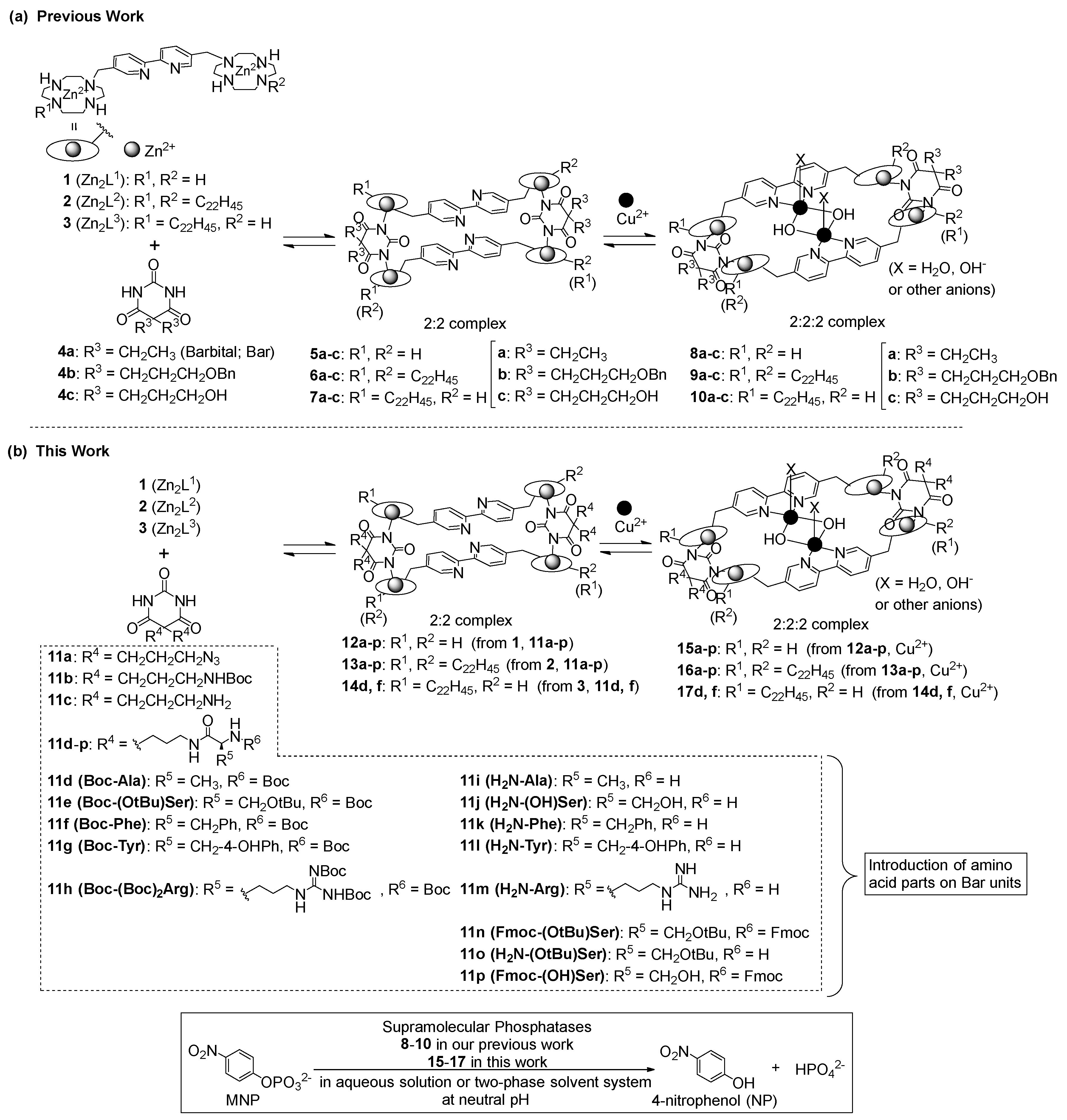
1. Introduction
2. Materials and Methods
2.1. General Information
2.2. Synthesis of Compounds
2.3. Hydrolysis of MNP
3. Results and Discussion
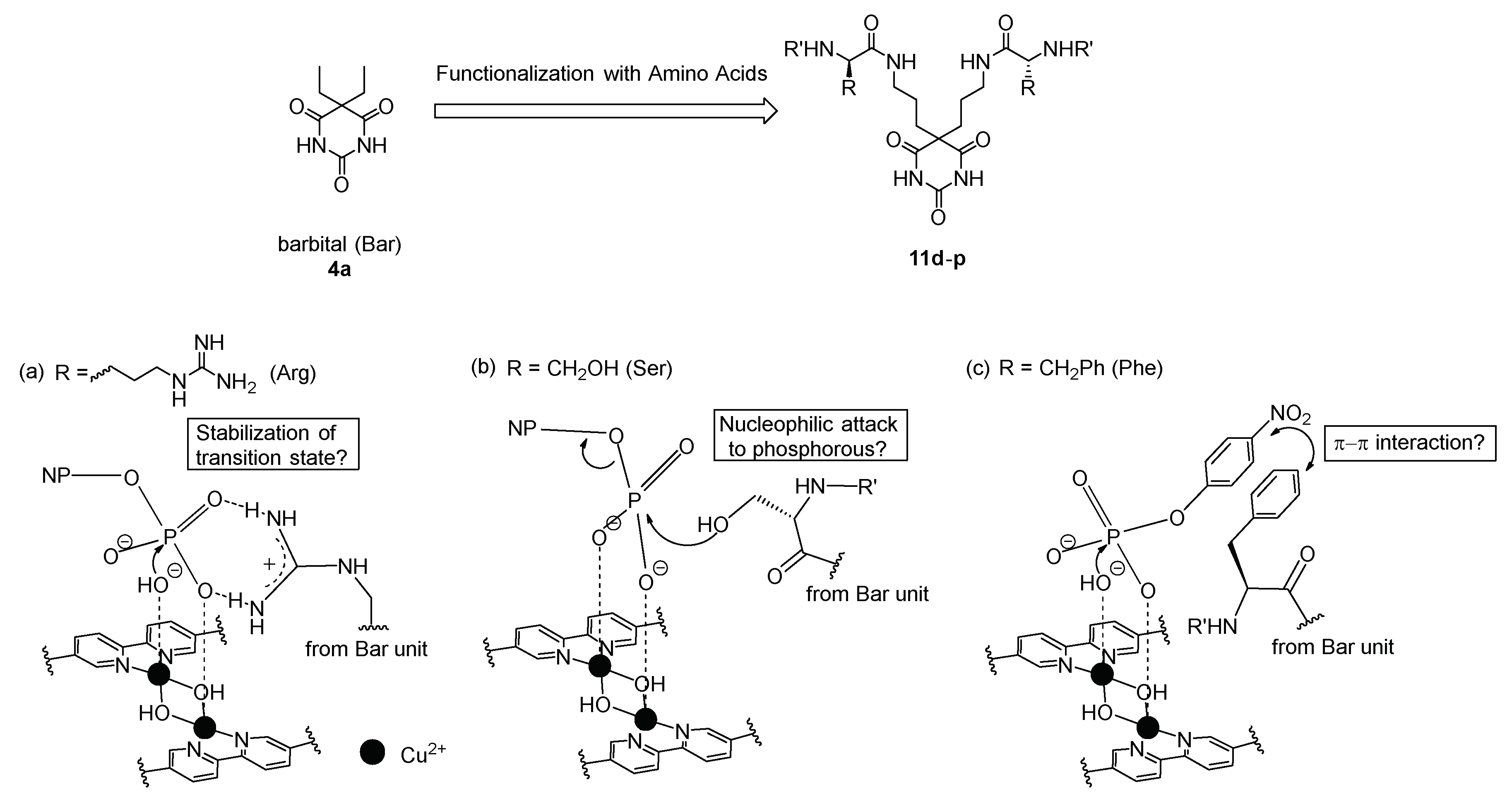
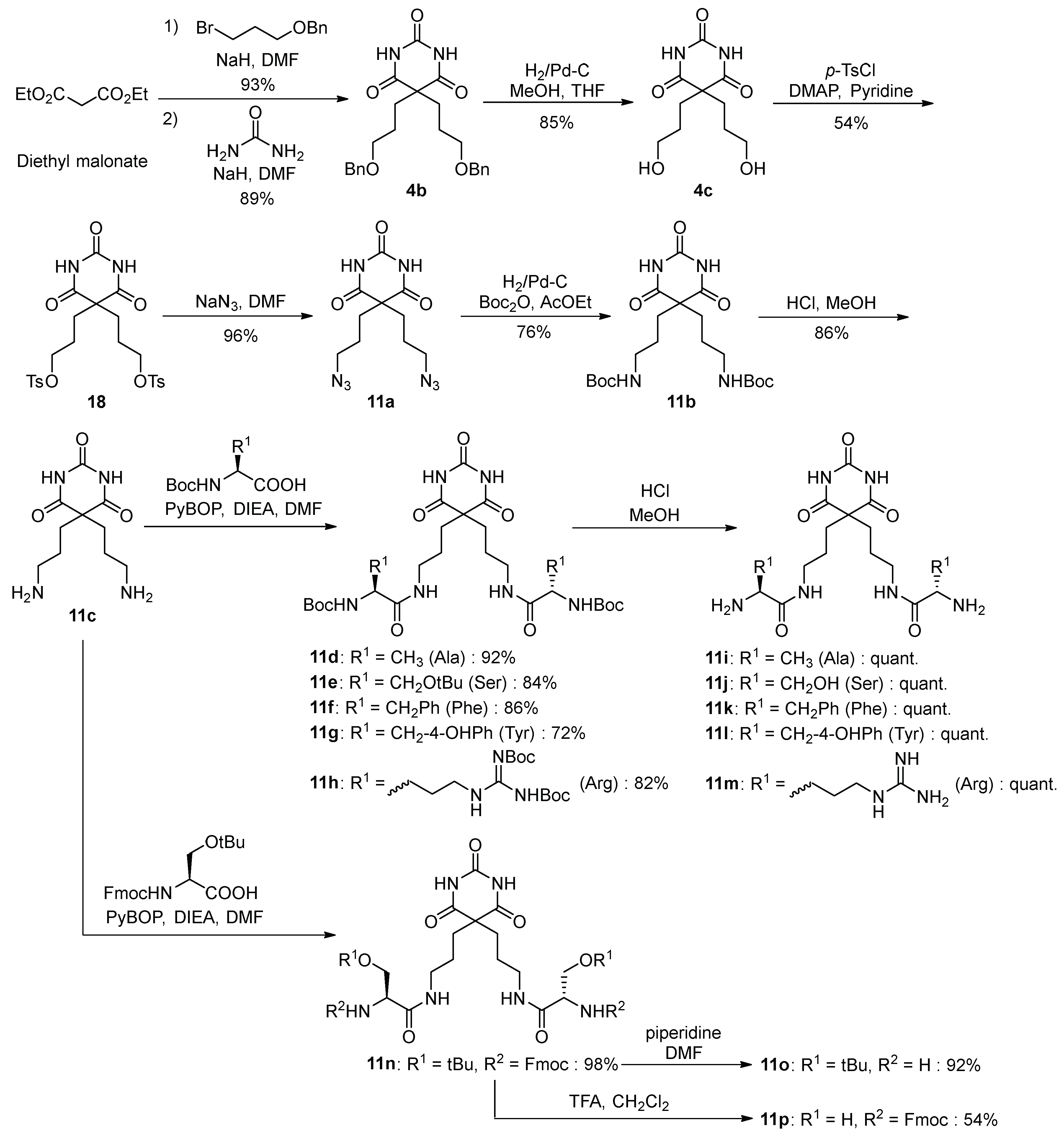
3.1. Synthesis of Barbital Derivatives
3.2. Complexation Behavior of 1 (Zn2L1) with Barbital Derivatives and Cu2+ by UV/Vis Titrations
3.3. Location of Complexes 13 and 16 in the Two-Phase Solvent System, as Determined by UV/Vis Spectra
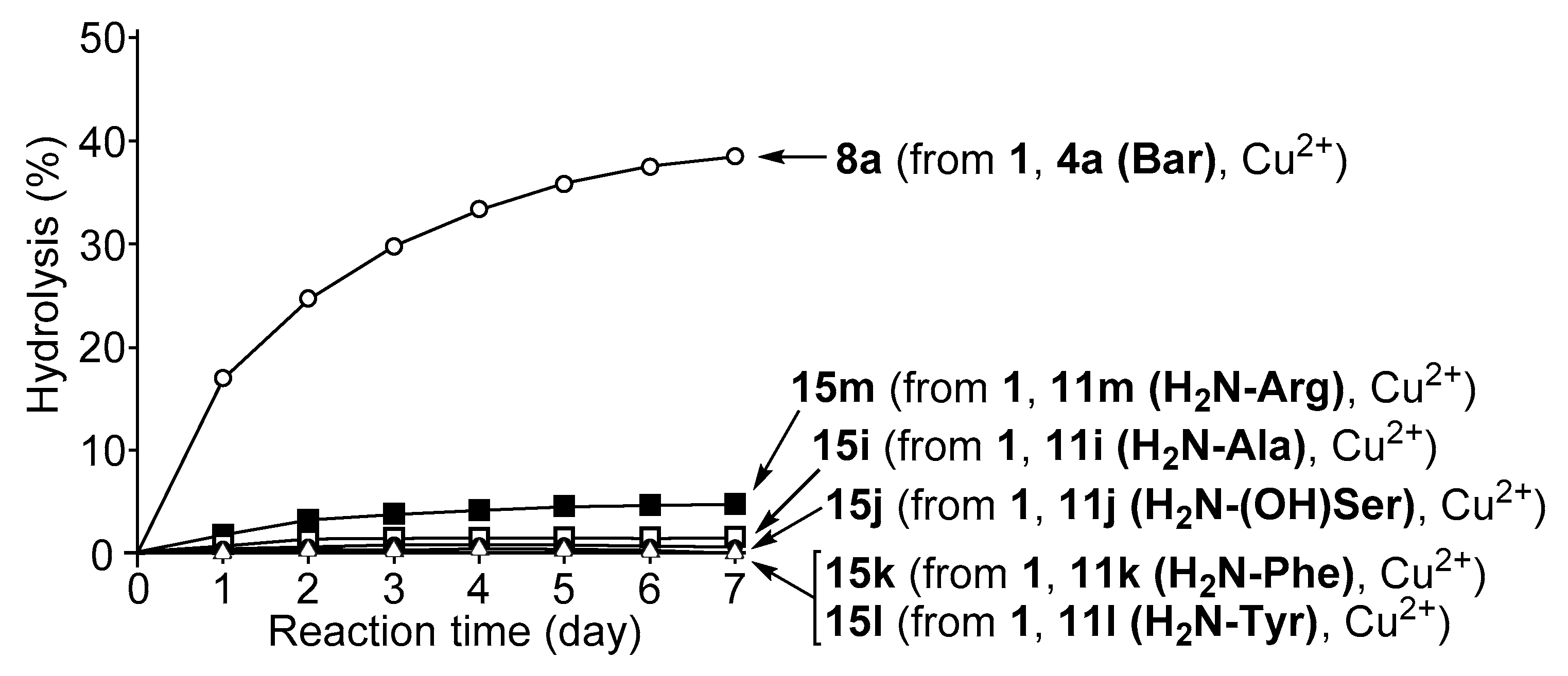
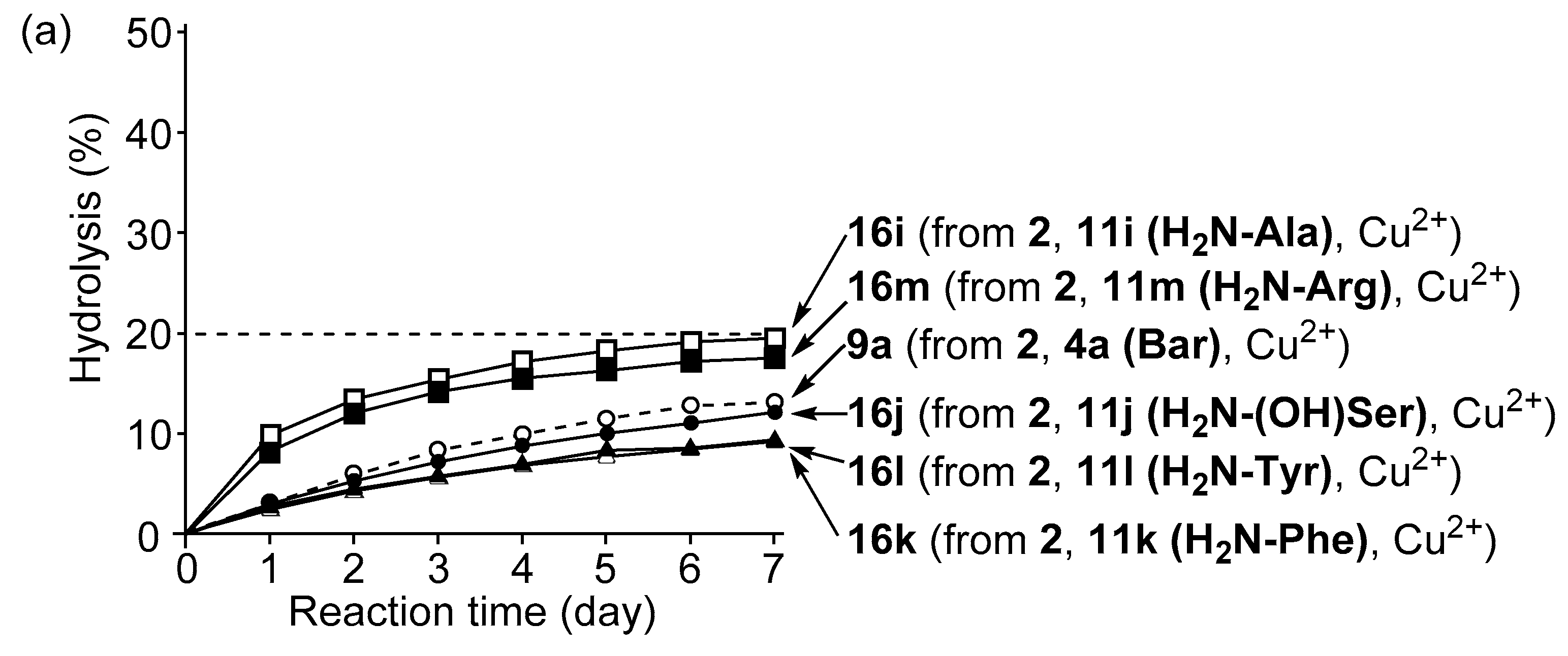
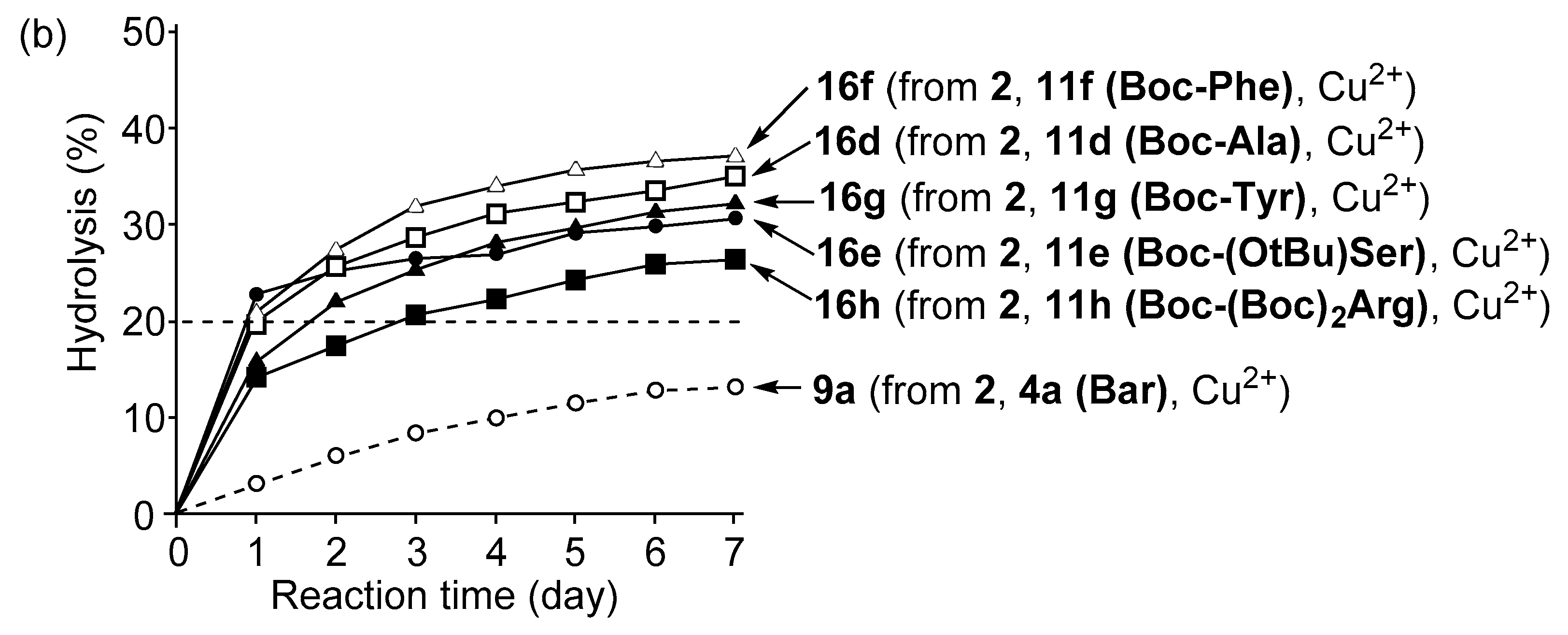
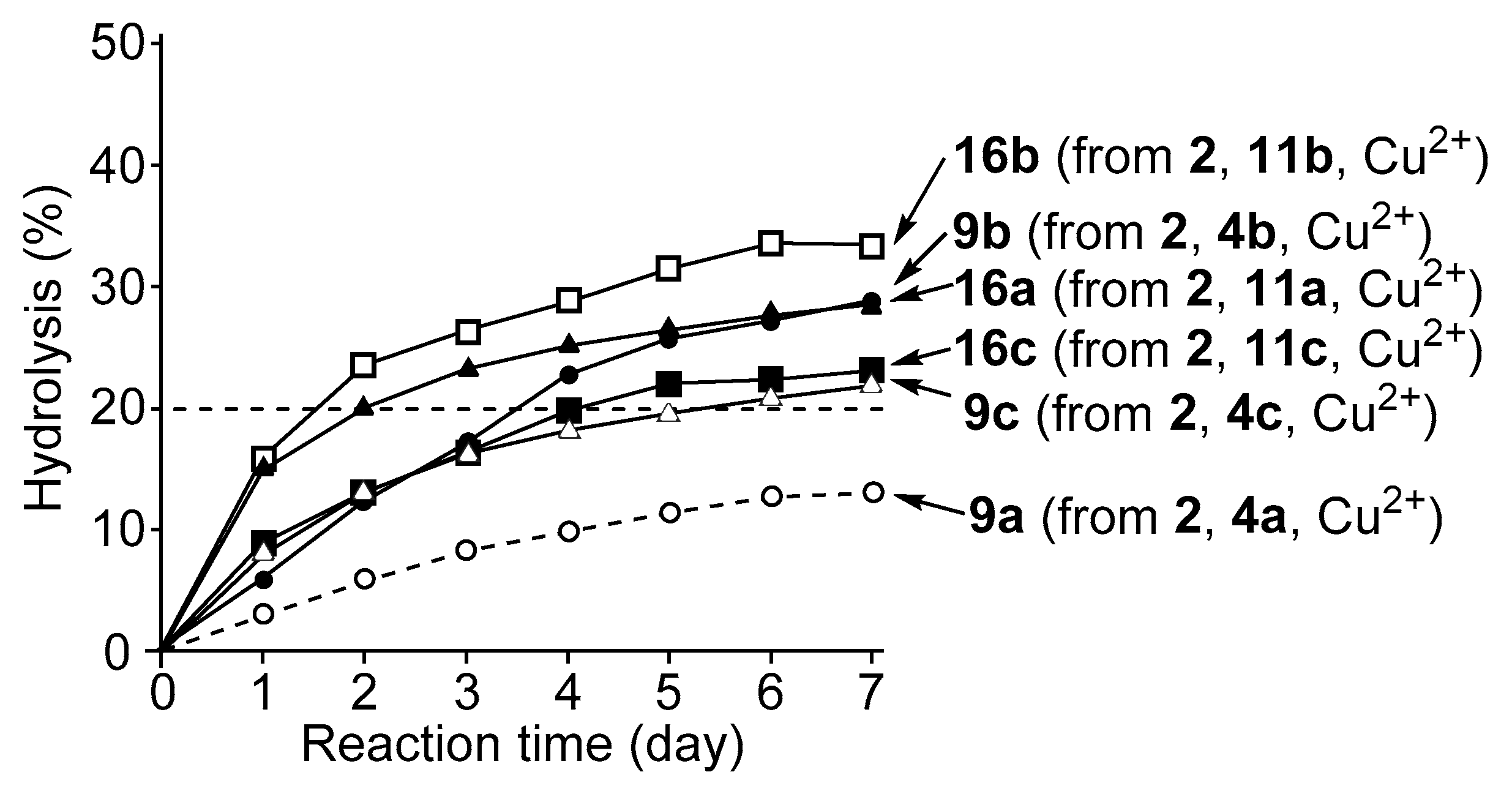
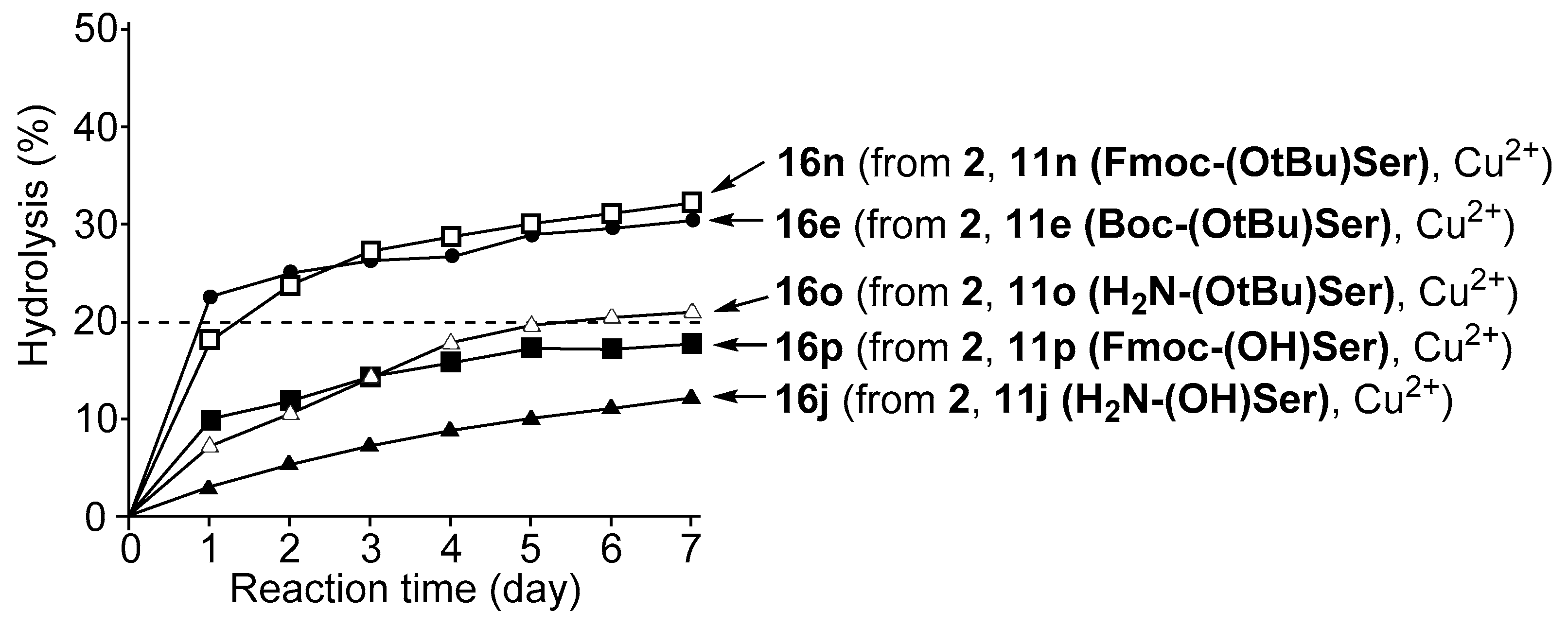
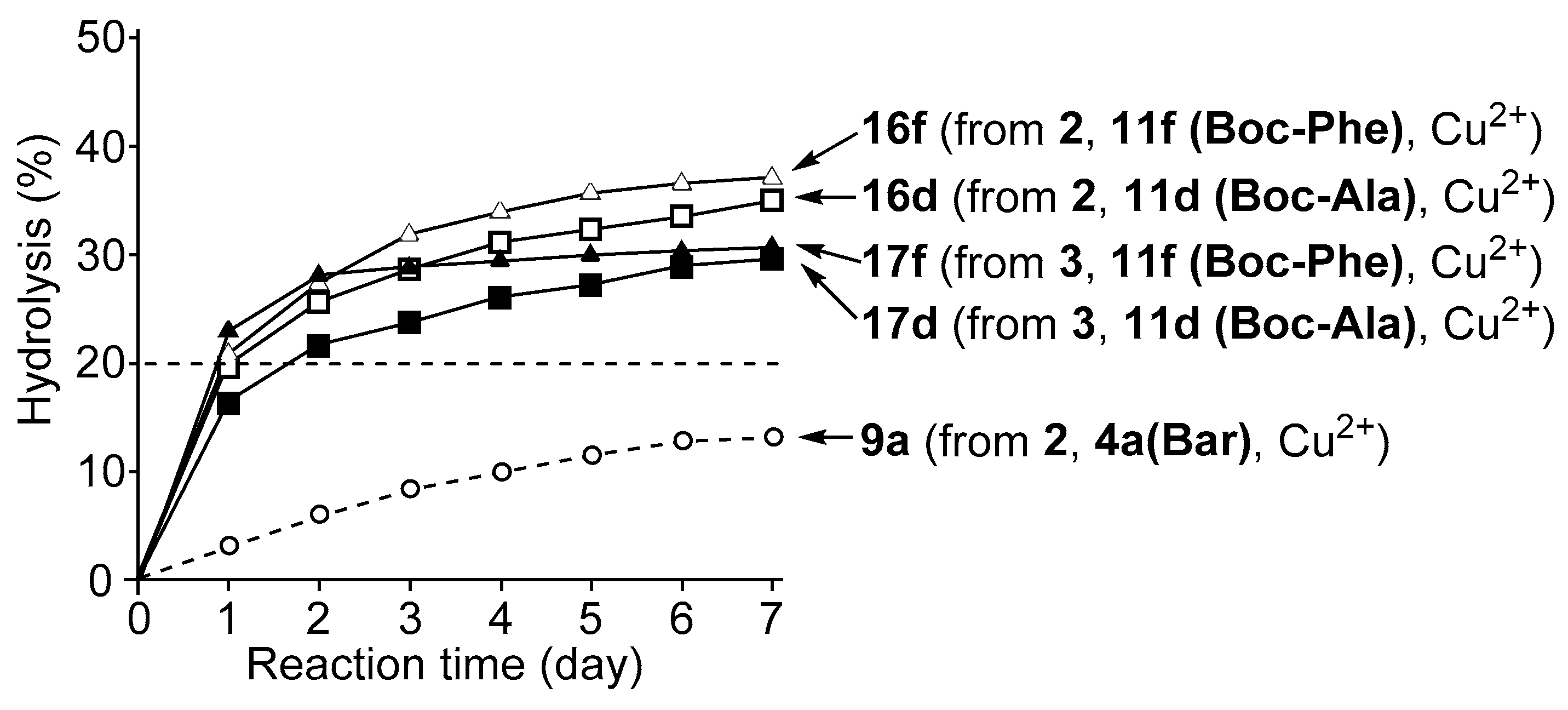
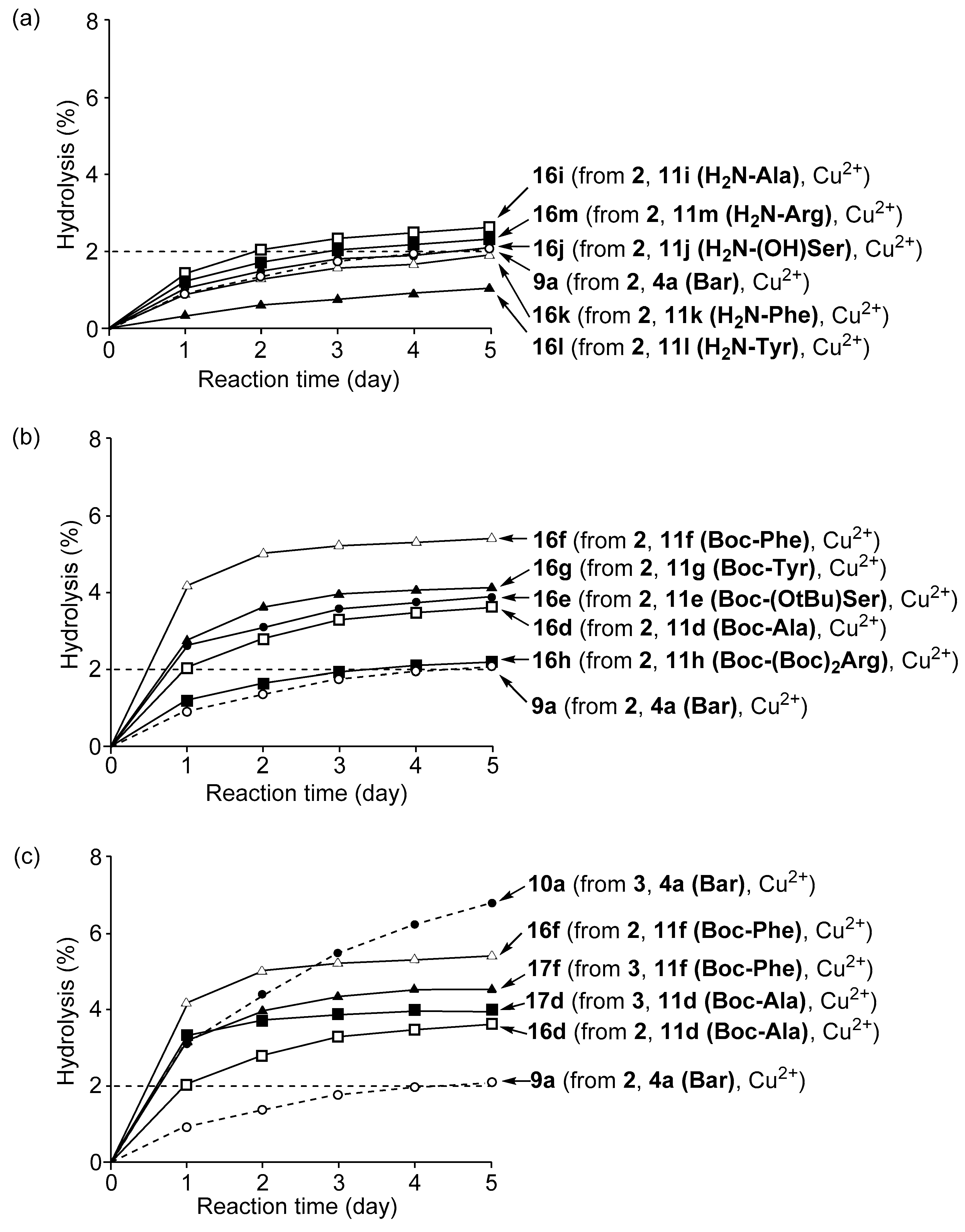
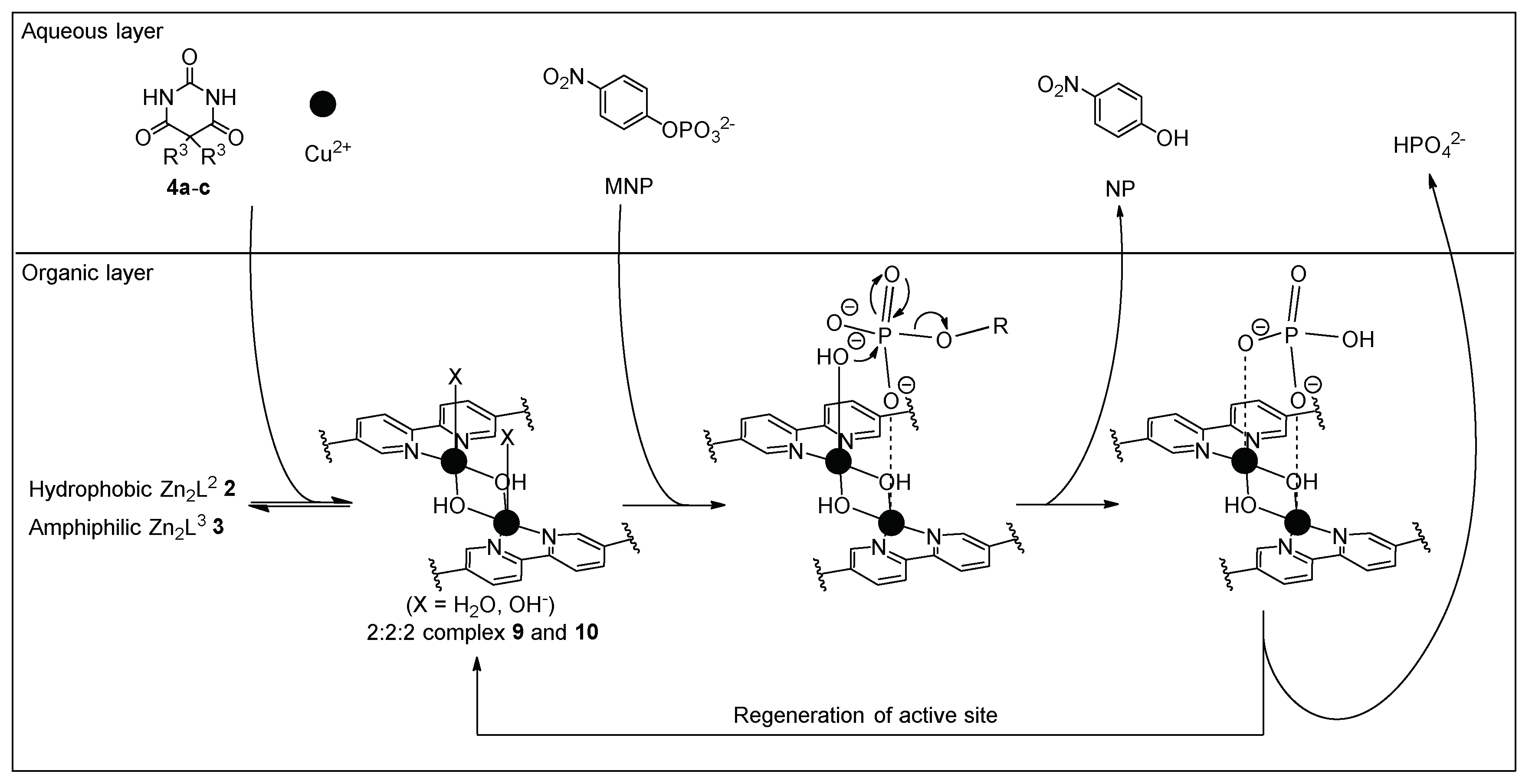
3.4. Hydrolysis of MNP by 2:2:2 Complexes in a Two-Phase Solvent System
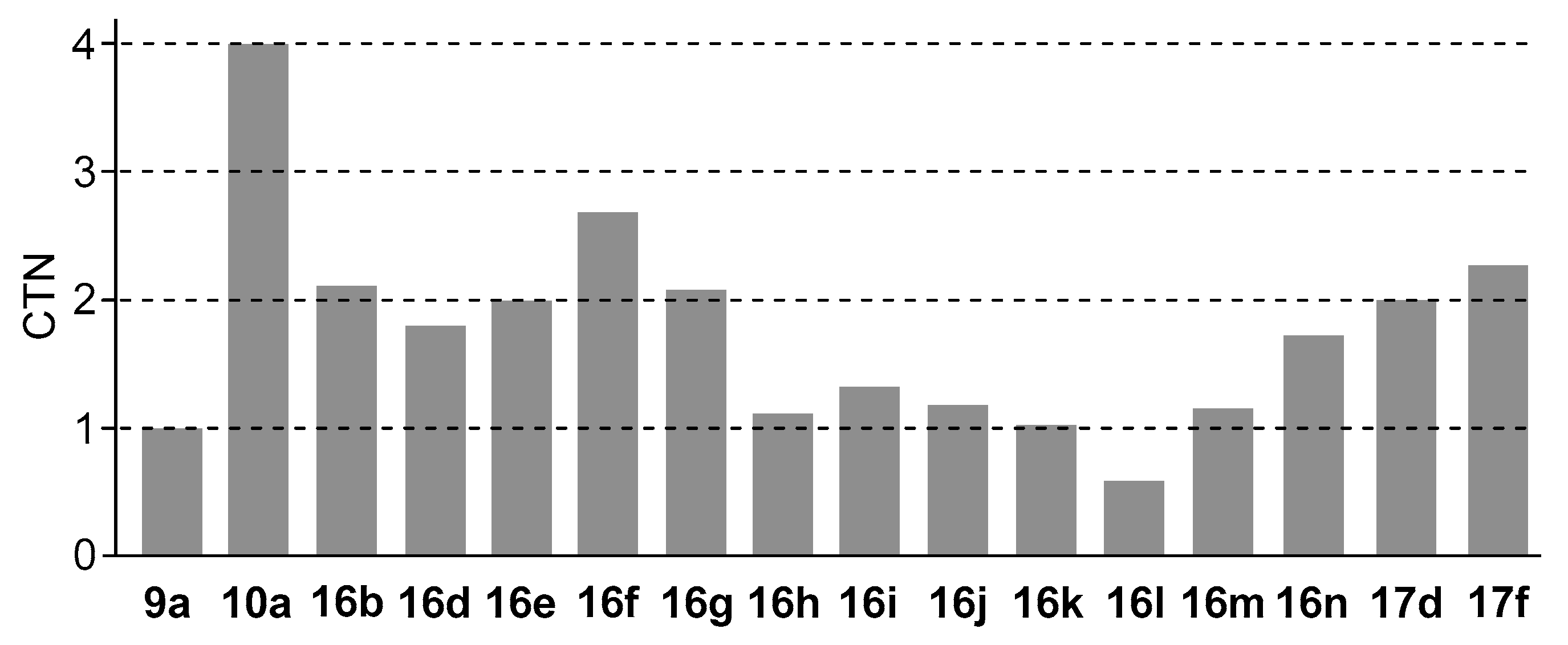
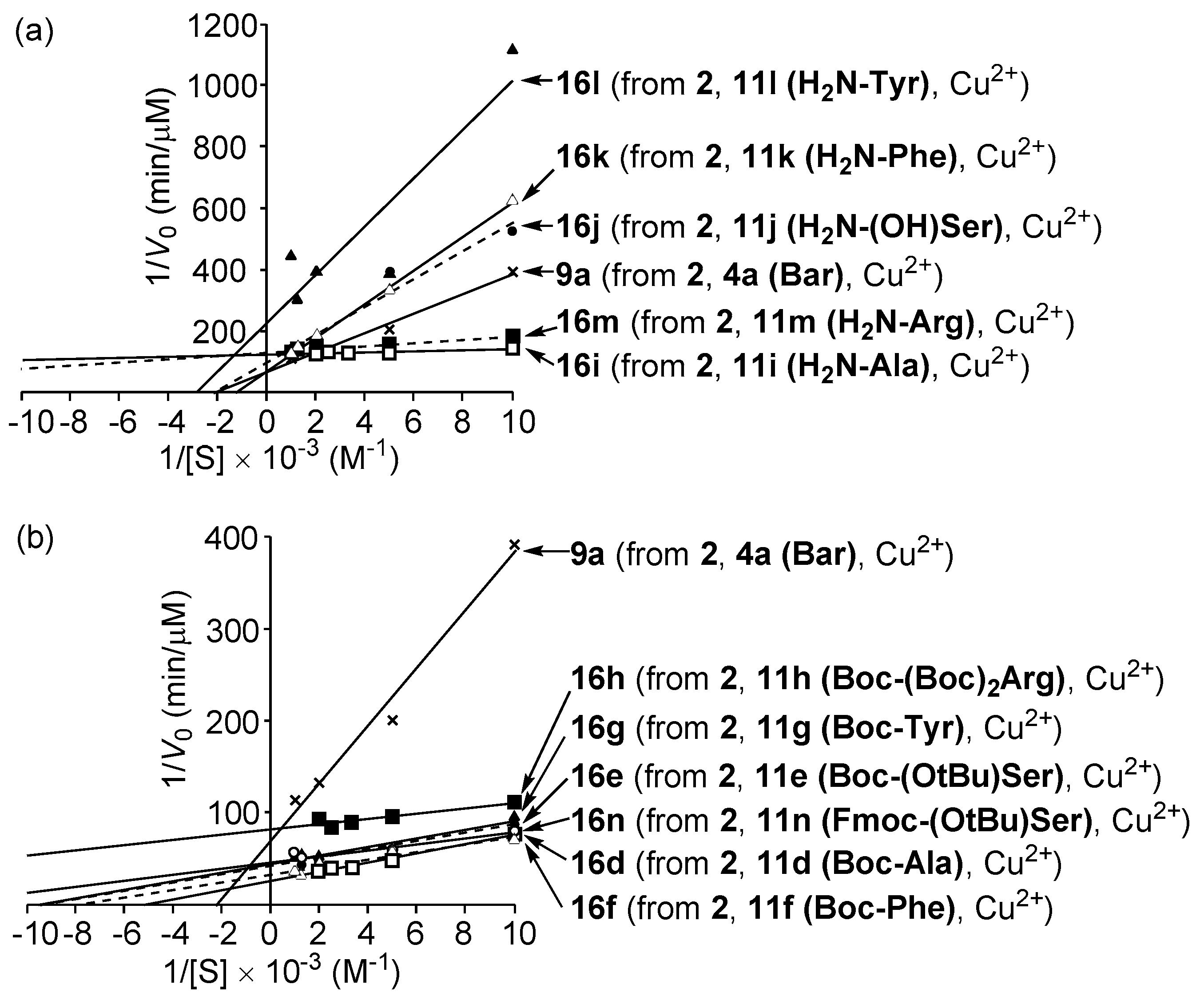
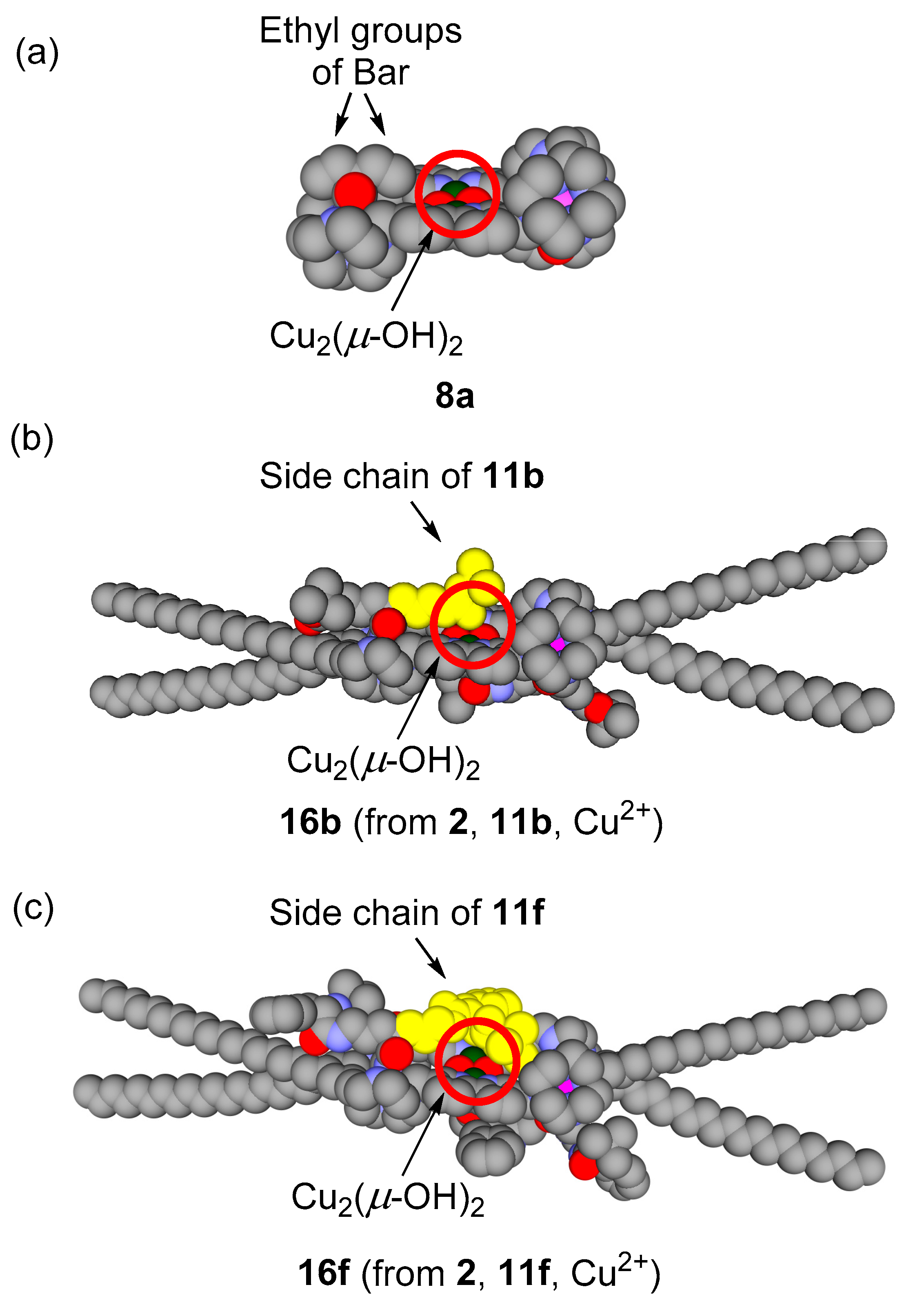
3.5. Michaelis–Menten Kinetics for Hydrolysis of MNP by 9, 16, and 17 in the Two-Phase Solvent System
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Woodgett, J. Protein Kinase Functions, Frontiers in Molecular Biology; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Tibes, R.; Trent, J.; Kurzrock, R. Tyrosine kinase inhibitors and the dawn of molecular cancer therapeutics. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 357–384. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.E. Structure and mechanism of alkaline phosphatase. Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 441–483. [Google Scholar] [CrossRef] [PubMed]
- Millan, J.L. Alkaline phosphatases structure, substrate specificity and functional relatedness to other members of a large superfamily of enzymes. Purinergic Signal. 2006, 2, 335–341. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.J.; Herschlag, D. Functional interrelationships in the alkaline phosphatase superfamily: Phosphodiesterase activity of Escherichia coli alkaline phosphatase. Biochemistry 2001, 40, 5691–5699. [Google Scholar] [PubMed]
- Denu, J.M.; Stuckey, J.A.; Saper, M.A.; Dixon, J.E. Form and function in protein dephosphorylation. Cell 1996, 87, 361–364. [Google Scholar] [CrossRef]
- Kramer, R. Bioinorganic models for the catalytic cooperation of metal ions and functional groups in nuclease and peptidase enzymes. Coord. Chem. Rev. 1999, 182, 243–261. [Google Scholar] [CrossRef]
- Cleland, W.W.; Hengge, A.C. Enzymatic mechanisms of phosphate and sulfate transfer. Chem. Rev. 2006, 106, 3252–3278. [Google Scholar] [CrossRef]
- Kim, E.E.; Wyckoff, H.W. Reaction mechanism of alkaline phosphatase based on crystal structures. J. Mol. Biol. 1991, 218, 449–464. [Google Scholar] [CrossRef]
- Kimura, E. Dimetallic hydrolases and their models. Curr. Opin. Chem. Biol. 2000, 4, 207–213. [Google Scholar] [CrossRef]
- Aoki, S.; Kimura, E. Bio-Coordination Chemistry in Comprehensive Coordination Chemistry II; Que, L., Jr., Tolman, W.B., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 8, pp. 601–640. [Google Scholar]
- Seo, J.S.; Sung, N.-D.; Hynes, R.C.; Chin, J. Structure and reactivity of a dinuclear cobalt(III) complex with a bridging phosphate monoester. Inorg. Chem. 1996, 35, 7472–7473. [Google Scholar] [CrossRef]
- Williams, N.H.; Lebuis, A.-M.; Chin, J. A structural and functional model of dinuclear metallophosphatases. J. Am. Chem. Soc. 1999, 121, 3341–3348. [Google Scholar] [CrossRef]
- Williams, N.H.; Takasaki, B.; Wall, M.; Chin, J. Structure and nuclease activity of simple dinuclear metal complexes: Quantitative dissection of the role of metal ions. Acc. Chem. Res. 1999, 32, 485–493. [Google Scholar] [CrossRef]
- Vance, D.H.; Czarnik, A.W. Functional group convergency in a binuclear dephosphorylation reagent. J. Am. Chem. Soc. 1993, 115, 12165–12166. [Google Scholar] [CrossRef]
- Koike, T.; Inoue, M.; Kimura, E.; Shiro, M. Novel properties of cooperative dinuclear zinc(II) ions: The selective recognition of phosphomonoesters and their P−O ester bond cleavage by a new dinuclear zinc(II) cryptate. J. Am. Chem. Soc. 1996, 118, 3091–3099. [Google Scholar] [CrossRef]
- Hettich, R.; Schneider, H.-J. Cobalt(III) polyamine complexes as catalysts for the hydrolysis of phosphate esters and of DNA. A measurable 10 million-fold rate increase. J. Am. Chem. Soc. 1997, 119, 5638–5647. [Google Scholar] [CrossRef]
- Zulkefeli, M.; Suzuki, A.; Shiro, M.; Hisamatsu, Y.; Kimura, E.; Aoki, S. Selective hydrolysis of phosphate monoester by supramolecular phosphatase formed by the self-assembly of a Bis(Zn2+-cyclen) complex, cyanuric acid, and copper in an aqueous solution (cyclen = 1, 4, 7, 10-Tetraazacyclododecane). Inorg. Chem. 2011, 50, 10113–10123. [Google Scholar] [CrossRef] [PubMed]
- Der, B.S.; Edwards, D.R.; Kuhlman, B. Catalysis by a de novo zinc-mediated protein interface: Implications for natural enzyme evolution and rational enzyme engineering. Biochemistry 2012, 51, 3933–3940. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, Y.; Zheng, X.; Phillips, D.L.; Zhao, C. Mechanistic investigation on the cleavage of phosphate monoester catalyzed by unsymmetrical macrocyclic dinuclear complexes: The selection of metal centers and intrinsic flexibility of the ligand. Inorg. Chem. 2014, 53, 3354–3361. [Google Scholar] [CrossRef]
- Xue, S.-S.; Zhao, M.; Ke, Z.-F.; Cheng, B.-C.; Su, H.; Cao, H.; Cao, K.C.; Wang, J.; Ji, L.-N.; Mao, Z.-W. Enantioselective hydrolysis of amino acid esters promoted by Bis(ß-cyclodextrin) copper complexes. Sci. Rep. 2016, 6, 22080. [Google Scholar] [CrossRef]
- Lin, Y.-W. Rational design of metalloenzymes: From single to multiple active sites. Coord. Chem. Rev. 2017, 336, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Zulkefeli, M.; Hisamatsu, Y.; Suzuki, A.; Miyazawa, Y.; Shiro, M.; Aoki, S. Supramolecular phosphatases formed by the self-assembly of the Bis(Zn2+-cyclen) complex, copper(II), and barbital derivatives in water. Chem. Asian J. 2014, 9, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Coon, M.M.; Rebek, J., Jr. Self-assembling capsules. Chem. Rev. 1997, 97, 1647–1668. [Google Scholar] [CrossRef]
- Fujita, M. Molecular Self-Assembly Organic Versus Inorganic Approaches; Springer: Berlin, Germany, 2000; Volume 96. [Google Scholar]
- Breit, B. Supramolecular approaches to generate libraries of chelating bidentate ligands for homogeneous catalysis. Angewandte Chem. Int. Ed. 2005, 44, 6816–6825. [Google Scholar] [CrossRef]
- Kleij, A.W.; Reek, J.N.H. Ligand-template directed assembly: An efficient approach for the supramolecular encapsulation of transition-metal catalysts. Chem. Eur. J. 2006, 12, 4218–4227. [Google Scholar] [CrossRef] [PubMed]
- Oshovsky, G.V.; Reinhoudt, D.N.; Verboom, W. Supramolecular chemistry in water. Angewandte Chem. Int. Ed. 2007, 46, 2366–2393. [Google Scholar] [CrossRef] [PubMed]
- Hannon, M.J. Supramolecular DNA recognition. Chem. Soc. Rev. 2007, 36, 280–295. [Google Scholar] [CrossRef]
- Van Leeuwen, P.W.N.M. Supramolecular Catalysis; Wiley-VCH: New York, NY, USA, 2008. [Google Scholar]
- Koblenz, T.S.; Wassenaar, J.; Reek, J.N.H. Reactivity within a confined self-assembled nanospace. Chem. Soc. Rev. 2008, 37, 247–262. [Google Scholar] [CrossRef]
- Suzuki, K.; Tominaga, M.; Kawano, M.; Fujita, M. Self-assembly of an M6L12 coordination cube. Chem. Commun. 2009, 1638–1640. [Google Scholar] [CrossRef]
- Northrop, B.H.; Zheng, Y.-R.; Chi, K.-W.; Stang, P.J. Self-organization in coordination-driven self-assembly. Acc. Chem. Res. 2009, 42, 1554–1563. [Google Scholar] [CrossRef]
- Leung, K.C.-F.; Chak, C.-P.; Lo, C.-M.; Wong, W.-Y.; Xuan, S.; Cheng, C.H.K. pH-Controllable supramolecular systems. Chem. Asian J. 2009, 4, 364–381. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, M.; Klosterman, J.K.; Fujita, M. Functional molecular flasks: New properties and reactions within discrete, self-assembled hosts. Angewandte Chem. Int. Ed. 2009, 48, 3418–3438. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, M.; Fujita, M. Development of unique chemical phenomena within nanometer-sized, self-assembled coordination hosts. Bull. Chem. Soc. Jpn. 2010, 83, 609–618. [Google Scholar] [CrossRef]
- Trabolsi, A.; Khashab, N.; Fahrenbach, A.C.; Friedman, D.C.; Colvin, M.T.; Coti, K.K.; Benitez, D.; Tkatchouk, E.; Olsen, J.-C.; Belowich, M.E.; et al. Radically enhanced molecular recognition. Nat. Chem. 2010, 2, 42–49. [Google Scholar] [CrossRef]
- Meeuwissen, J.; Reek, J.N.H. Supramolecular catalysis beyond enzyme mimics. Nat. Chem. 2010, 2, 615–621. [Google Scholar] [CrossRef]
- Ma, Z.; Moulton, B. Recent advances of discrete coordination complexes and coordination polymers in drug delivery. Coord. Chem. Rev. 2011, 255, 1623–1641. [Google Scholar] [CrossRef]
- Aoki, S.; Zulkefeli, M.; Hisamatsu, Y.; Kitamura, M. Supramolecular host and catalysts formed by the synergistic molecular assembly of multinuclear zinc(II) complexes in aqueous solution. In Synergy in Supramolecular Chemistry; Nabeshima, T., Ed.; CRC: Boca Raton, FL, USA, 2015; pp. 33–56. [Google Scholar]
- Kimura, E.; Koike, T.; Aoki, S. Evolution of ZnII-macrocyclic polyamines to biological probes and supramolecular assembly elements. In Macrocyclic and Supramolecular Chemistry: How Izatt-Christensen Award Winners Shaped the Field; Izatt, R.M., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016; pp. 417–445. [Google Scholar]
- Gibb, C.L.D.; Gibb, B.C. Well-defined, organic nanoenvironments in water: The hydrophobic effect drives a capsular assembly. J. Am. Chem. Soc. 2004, 126, 11408–11409. [Google Scholar] [CrossRef] [PubMed]
- Slagt, V.F.; Kamer, P.C.J.; Van Leeuwen, P.W.N.M.; Reek, J.N.H. Encapsulation of transition metal catalysts by ligand-template directed assembly. J. Am. Chem. Soc. 2004, 126, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Kuil, M.; Soltner, T.; Van Leeuwen, P.W.N.M.; Reek, J.N.H. High-precision catalysts: Regioselective hydroformylation of internal alkenes by encapsulated rhodium complexes. J. Am. Chem. Soc. 2006, 128, 11344–11345. [Google Scholar] [CrossRef] [PubMed]
- Neelakandan, P.P.; Hariharan, M.; Ramaiah, D. A supramolecular ON−OFF−ON fluorescence assay for selective recognition of GTP. J. Am. Chem. Soc. 2006, 128, 11334–11335. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, Y.; Yamaguchi, T.; Yoshizawa, M.; Fujita, M. Unusual [2+4] and [2+2] cycloadditions of arenes in the confined cavity of self-assembled cages. J. Am. Chem. Soc. 2007, 129, 7000–7001. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.; Kaanumalle, L.S.; Jockusch, S.; Gibb, C.L.D.; Gibb, B.C.; Turro, N.J.; Ramamurthy, V. Controlling photoreactions with restricted spaces and weak intermolecular forces: Exquisite selectivity during oxidation of olefins by singlet oxygen. J. Am. Chem. Soc. 2007, 129, 4132–4133. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Yoshizawa, M.; Sato, S.; Fujita, M. Minimal nucleotide duplex formation in water through enclathration in self-assembled hosts. Nat. Chem. 2009, 1, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Gianneschi, N.C.; Nguyen, S.T.; Mirkin, C.A. Signal amplification and detection via a supramolecular allosteric catalyst. J. Am. Chem. Soc. 2005, 127, 1644–1645. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Cho, S.H.; Mulfort, K.L.; Tiede, D.M.; Hupp, J.T.; Nguyen, S.T. Cavity-tailored, self-sorting supramolecular catalytic boxes for selective oxidation. J. Am. Chem. Soc. 2008, 130, 16828–16829. [Google Scholar] [CrossRef]
- Murase, T.; Horiuchi, S.; Fujita, M. Naphthalene Diels−Alder in a self-assembled molecular flask. J. Am. Chem. Soc. 2010, 132, 2866–2867. [Google Scholar] [CrossRef]
- Hisamatsu, Y.; Miyazawa, Y.; Yoneda, K.; Miyauchi, M.; Zulkefeli, M.; Aoki, S. Supramolecular complexes formed by the self-assembly of hydrophobic Bis(Zn2+-cyclen) complexes, copper, and Di- or Triimide units for the hydrolysis of phosphate Mono- and diesters in two-phase solvent systems (Cyclen = 1, 4, 7, 10-Tetraazacyclododecane). Chem. Pharm. Bull. 2016, 64, 451–464. [Google Scholar] [CrossRef]
- Rahman, A.B.; Imafuku, Y.; Miyazawa, Y.; Kalfe, A.; Sakai, H.; Saga, Y.; Aoki, S. Catalytic hydrolysis of phosphate monoester by supramolecular phosphatases formed from a monoalkylated dizinc(II) complex, cyclic diimide units, and copper(II) in two-phase solvent system. Inorg. Chem. 2019, 58, 5603–5616. [Google Scholar] [CrossRef]
- Nilsson, B.L.; Soellner, M.B.; Raines, R.T. Chemical synthesis of proteins. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 91–118. [Google Scholar] [CrossRef]
- Van Berkel, S.S.; Van Eldijk, M.B.; Van Hest, J.C.M. Staudinger ligation as a method for bioconjugation. Angewandte Chem. Int. Ed. 2011, 50, 8806–8827. [Google Scholar] [CrossRef]
- Sagel, I.H. Biochemical Calculations, 2nd ed.; John Wiley & Sons: New York, NY, USA, 1976. [Google Scholar]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| - | In H2O Layer (%) | In CHCl3 Layer (%) |
|---|---|---|
| 5aa | >98 | <2 |
| 6a | 1 | 99 |
| 7aa | 35 | 65 |
| 13d | 2 | 98 |
| 13f | 4 | 96 |
| 13i | <1 | >99 |
| 13k | <1 | >99 |
| Entry | Cat. a | Vmax (μM min−1) | Km (μM) | k (min−1) b | Ki (μM) | Km/Ki | CTN c |
|---|---|---|---|---|---|---|---|
| 1 | 8ad | (8.9 ± 0.2) × 10−2 d | (4.1 ± 0.3) × 102 d | (2.2 ± 0.2) × 10−4 d | ca. 15 (mixed-type) d | ca. 27 | 0.4 |
| 2 | APe | 1.3 ± 0.1 e | 7 ± 4 e | (2.9 ± 1.8) × 10−1 e | 3 ± 1 (competitive) e | ca. 2.3 | >103 f |
| 3 | 9ag | (1.4 ± 0.4) × 10−2 g | (5.4 ± 0.5) × 102 g | (2.7 ± 1.0) × 10−5 g | ca. 15 (competitive) g,h | ca. 36 | 1.0 |
| 4 | 10ah | (6.8 ± 0.3) × 10−2 h | (3.8 ± 0.2) × 102 h | (1.8 ± 0.2) × 10−4 h | ca. 80 (mixed-type) h | ca. 4.8 h | ~4 |
| 5 | 16bi | (3.6 ± 0.2) × 10−2 | (2.5 ± 0.3) × 102 | (1.4 ± 0.3) × 10−4 | n.d. j | n.d. j | 2.1 |
| 6 | 16di | (3.9 ± 0.2) × 10−2 | (1.9 ± 0.1) × 102 | (2.1 ± 0.2) × 10−4 | 16 (competitive) | ca. 12 | 1.8 |
| 7 | 16ei | (2.4 ± 0.2) × 10−2 | (1.1 ± 0.1) × 102 | (2.2 ± 0.4) × 10−4 | n.d. j | n.d. j | 2.0 |
| 8 | 16fi | (2.9 ± 0.1) × 10−2 | (1.2 ± 0.1) × 102 | (2.4 ± 0.3) × 10−4 | 23 (competitive) | ca. 5.4 | 2.7 |
| 9 | 16gi | (2.3 ± 0.2) × 10−2 | (1.0 ± 0.1) × 102 | (2.4 ± 0.4) × 10−4 | n.d. j | n.d. j | 2.1 |
| 10 | 16hi | (1.2 ± 0.1) × 10−2 | 33 ± 1 | (3.6 ± 0.5) × 10−4 | n.d. j | n.d. j | 1.1 |
| 11 | 16ii | (8.1 ± 0.2) × 10−3 | 13 ± 1 | (6.0 ± 0.4) × 10−4 | 0.67 (competitive) | ca. 20 | 1.3 |
| 12 | 16ji | (1.0 ± 0.1) × 10−2 | (4.7 ± 0.2) × 102 | (2.2 ± 0.2) × 10−5 | n.d. j | n.d. j | 1.2 |
| 13 | 16ki | (1.4 ± 0.1) × 10−2 | (7.6 ± 0.2) × 102 | (1.9 ± 0.2) × 10−5 | n.d. j | n.d. j | 1.0 |
| 14 | 16li | (4.5 ± 0.2) × 10−3 | (3.5 ± 0.1) × 102 | (1.3 ± 0.1) × 10−5 | n.d. j | n.d. j | 0.6 |
| 15 | 16mi | (7.8 ± 0.3) × 10−3 | 40 ± 1 | (2.0 ± 0.1) × 10−4 | n.d. j | n.d. j | 1.2 |
| 16 | 16ni | (2.2 ± 0.2) × 10−2 | 72 ± 2 | (3.0 ± 0.3) × 10−4 | n.d. j | n.d. j | 1.7 |
| 17 | 17di | (2.5 ± 0.2) × 10−2 | (1.2 ± 0.1) × 102 | (2.2 ± 0.4) × 10−4 | 17 (mixed-type) | ca. 7.4 | 2.0 |
| 18 | 17fi | (2.3 ± 0.2) × 10−2 | 47 ± 2 | (5.0 ± 0.6) × 10−4 | 5.7 (competitive) | ca. 8.3 | 2.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyazawa, Y.; Rahman, A.B.; Saga, Y.; Imafuku, H.; Hisamatsu, Y.; Aoki, S. Catalytic Hydrolysis of Phosphate Monoester by Supramolecular Complexes Formed by the Self-Assembly of a Hydrophobic Bis(Zn2+-cyclen) Complex, Copper, and Barbital Units That Are Functionalized with Amino Acids in a Two-Phase Solvent System. Micromachines 2019, 10, 452. https://doi.org/10.3390/mi10070452
Miyazawa Y, Rahman AB, Saga Y, Imafuku H, Hisamatsu Y, Aoki S. Catalytic Hydrolysis of Phosphate Monoester by Supramolecular Complexes Formed by the Self-Assembly of a Hydrophobic Bis(Zn2+-cyclen) Complex, Copper, and Barbital Units That Are Functionalized with Amino Acids in a Two-Phase Solvent System. Micromachines. 2019; 10(7):452. https://doi.org/10.3390/mi10070452
Chicago/Turabian StyleMiyazawa, Yuya, Akib Bin Rahman, Yutaka Saga, Hiroki Imafuku, Yosuke Hisamatsu, and Shin Aoki. 2019. "Catalytic Hydrolysis of Phosphate Monoester by Supramolecular Complexes Formed by the Self-Assembly of a Hydrophobic Bis(Zn2+-cyclen) Complex, Copper, and Barbital Units That Are Functionalized with Amino Acids in a Two-Phase Solvent System" Micromachines 10, no. 7: 452. https://doi.org/10.3390/mi10070452
APA StyleMiyazawa, Y., Rahman, A. B., Saga, Y., Imafuku, H., Hisamatsu, Y., & Aoki, S. (2019). Catalytic Hydrolysis of Phosphate Monoester by Supramolecular Complexes Formed by the Self-Assembly of a Hydrophobic Bis(Zn2+-cyclen) Complex, Copper, and Barbital Units That Are Functionalized with Amino Acids in a Two-Phase Solvent System. Micromachines, 10(7), 452. https://doi.org/10.3390/mi10070452